Size Distribution and Characteristics of Chitin Microgels Prepared via Emulsified Reverse-Micelles


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Extraction of Chitin
2.3. Preparation of Microgels
2.4. Characterization
2.4.1. X-Ray Fluorescence Spectroscopy (XRF)
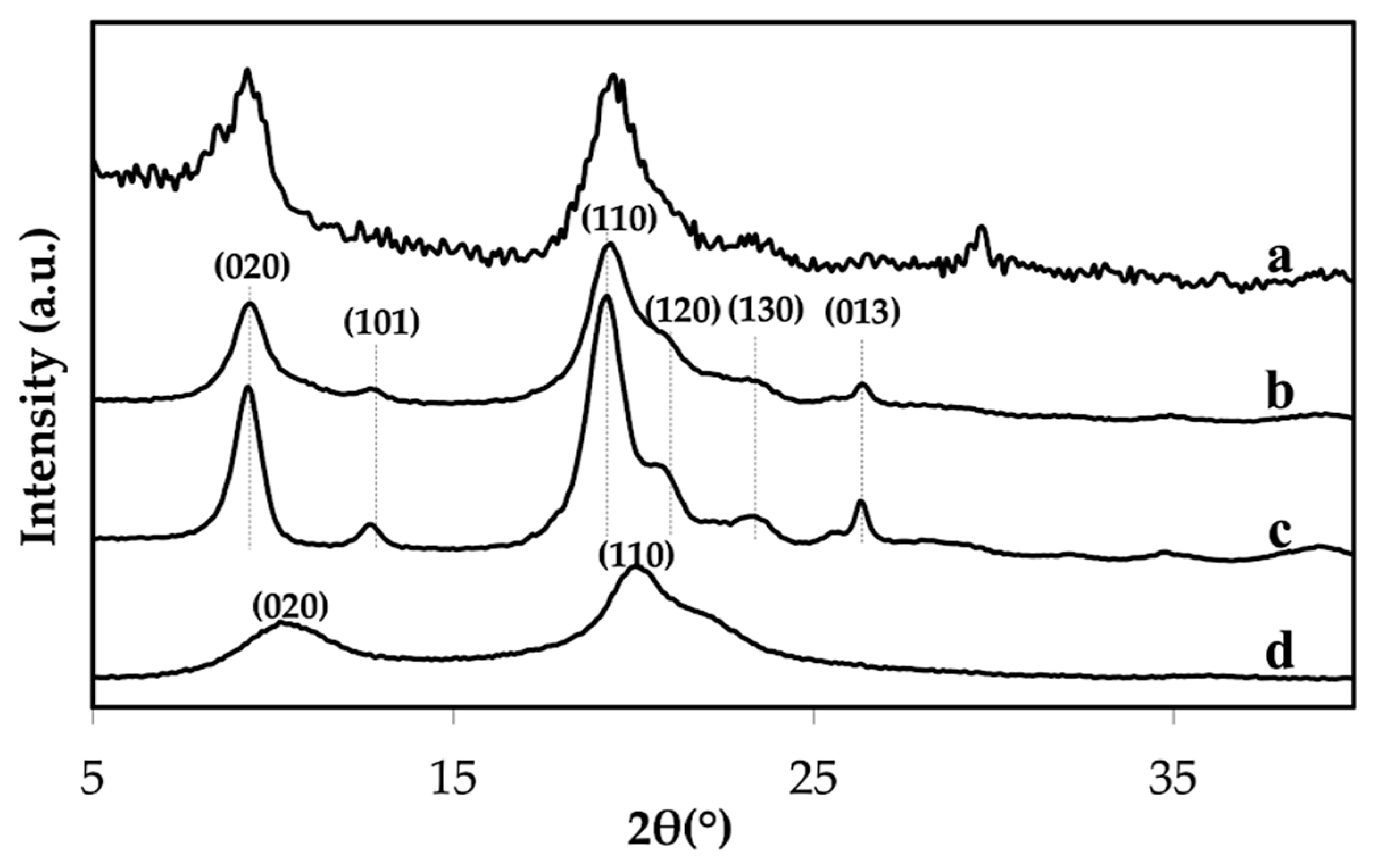
2.4.2. X-Ray Powder Diffraction (XRD)
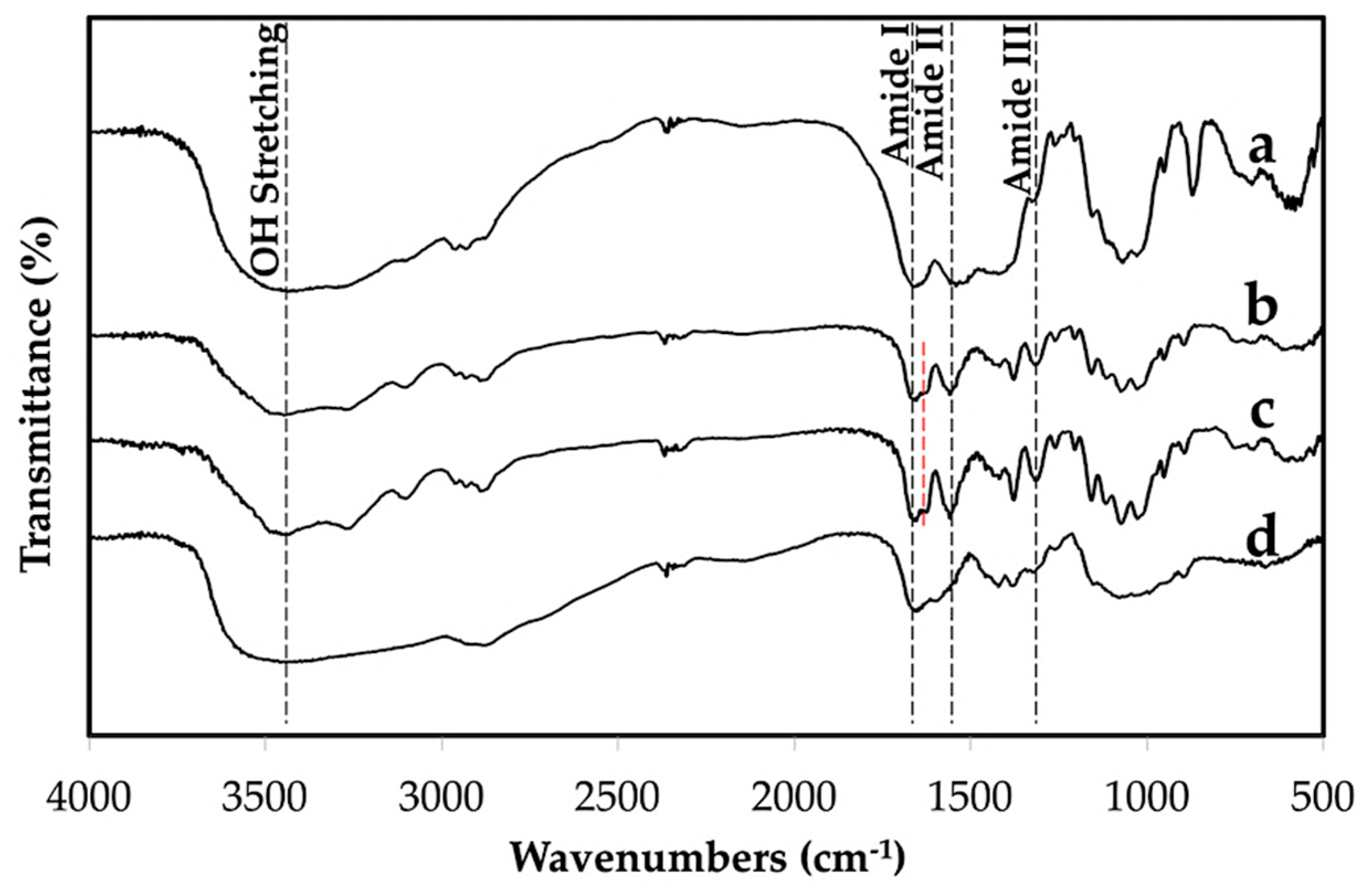
2.4.3. Fourier Transform Infrared Spectroscopy (FTIR)
