Efficient Calculation Methods for the Diffusion Coefficient of Interstitial Solutes in Dilute Alloys


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
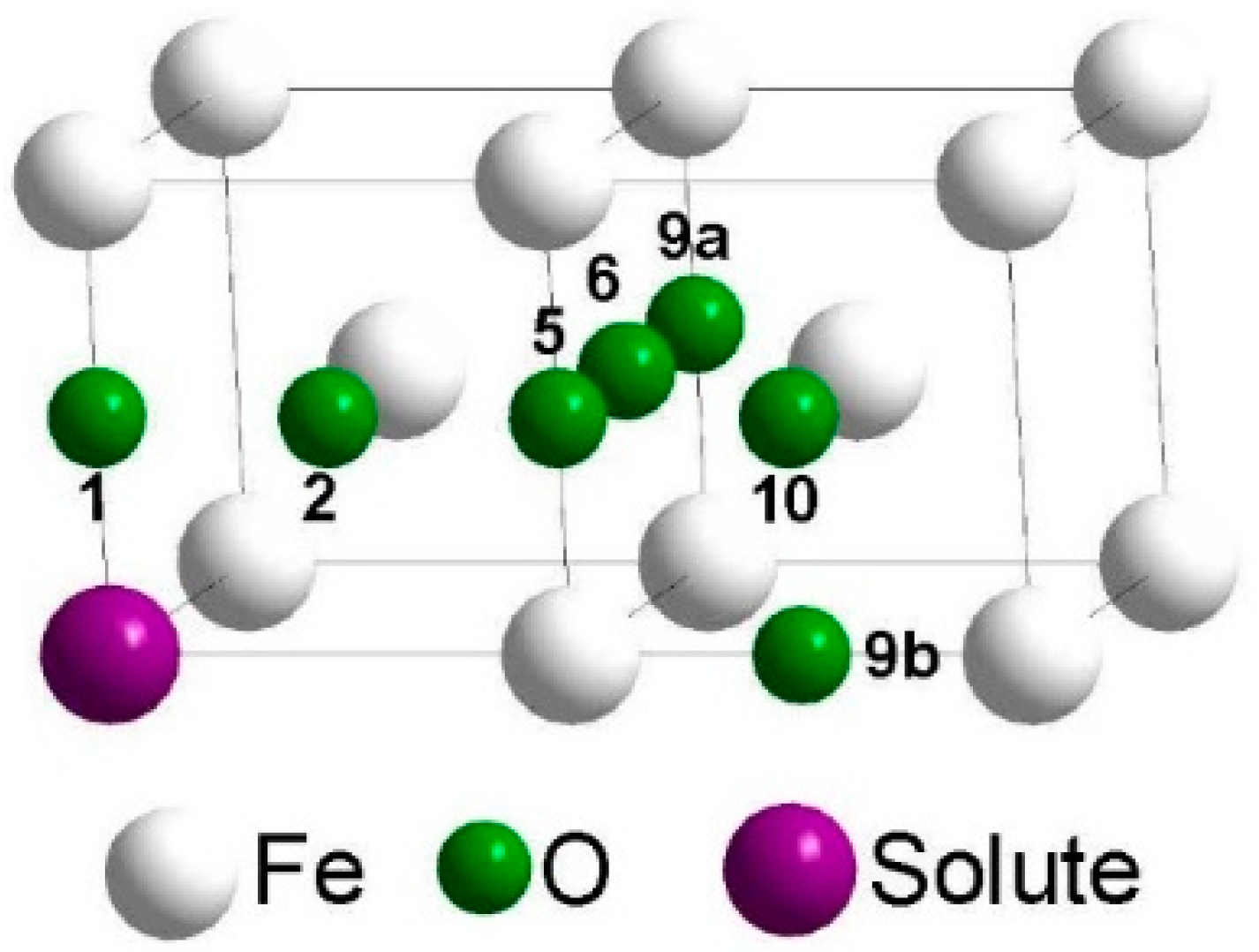
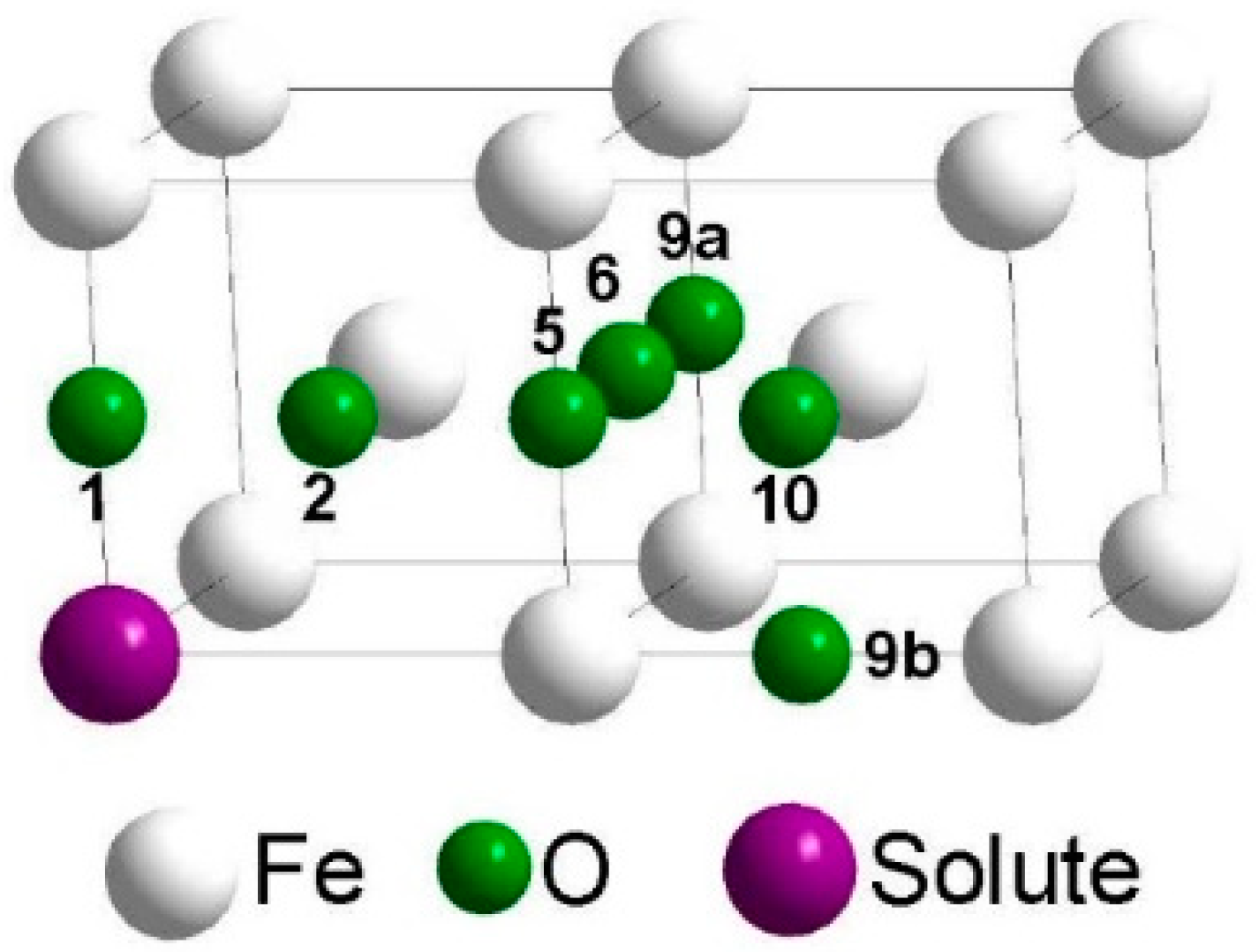
2. Calculation Methods
3. Results and Discussion
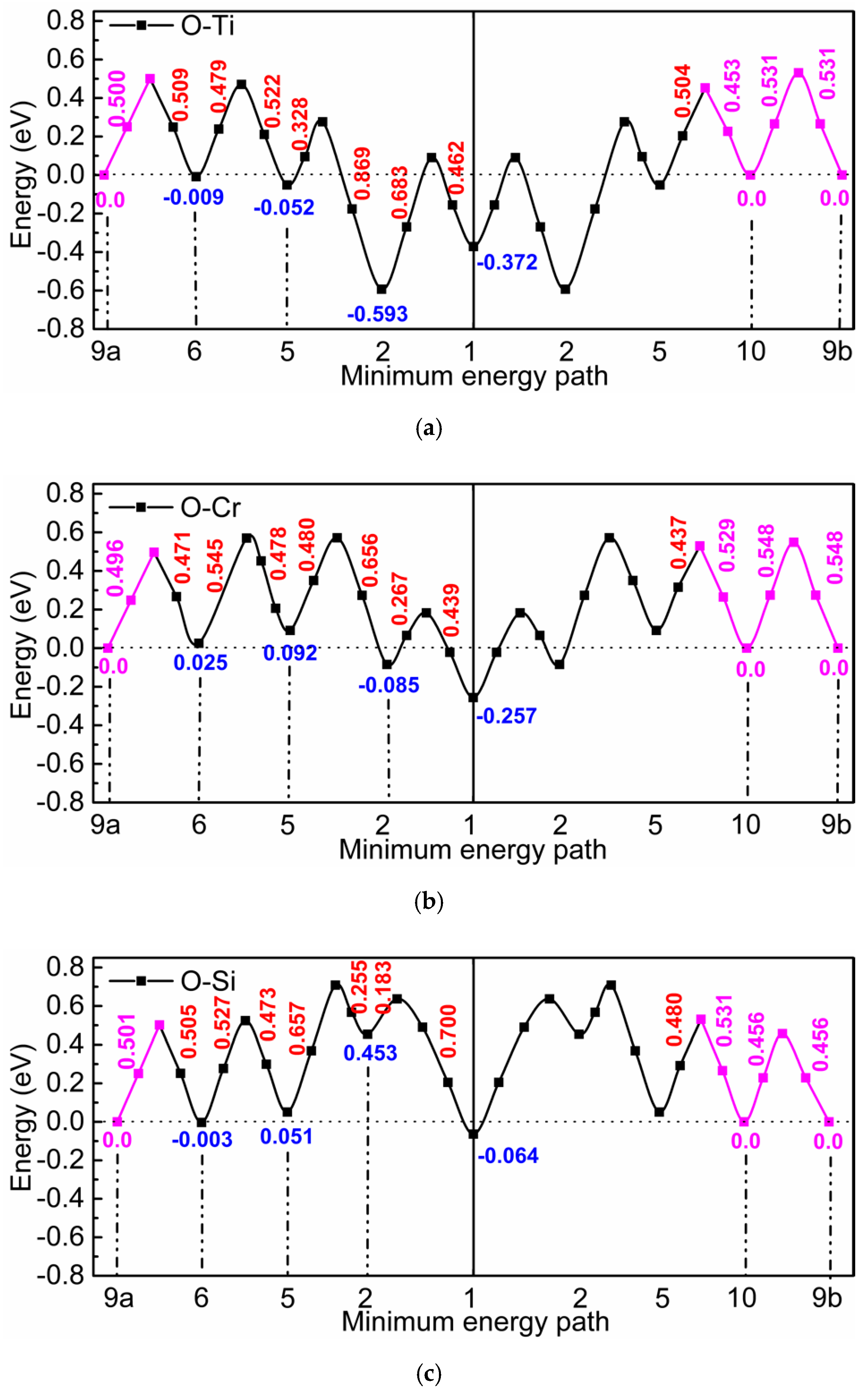
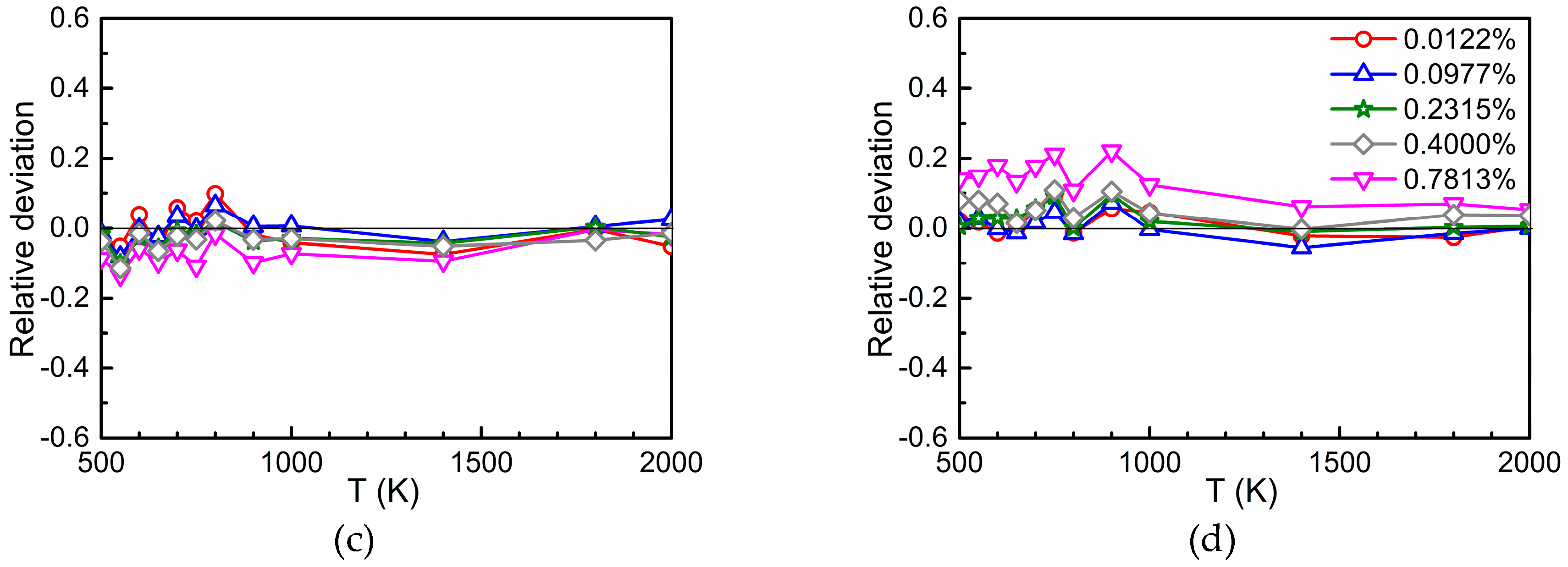
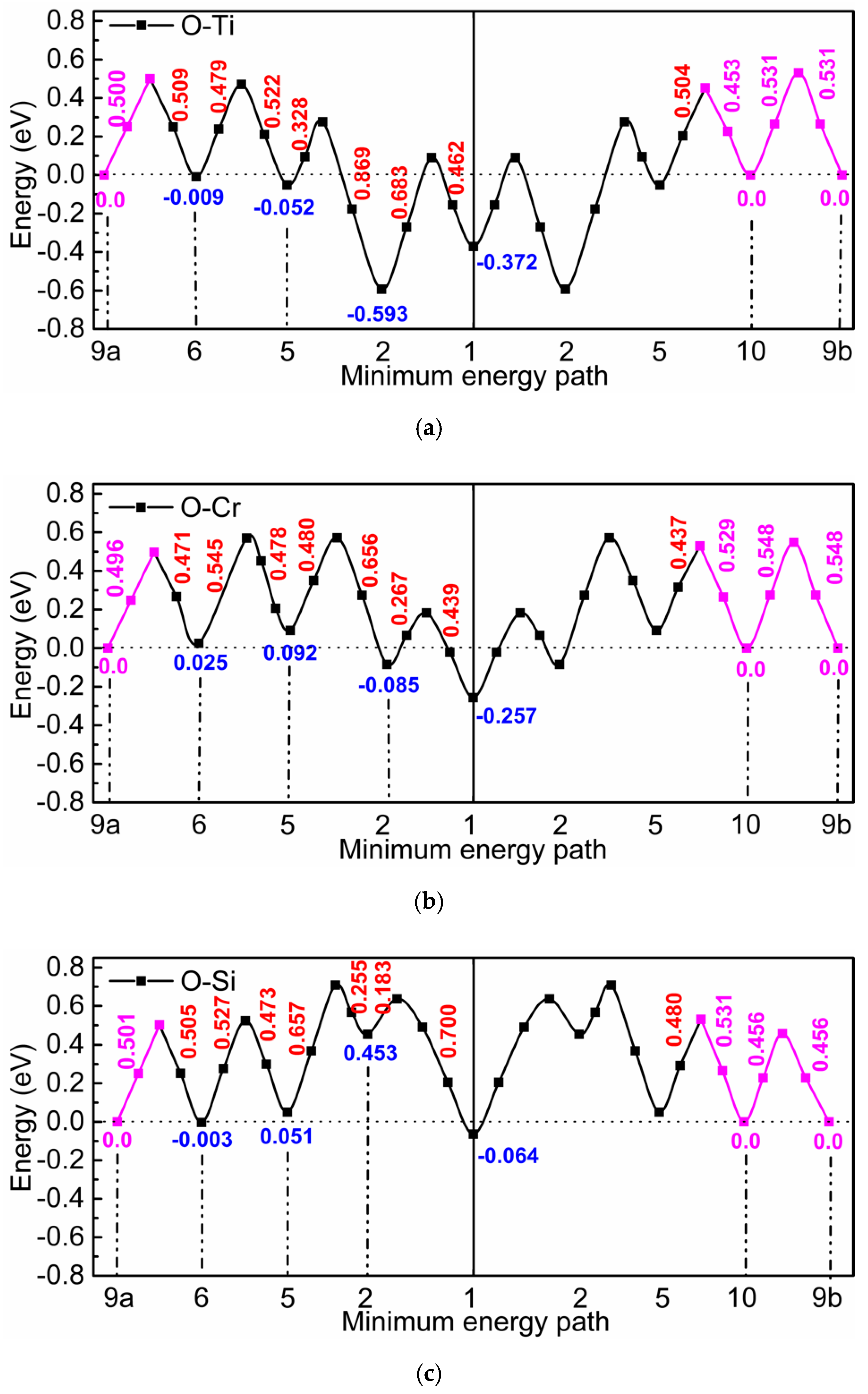
3.1. The Value of
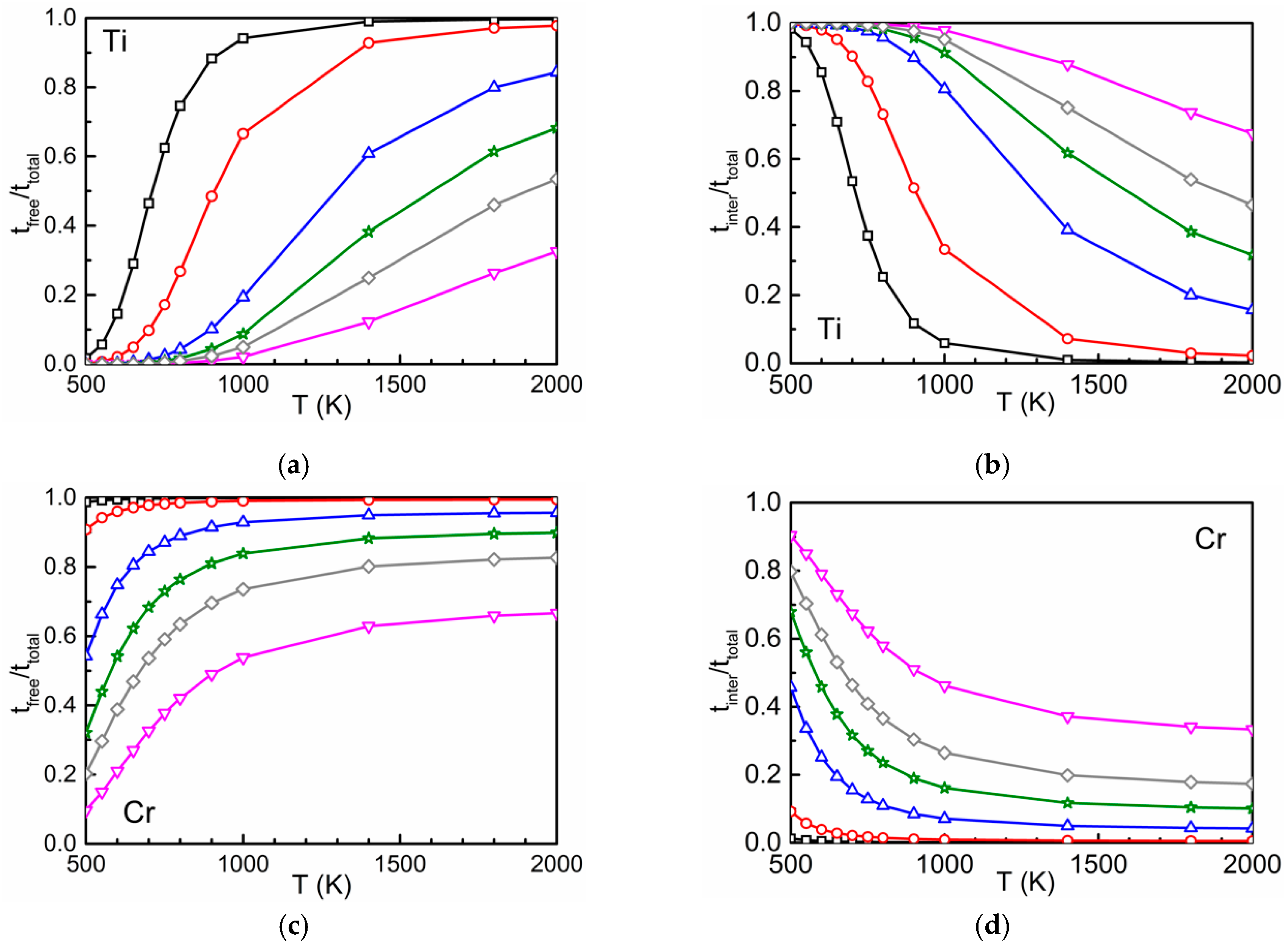
