Representation of the Structure—A Key Point of Building QSAR/QSPR Models for Ionic Liquids



Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Data and Molecular Descriptors
2.2. Model Development
2.3. Validation Process
2.4. Applicability Domain
3. Results and Discussion
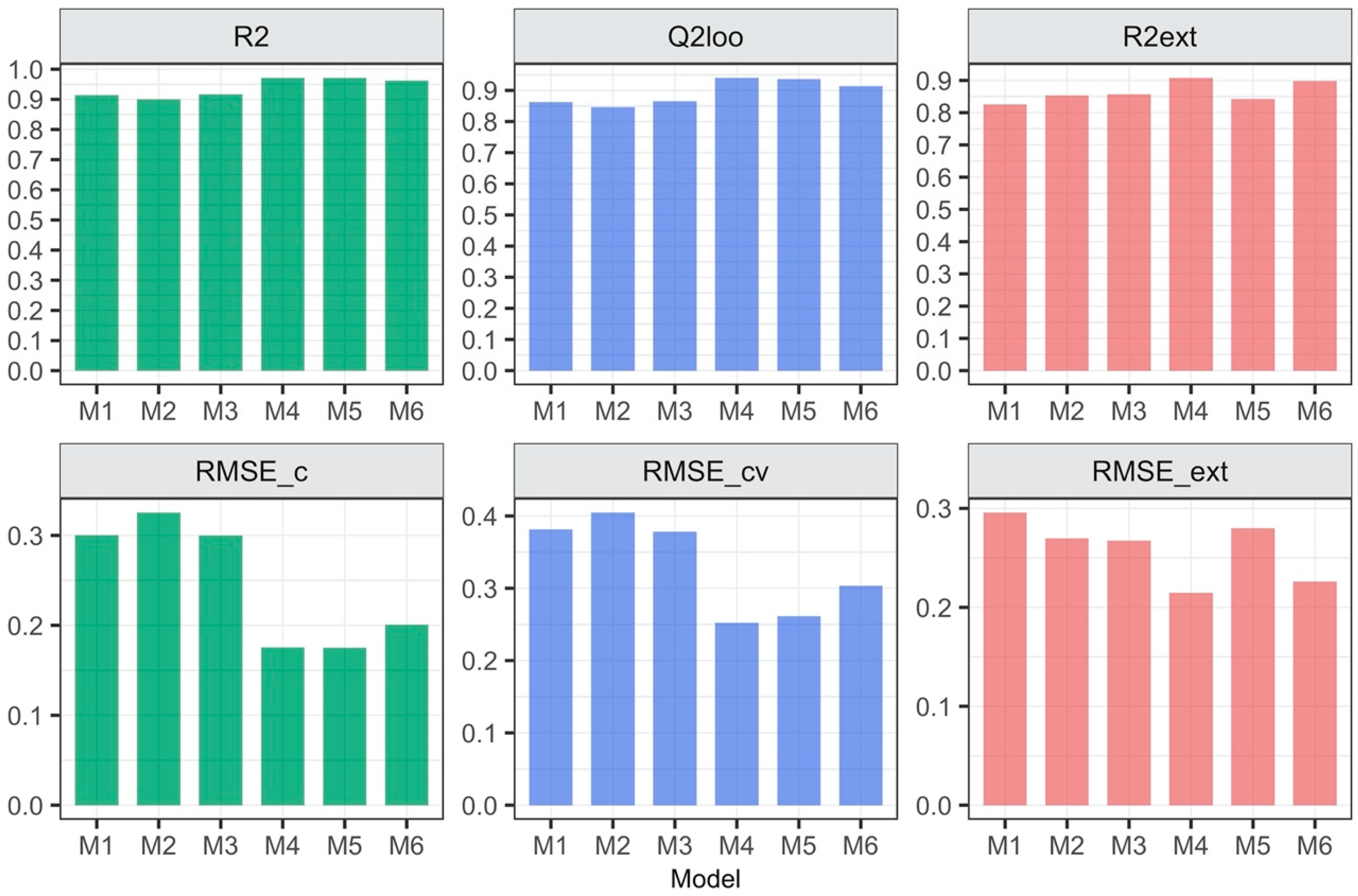
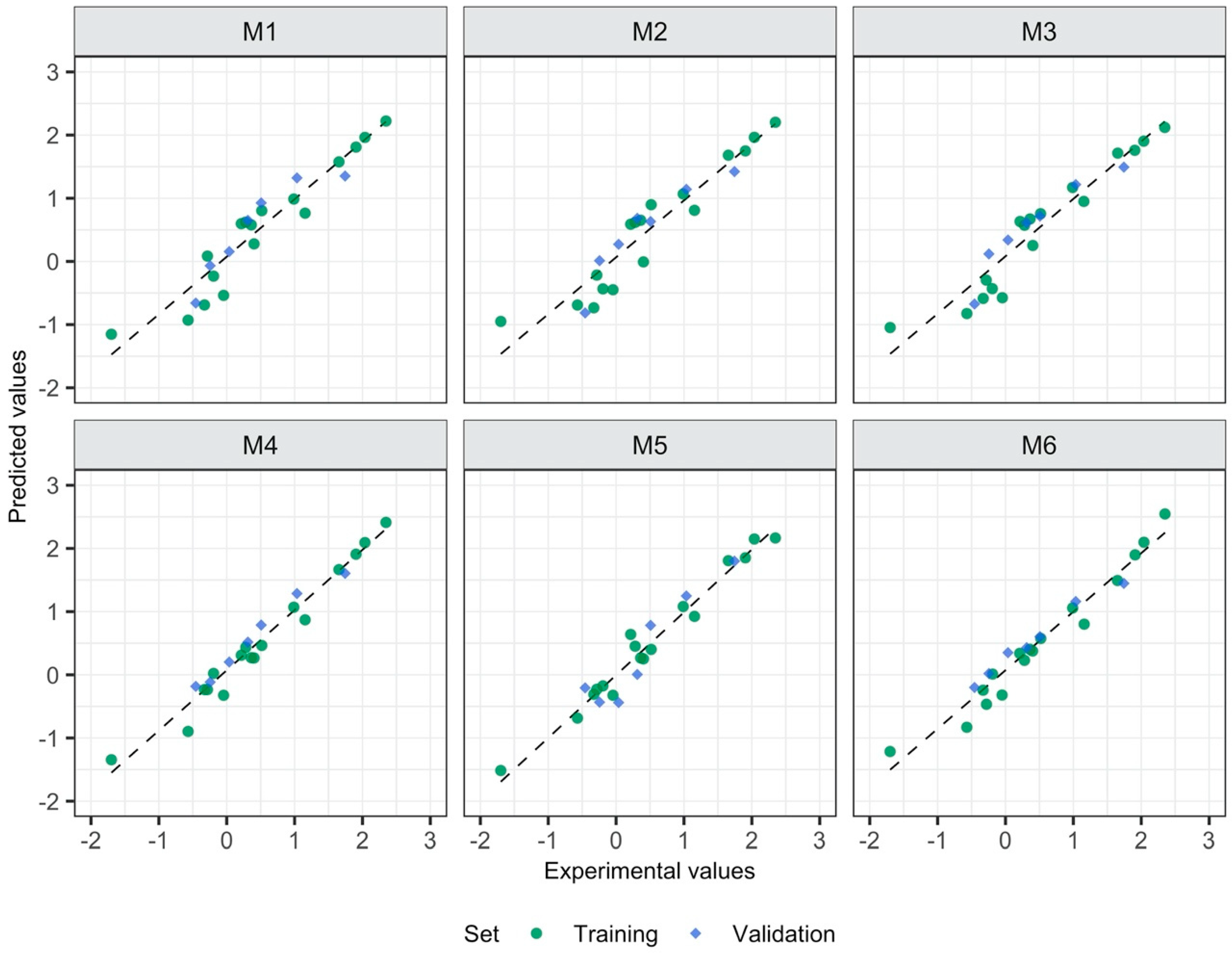
3.1. Relationship between the Form of Structure Representation and the Model Quality
3.2. Influence of Structure Representation on the Variable Selection
3.3. Influence of the Presence of the Second ion on Reliability of the Applicability Domain Assessment
3.4. Other Practical Aspects of ILs Modeling with QSAR/QSPR
4. Conclusions
- 2D descriptors are suitable to build reliable QSAR models;
- The strategy in which the 2D descriptors were calculated for the whole ionic liquid allowed to build the model with the highest quality;
- More precise description of the ionic liquid’s structure (through 3D descriptors calculated for ions or geometry optimization of ionic pairs followed by descriptors calculation) does not guarantee the better accuracy and predictive ability of the developed model;
- Models based on 2D descriptors are easier to apply and reproduce, even by non-experts in computational chemistry, which could lead to an increase of the application of in silico methods in various R&D areas.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Das, R.N.; Roy, K. Advances in QSPR/QSTR models of ionic liquids for the design of greener solvents of the future. Mol. Divers. 