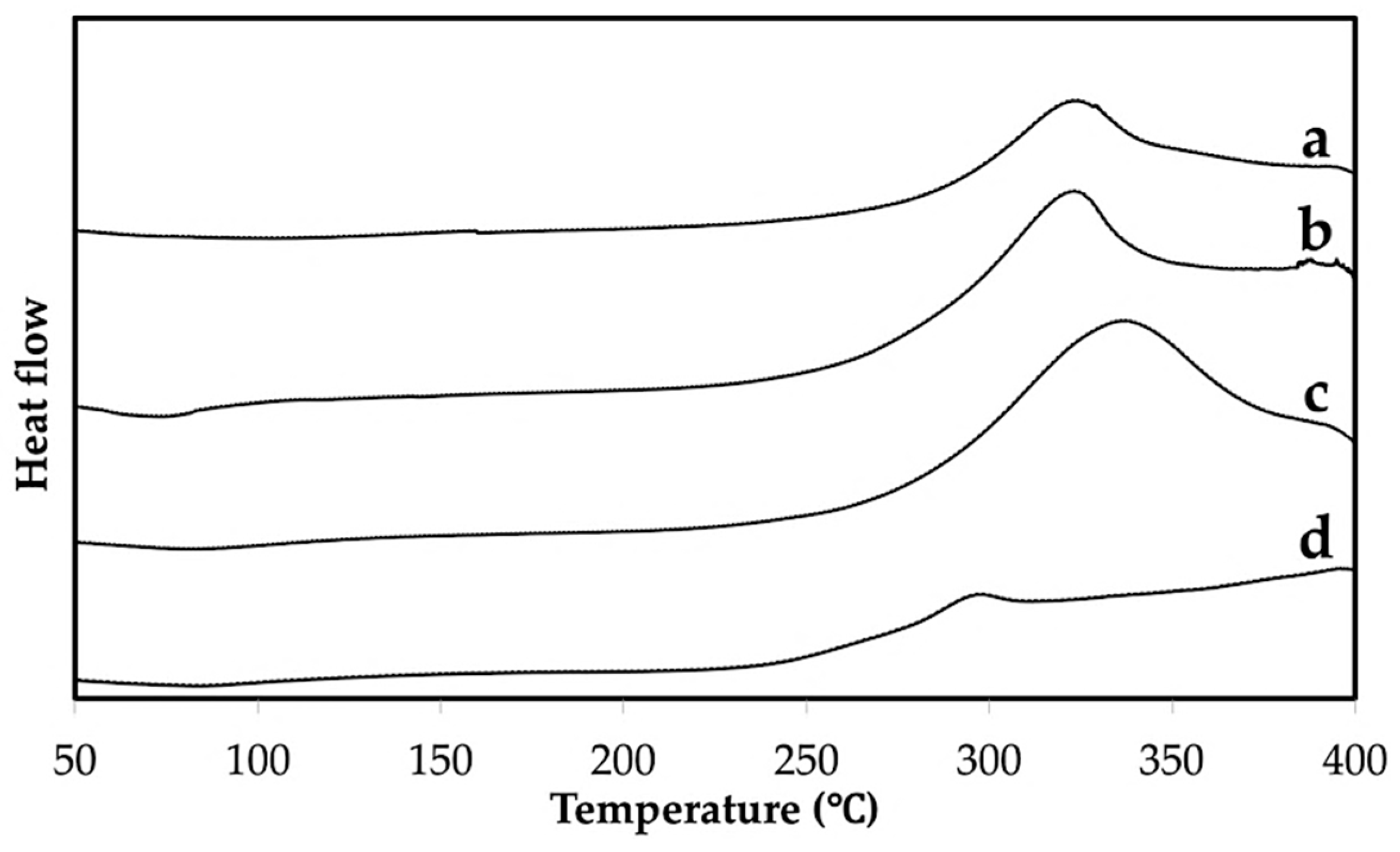
2.4.4. Differential Scanning Calorimetry (DSC)
2.4.5. Dynamic Light Scattering (DLS)