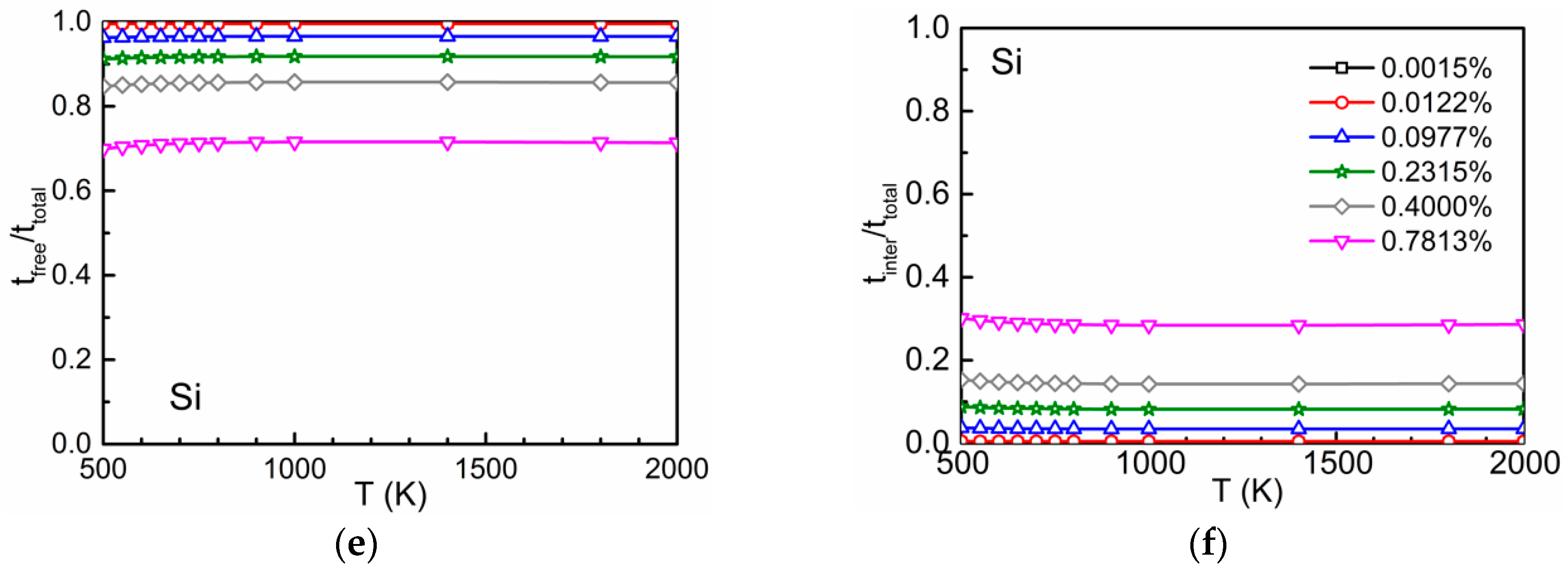
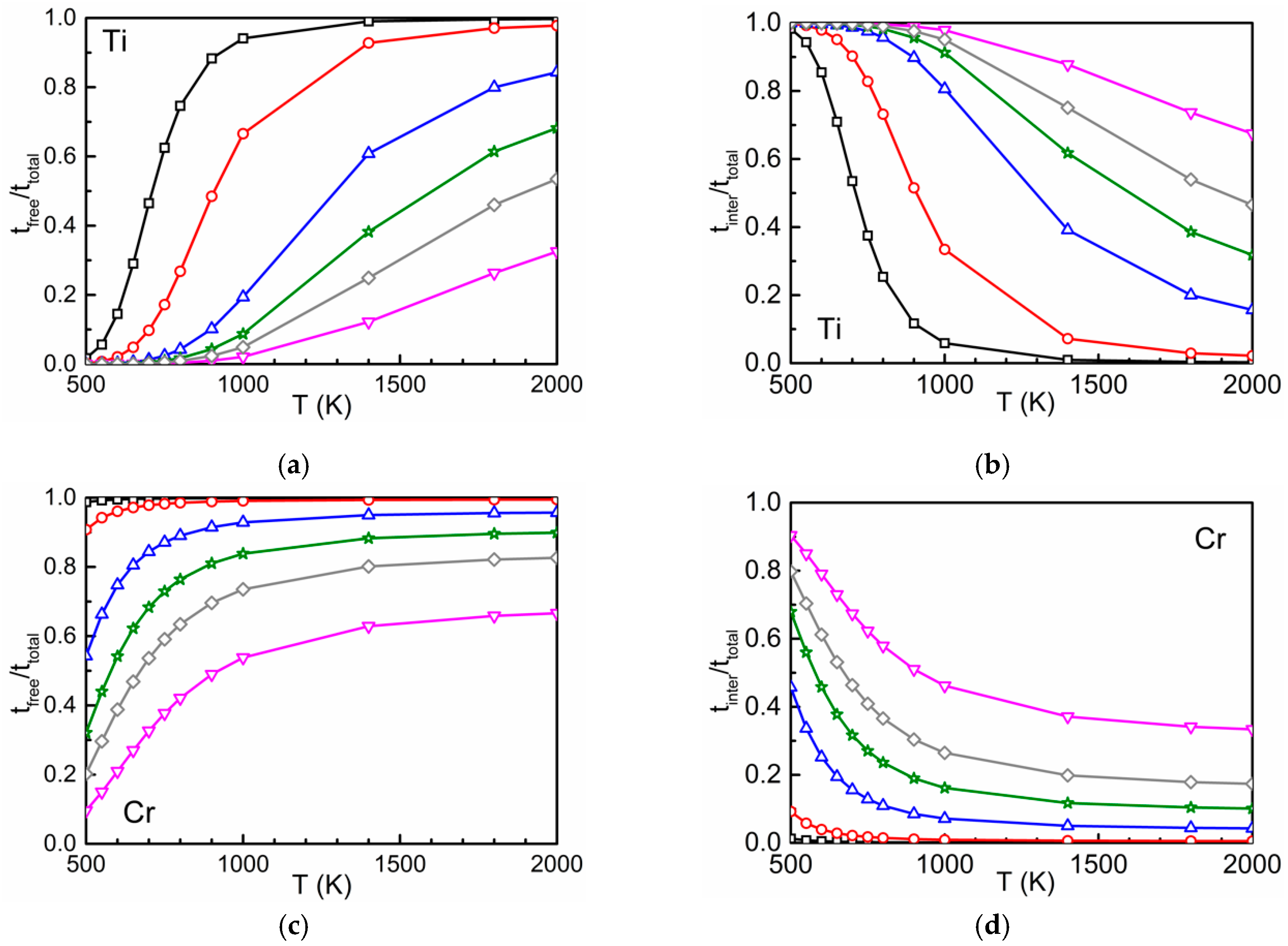
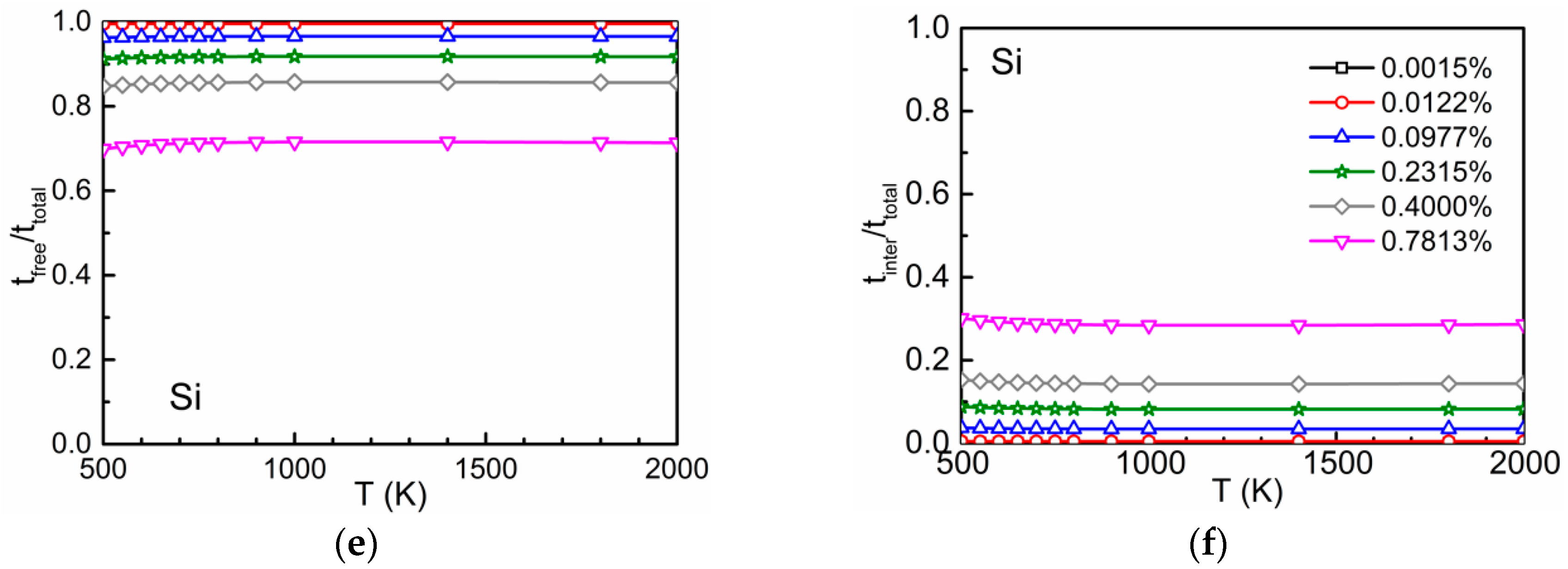
3.2. Time Ratios
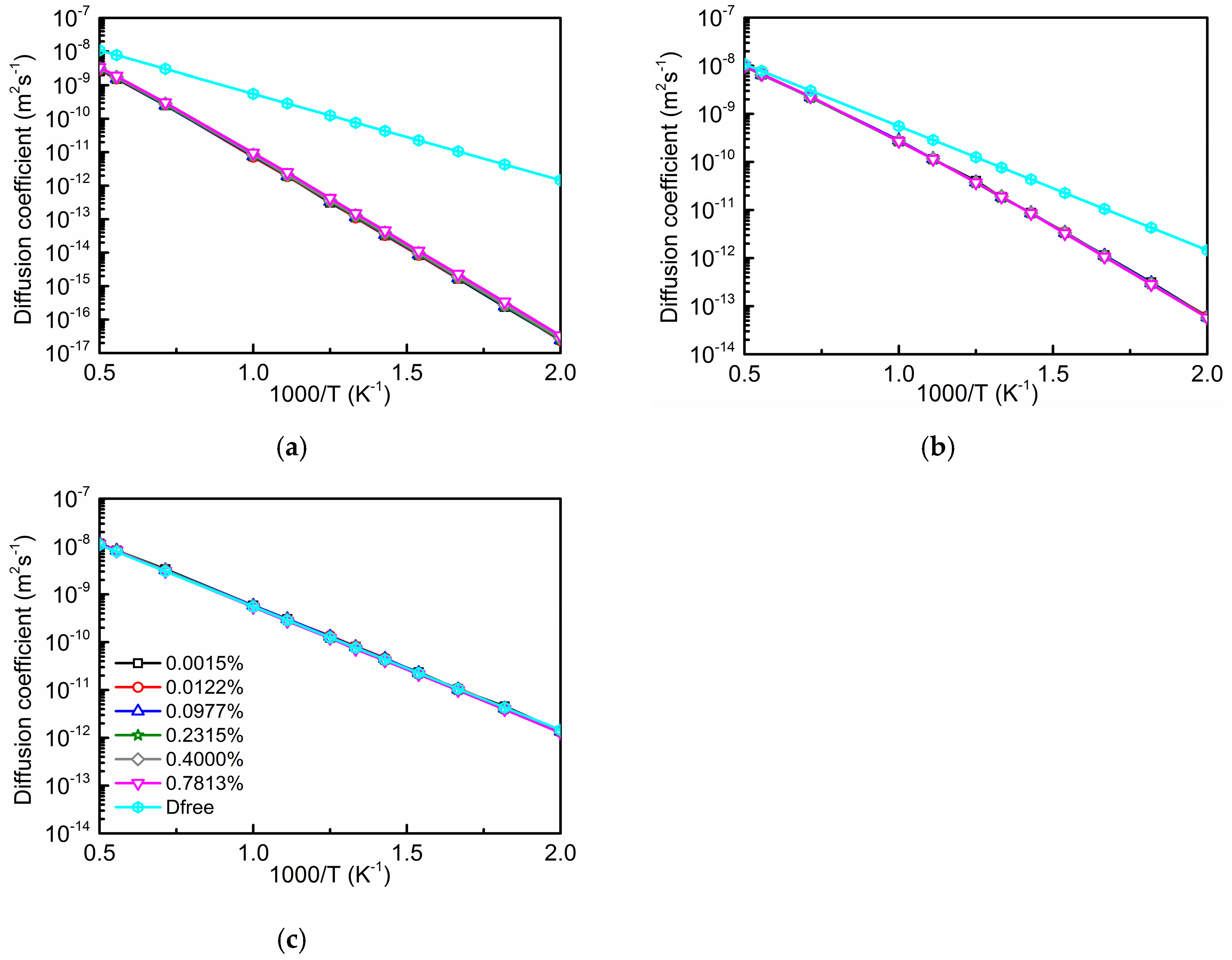
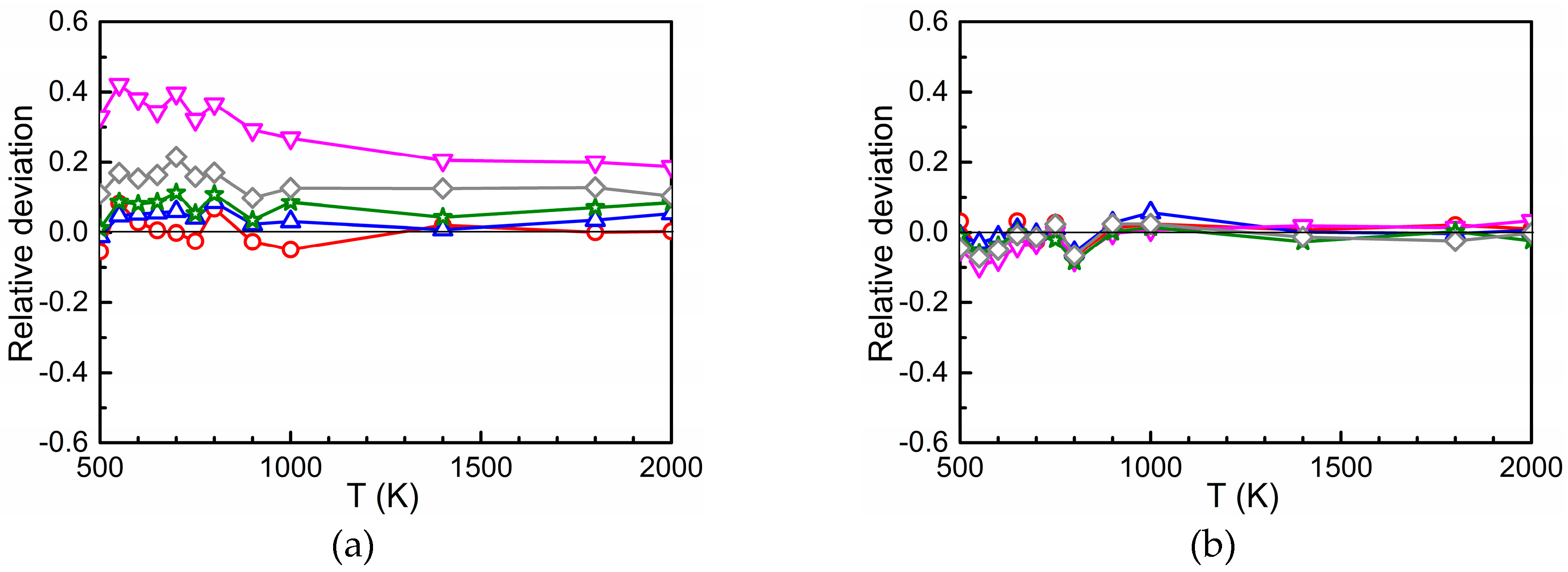
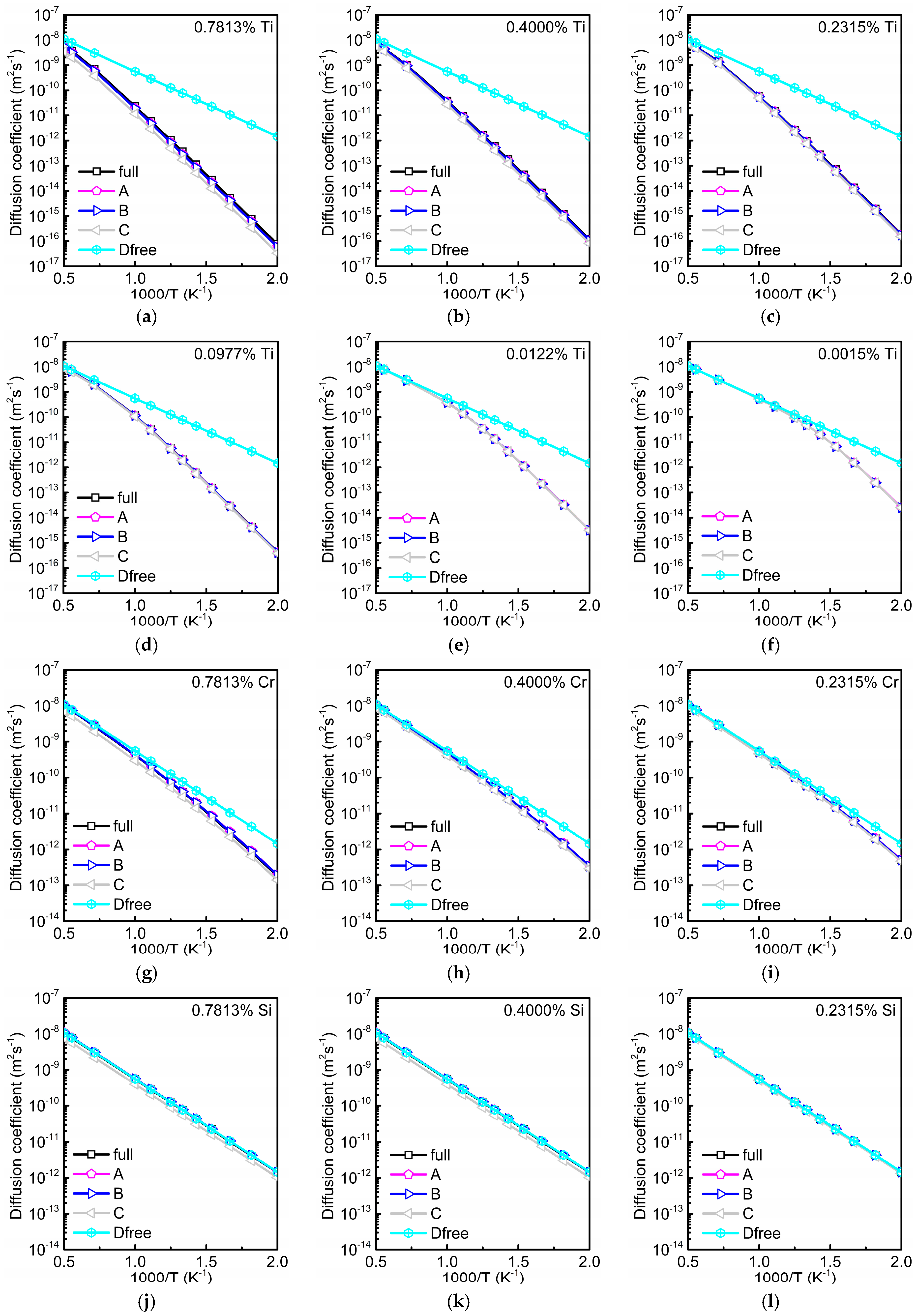
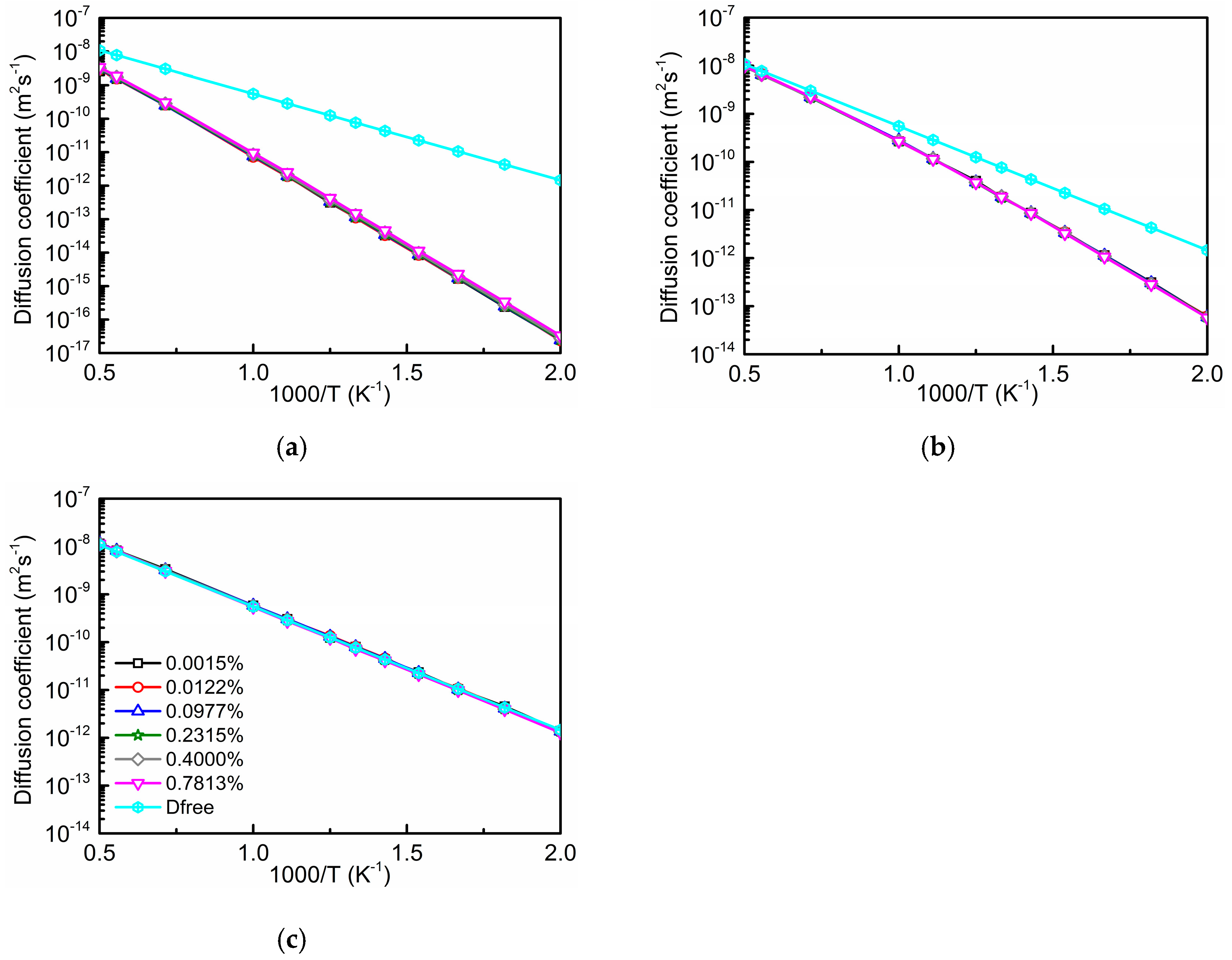
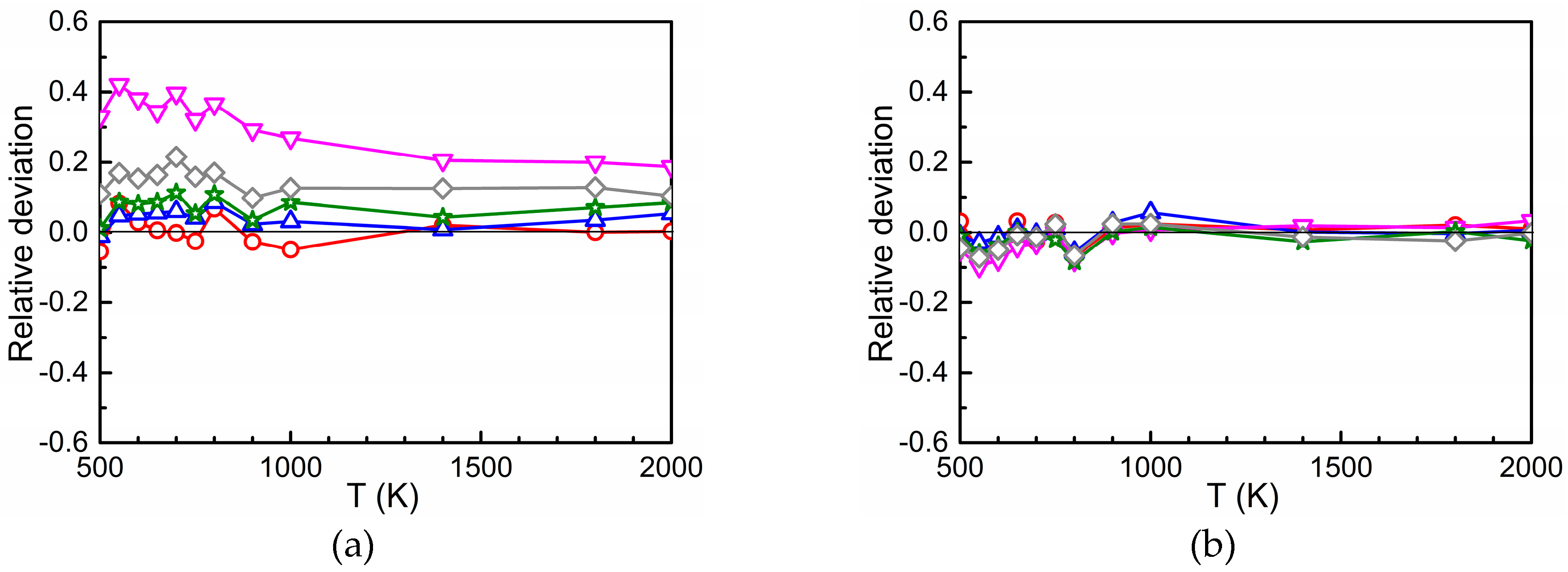
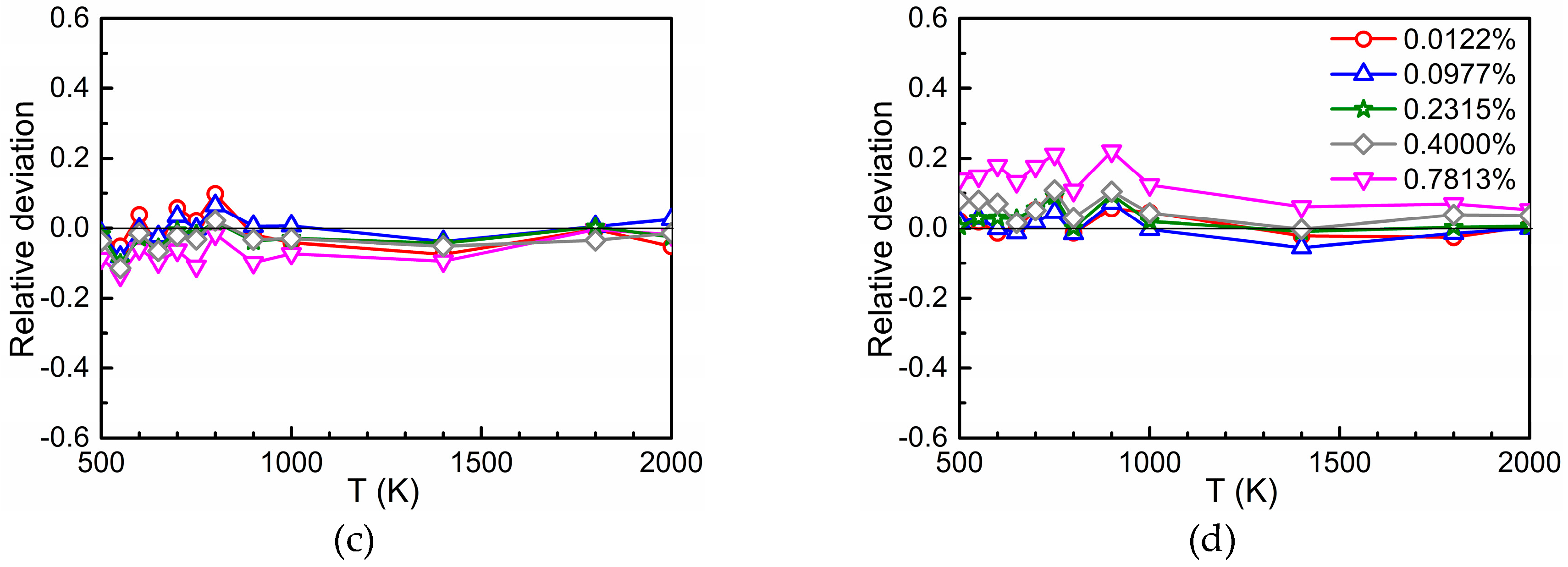
3.3. Total Diffusion Coefficient
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mehrer, H. Diffusion