2013, 17, 151–196. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.T.D. Quantitative Structure–Activity Relationships (QSARs)—Applications and methodology. In Recent Advances in QSAR Studies. Methods and Applications; Puzyn, T., Leszczynski, J., Cronin, M.T.D., Eds.; Springer: Dordrecht, The Netherlands; New York, NY, USA, 2010; pp. 3–11. ISBN 97814020978291402097824. [Google Scholar]
- Peric, B.; Sierra, J.; Martí, E.; Cruañas, R.; Garau, M.A. Quantitative structure-activity relationship (QSAR) prediction of (eco)toxicity of short aliphatic protic ionic liquids. Ecotoxicol. Environ. Saf. 2015, 115, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Das, R.N.; Popelier, P.L.A. Quantitative structure-activity relationship for toxicity of ionic liquids to Daphnia magna: Aromaticity vs. lipophilicity. Chemosphere 2014, 112, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-K.K.; Liang, M.-J.J.; Wu, T.-Y.Y.; Wang, H.P. A high correlate and simplified QSPR for viscosity of imidazolium-based ionic liquids. Fluid Phase Equilib. 2013, 350, 37–42. [Google Scholar] [CrossRef]
- Rybinska, A.; Sosnowska, A.; Grzonkowska, M.; Barycki, M.; Puzyn, T. Filling environmental data gaps with QSPR for ionic liquids: Modeling n-octanol/water coefficient. J. Hazard. Mater. 2016, 303, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Das, R.N. Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Elsevier Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2015; ISBN 01280150559780128015056. [Google Scholar]