2.4.6. Optical Microscopy, Scanning Electron Microscopy (SEM), and Transmission Electron Microscopy (TEM)
2.4.7. Zeta-Potential
2.4.8. Brunauer-Emmett-Teller (BET)
3. Results and Discussion
3.1. Properties of the Extracted Chitin
3.2. Reverse Micelle Emulsification for the Fabrication of Chitin Microgels
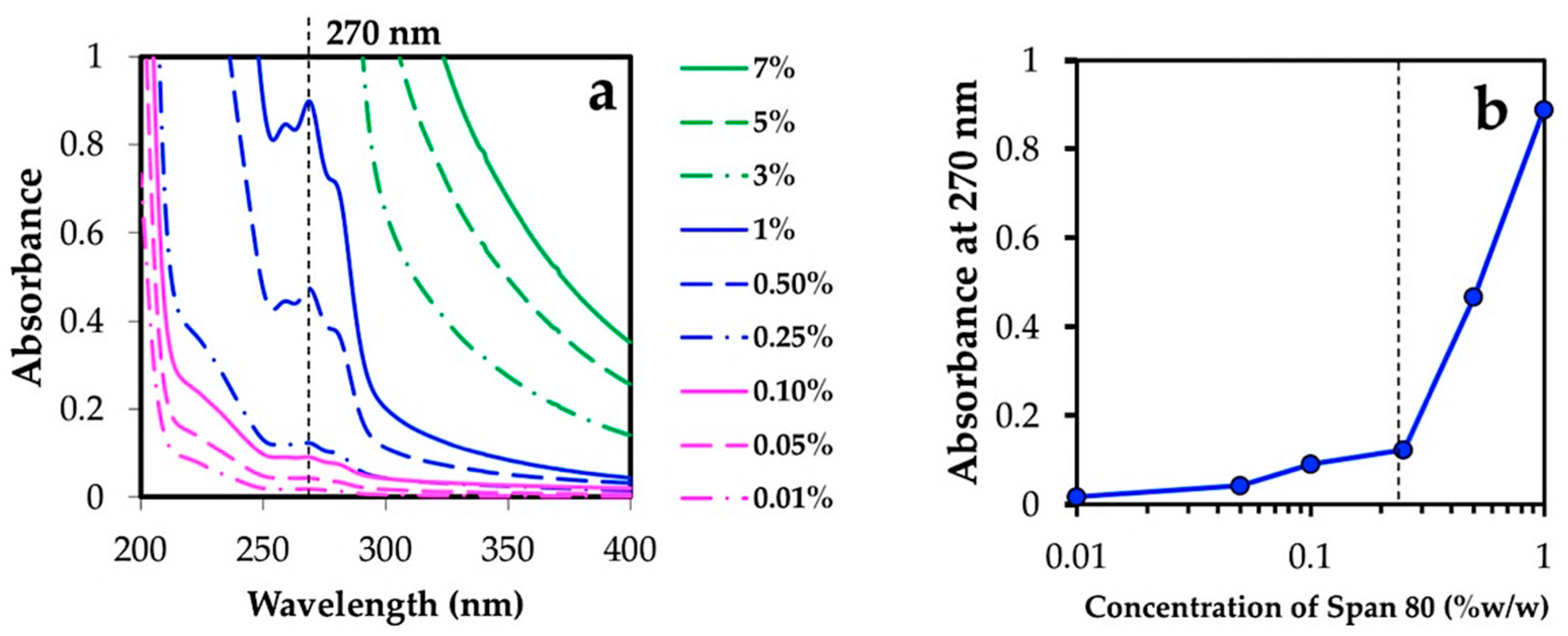
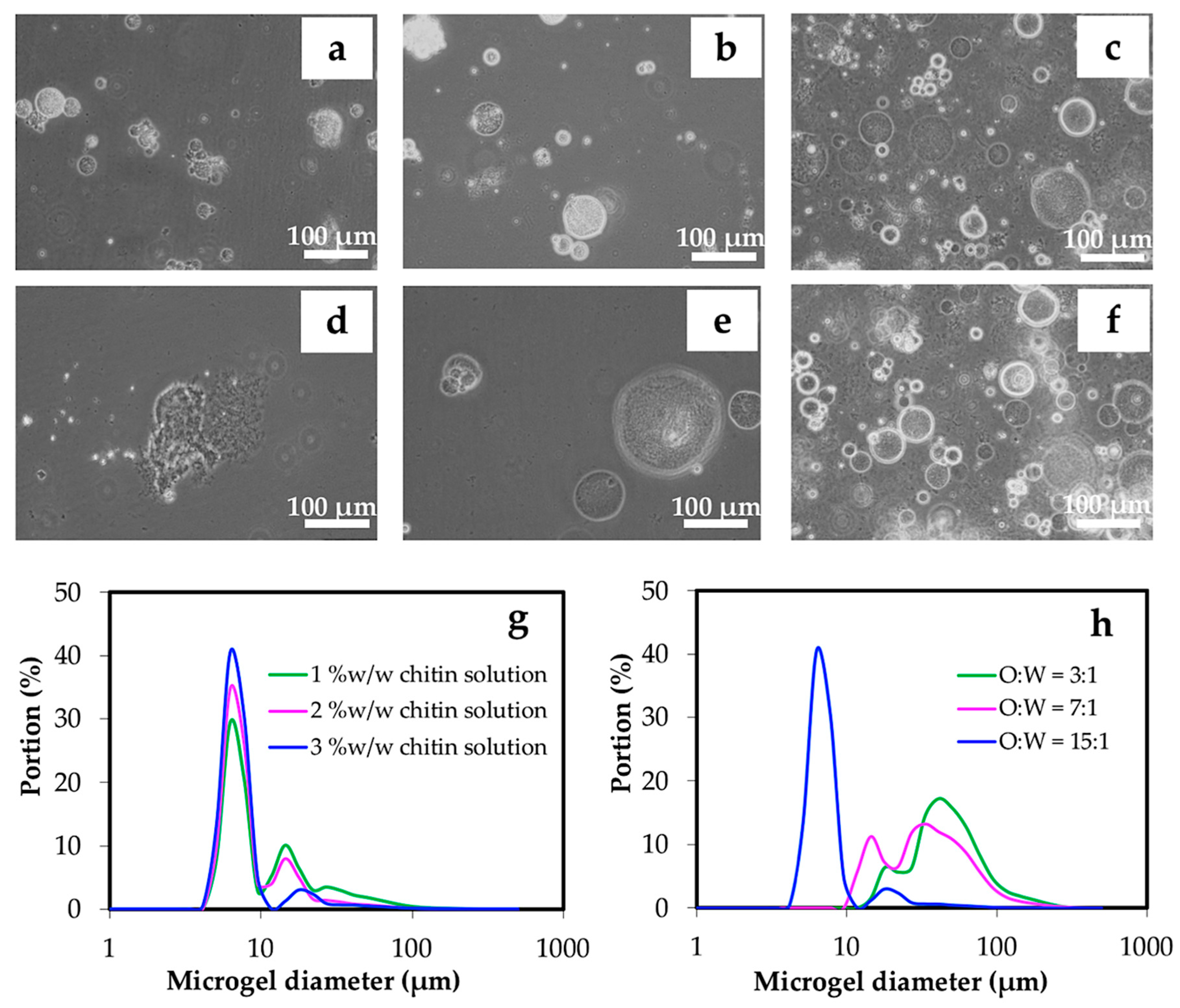
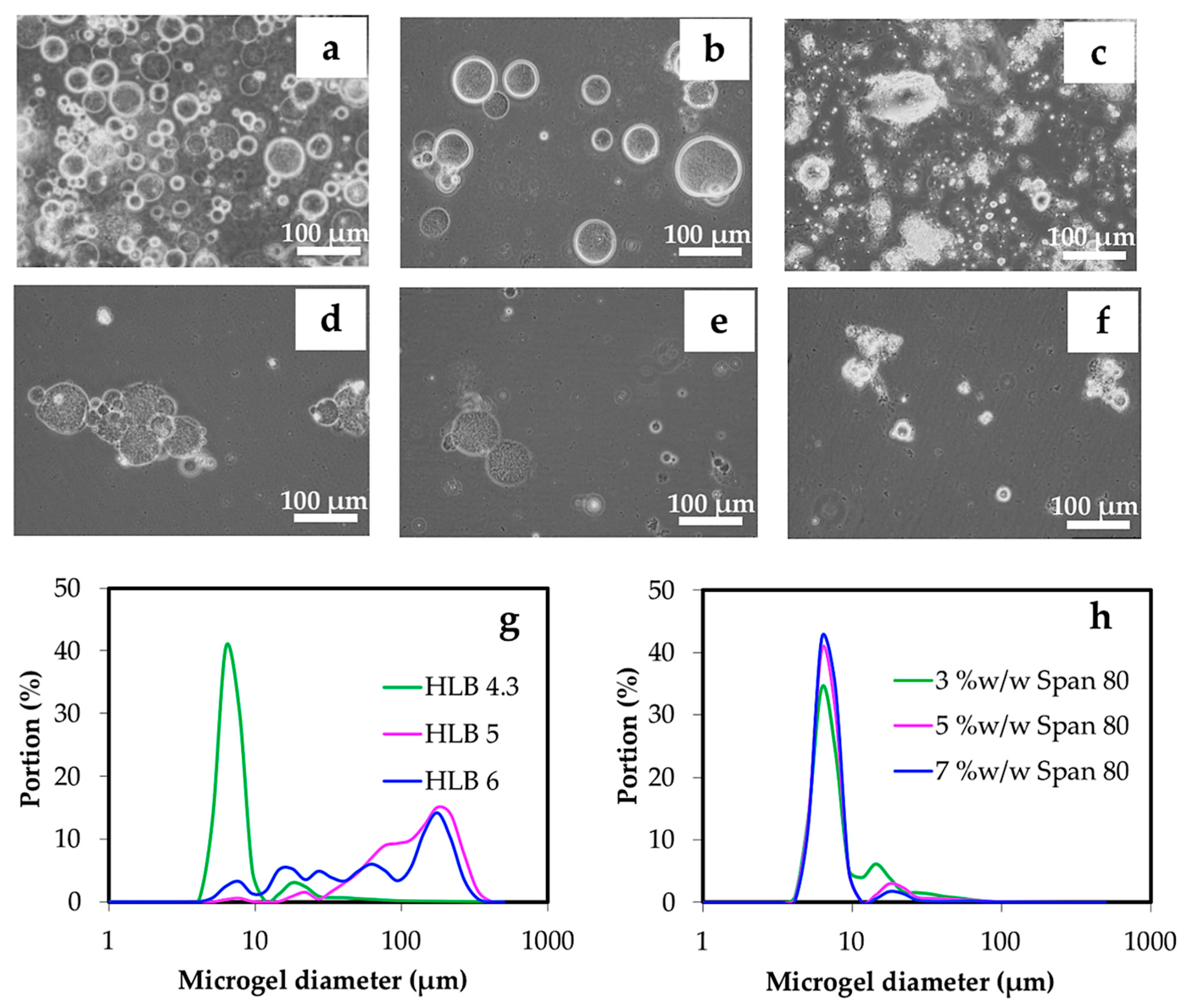