Mechanisms. In Diffusion in Solids: Fundamentals, Methods, Materials, Diffusion-Controlled Processes; Springer: Berlin/Heidelberg, Germany, 2007; pp. 95–103. [Google Scholar]
- Wang, X.; Posselt, M.; Faßbender, J. Influence of substitutional atoms on the diffusion of oxygen in dilute iron alloys. Phys. Rev. B 2018, 98, 064103. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Methfessel, M.P.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef]
- Barouh, C.; Schuler, T.; Fu, C.C.; Jourdan, T. Predicting vacancy-mediated diffusion of interstitial solutes in α-Fe. Phys. Rev. B 2015, 92, 104102. [Google Scholar] [CrossRef]
- Arokiam, A.C.; Barashev, A.V.; Bacon, D.J.; Osetsky, Y.N. Simulation of copper atom diffusion via the vacancy mechanism in a dilute Fe-Cu alloy. Phys. Rev. B 2005, 71, 174205. [Google Scholar] [CrossRef]
- Takada, J.; Adachi, M. Determination of diffusion coefficient of oxygen in α-iron from internal oxidation measurements in Fe-Si alloys. J. Mater. Sci. 1986, 21, 2133–2137. [Google Scholar] [CrossRef]
- Takada, J.; Yamamoto, S.; Kikuchi, S.; Adachi, M. Internal oxidation of Fe-Al alloys in the α-phase region. Oxid. Met. 1986, 25, 93–105. [Google Scholar] [CrossRef]
- Takada, J.; Yamamoto, S.; Adachi, M. Diffusion coefficient of oxygen in alpha-iron determined by internal oxidation technique. Z. Metallkde. 1986, 77, 6–11. [Google Scholar]
- Schuler, T.; Barouh, C.; Nastar, M.; Fu, C.C. Equilibrium vacancy