- Barycki, M.; Sosnowska, A.; Puzyn, T. Which structural features stand behind micelization of ionic liquids? Quantitative Structure-Property Relationship studies. J. Colloid Interface Sci. 2017, 487, 475–483. [Google Scholar] [CrossRef]
- Rybinska, A.; Sosnowska, A.; Barycki, M.; Puzyn, T. Geometry optimization method versus predictive ability in QSPR modeling for ionic liquids. J. Comput. Aided. Mol. Des. 2016, 30, 165–176. [Google Scholar] [CrossRef]
- Muratov, E.N.; Varlamova, E.V.; Artemenko, A.G.; Polishchuk, P.G.; Kuz’min, V.E. Existing and Developing Approaches for QSAR Analysis of Mixtures. Mol. Inform. 2012, 31, 202–221. [Google Scholar] [CrossRef]
- Ghanem, O.B.; Shah, S.N.; Lévêque, J.-M.; Mutalib, M.I.A.; El-Harbawi, M.; Khan, A.S.; Alnarabiji, M.S.; Al-Absi, H.R.H.; Ullah, Z. Study of the antimicrobial activity of cyclic cation-based ionic liquids via experimental and group contribution QSAR model. Chemosphere 2018, 195, 21–28. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. DRAGON software: an easy approach to molecular descriptor. Commun. Math. Comput. Chem. 2006, 56, 237–248. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Mannhold, R., Kubinyi, H., Folkers, G., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; ISBN 978-3-527-31852-0. [Google Scholar]
- Hebbali, A. olsrr: Tools for Building OLS Regression Models. Available online: https://cran.r-project.org/web/packages/olsrr/index.html. (accessed on 5 April 2020).
- OECD. OECD Principles for the Validation, for Regulatory Purposes, of Quantitative Structure-Activity Relationship Models; OECD: Paris, France, 2004. [Google Scholar]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “r(m)(2)” metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Bodai, Z.; Heberger, K. Estimation of influential points in any data set from coefficient of determination and its leave-one-out cross-validated counterpart. J. Comput. Aided. Mol. Des. 2013, 27, 837–844. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicabilty domain estimation by projection of the training set descriptor space: A review. Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Roy, P.P.; Kovarich, S.; Yap, C.W.; Papa, E. QSAR Modeling is not “Push a Button and Find a Correlation”: A Case Study of Toxicity of (Benzo-)triazoles on Algae. Mol. Inform. 2012, 31, 817–835. [Google Scholar] [CrossRef]
- Pogliani, L. Modeling with Molecular Pseudoconnectivity Descriptors. A Useful Extension of the Intrinsic I-State Concept. J. Phys. Chem. A 2000, 104, 9029–9045. [Google Scholar] [CrossRef]
- Ghanem, O.B.; Mutalib, M.; El-Harbawi, M.; Gonfa, G.; Kait, C.F.; Alitheen, N.B.M.; Lévêque, J.-M. Effect of imidazolium-based ionic liquids on bacterial growth inhibition investigated via experimental and QSAR modelling studies. J. Hazard. Mater. 2015, 297, 198–206. [Google Scholar] [CrossRef]
- Cho, C.-W.; Park, J.-S.; Stolte, S.; Yun, Y.-S. Modelling for antimicrobial activities of ionic liquids towards Escherichia coli, Staphylococcus aureus and Candida albicans using linear free energy relationship descriptors. J. Hazard. Mater. 2016, 311, 168–175. [Google Scholar] [CrossRef]
- Cornmell, R.J.; Winder, C.L.; Tiddy, G.J.T.; Goodacre, R.; Stephens, G. Accumulation of ionic liquids in Escherichia coli cells. Green Chem. 2008, 10, 836. [Google Scholar] [CrossRef]
- Forrest, S. Genetic algorithms: principles of natural selection applied to computation. Science 1993, 261, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Chiappe, C.; Focardi, S.E.; Pretti, C.; Renzi, M. Theoretical descriptor for the correlation of aquatic toxicity of ionic liquids by quantitative structure-toxicity relationships. Chem. Eng. J. 2011, 175, 17–23. [Google Scholar] [CrossRef]
- Nekoeinia, M.; Yousefinejad, S.; Abdollahi-Dezaki, A. Prediction of E-N(T) Polarity Scale of Ionic Liquids Using a QSPR Approach. Ind. Eng. Chem. Res. 2015, 54, 12682–12689. [Google Scholar] [CrossRef]
- Torrecilla, J.S.; Palomar, J.; Lemus, J.; Rodríguez, F. A quantum-chemical-based guide to analyze/quantify the cytotoxicity of ionic liquids. Green Chem. 2010, 12, 123–134. [Google Scholar] [CrossRef]