concentration driven by undetectable impurities. Phys. Rev. Lett. 2015, 115, 015501. [Google Scholar] [CrossRef] [PubMed]
- Simonovic, D.; Ande, C.K.; Duff, A.I.; Syahputra, F.; Sluiter, M.H.F. Diffusion of carbon in bcc Fe in the presence of Si. Phys. Rev. B 2010, 81, 054116. [Google Scholar] [CrossRef]
- Liu, P.; Xing, W.; Cheng, X.; Li, D.; Li, Y.; Chen, X.-Q. Effects of dilute substitutional solutes on interstitial carbon in α-Fe: Interactions and associated carbon diffusion from first-principles calculations. Phys. Rev. B 2014, 90, 024103. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Faßbender, J.; Posselt, M. Efficient Calculation Methods for the Diffusion Coefficient of Interstitial Solutes in Dilute Alloys. Materials 2019, 12, 1491. https://doi.org/10.3390/ma12091491
Wang X, Faßbender J, Posselt M. Efficient Calculation Methods for the Diffusion Coefficient of Interstitial Solutes in Dilute Alloys. Materials. 2019; 12(9):1491. https://doi.org/10.3390/ma12091491
Chicago/Turabian StyleWang, Xiaoshuang, Jürgen Faßbender, and Matthias Posselt. 2019. "Efficient Calculation Methods for the Diffusion Coefficient of Interstitial Solutes in Dilute Alloys" Materials 12, no. 9: 1491. https://doi.org/10.3390/ma12091491
APA StyleWang, X., Faßbender, J., & Posselt, M. (2019). Efficient Calculation Methods for the Diffusion Coefficient of Interstitial Solutes in Dilute Alloys. Materials, 12(9), 1491. https://doi.org/10.3390/ma12091491