- Wang, C.; Wei, Z.; Wang, L.; Sun, P.; Wang, Z. Assessment of bromide-based ionic liquid toxicity toward aquatic organisms and QSAR analysis. Ecotoxicol. Environ. Saf. 2015, 115, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, B. A review on created QSPR models for predicting ionic liquids properties and their reliability from chemometric point of view. J. Mol. Liq. 2020, 297, 112013. [Google Scholar] [CrossRef]
- Abramenko, N.; Kustov, L.; Metelytsia, L.; Kovalishyn, V.; Tetko, I.; Peijnenburg, W. A review of recent advances towards the development of QSAR models for toxicity assessment of ionic liquids. J. Hazard. Mater. 2020, 384, 121429. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Model’s ID | Type of Descriptors | Descriptors Calculated for: | Equation for Predicting logEC50 [mM] |
|---|---|---|---|
| M1 | 2D | Separate ions | logEC50 = 2.49−0.14 Psi_i_0A − 0.001 SMTIVC |
| M2 | 3D | Separate ions | logEC50 = 2.52−0.12 L1mC − 0.19 L1iA |
| M3 | 2D, 3D | Separate ions | logEC50 = 2.304−0.142 Psi_i_0A − 0.006 QZZmC |
| M4 | 2D | Ionic pair | logEC50 = 4.15−0.001 GMTI − 0.09 MDDD − 0.16 AMW |
| M5 | 3D | Ionic pair | logEC50 = 6.91−0.24 L/Bw − 1.05 RTv + 0.53 L3u |
| M6 | 2D, 3D | Ionic pair | logEC50 = 3.49−0.001 GMTI − 3.21 E1e + 0.04 DISPm |
| Model | Number of All Variables | Anions’ Descriptors | Cations’ Descriptors |
|---|---|---|---|
| M1 | 572 | 278 | 294 |
| M2 | 813 | 352 | 461 |
| M3 | 1385 | 630 | 755 |
| M4 | 298 | 0 | 0 |
| M5 | 414 | 0 | 0 |
| M6 | 712 | 0 | 0 |
| IL | L1iA | L1mC | |
|---|---|---|---|
| [C4mpy] [SCN] | 1.38 | 6.78 | The same anion |
| [C8mim] [SCN] | 1.38 | 18.4 | |
| [C4mpyrr] [NTf2] | 5.67 | 4.48 | The same cation |
| [C4mpyrr] [Br] | 0.00 | 4.48 |
| IL | L/Bw | RTv | L3u |
|---|---|---|---|
| [C4mim] [NTf2] | 1.98 | 6.69 | 2.29 |
| [C4py] [NTf2] | 1.68 | 6.65 | 1.39 |
| [C4mpyrr] [NTf2] | 2.3 | 6.75 | 2.13 |
| [C4mpip] [NTf2] | 2.03 | 6.82 | 1.39 |
| [C8mim] [NTf2] | 2.85 | 7.20 | 2.13 |
| [C8py] [NTf2] | 2.2 | 7.16 | 1.56 |
| [C8mpyrr] [NTf2] | 2.05 | 6.93 | 1.28 |
| [C8mpip] [NTf2] | 2.23 | 7.34 | 1.90 |
| Range | 1.17 | 0.69 | 1.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybińska-Fryca, A.; Sosnowska, A.; Puzyn, T. Representation of the Structure—A Key Point of Building QSAR/QSPR Models for Ionic Liquids. Materials 2020, 13, 2500. https://doi.org/10.3390/ma13112500
Rybińska-Fryca A, Sosnowska A, Puzyn T. Representation of the Structure—A Key Point of Building QSAR/QSPR Models for Ionic Liquids. Materials. 2020; 13(11):2500. https://doi.org/10.3390/ma13112500
Chicago/Turabian StyleRybińska-Fryca, Anna, Anita Sosnowska, and Tomasz Puzyn. 2020. "Representation of the Structure—A Key Point of Building QSAR/QSPR Models for Ionic Liquids" Materials 13, no. 11: 2500. https://doi.org/10.3390/ma13112500
APA StyleRybińska-Fryca, A., Sosnowska, A., & Puzyn, T. (2020). Representation of the Structure—A Key Point of Building QSAR/QSPR Models for Ionic Liquids. Materials, 13(11), 2500. https://doi.org/10.3390/ma13112500