3.2.1. Effect of Water, Oil Phase, and Surfactant
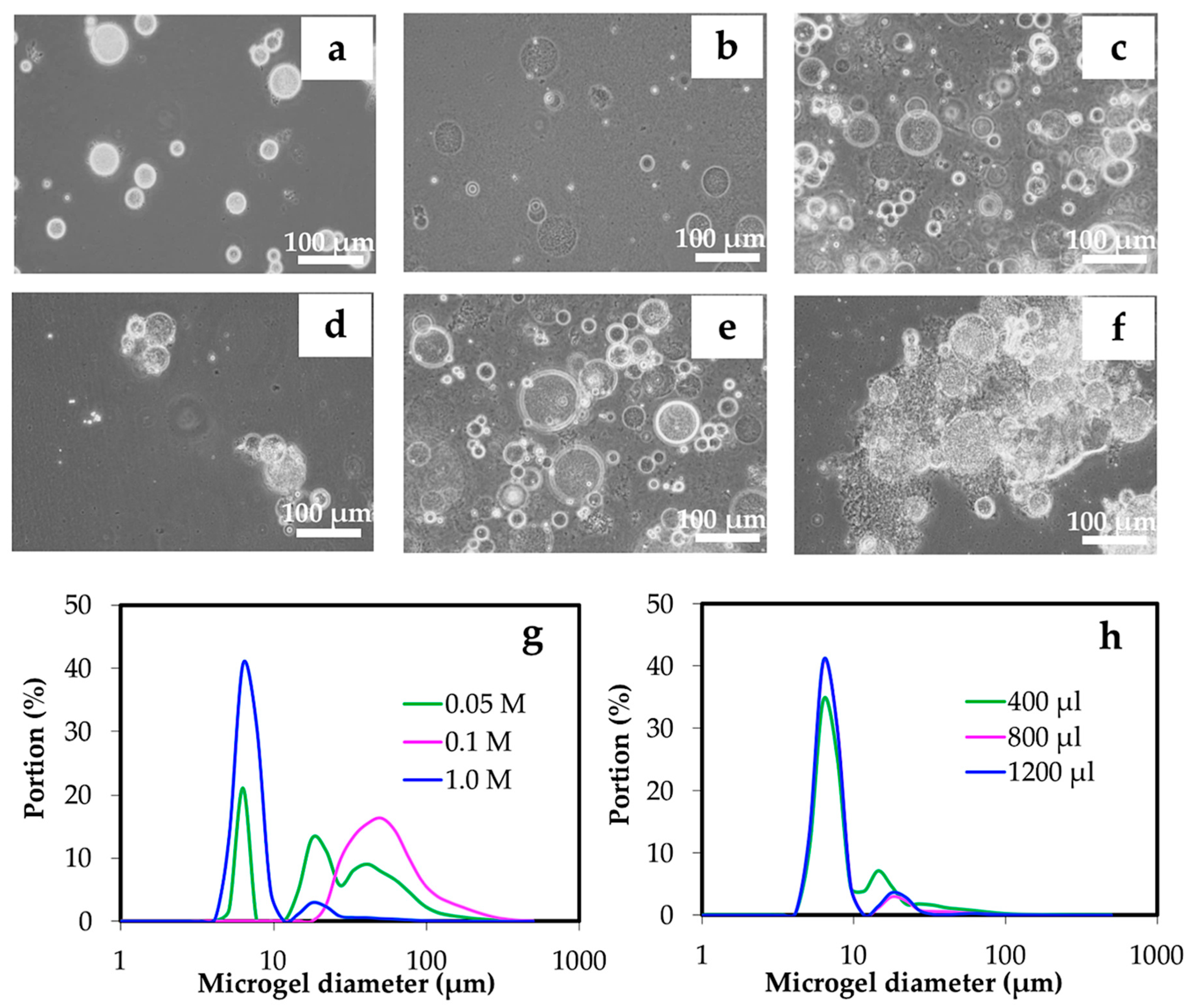
3.2.2. Effect of Gelation
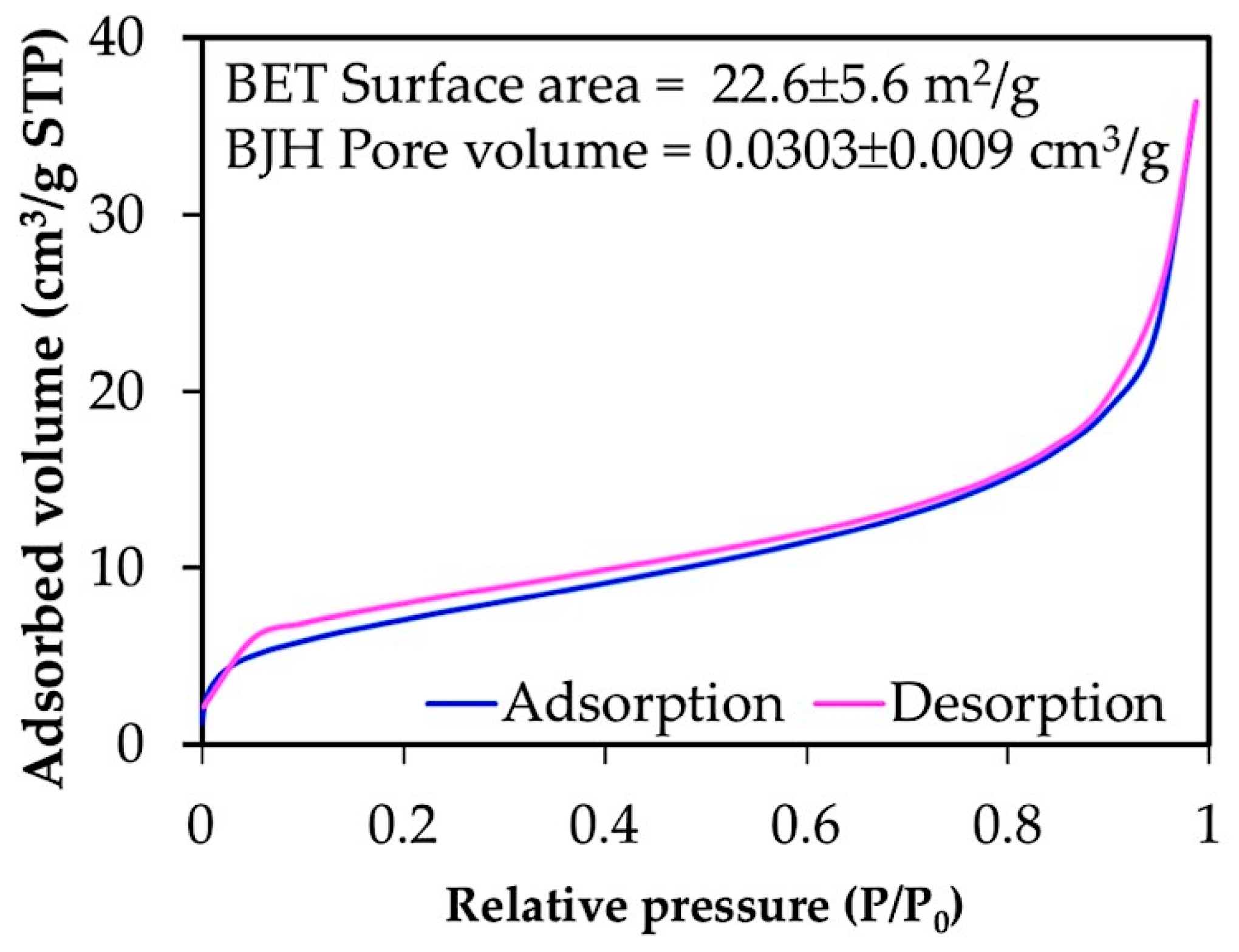
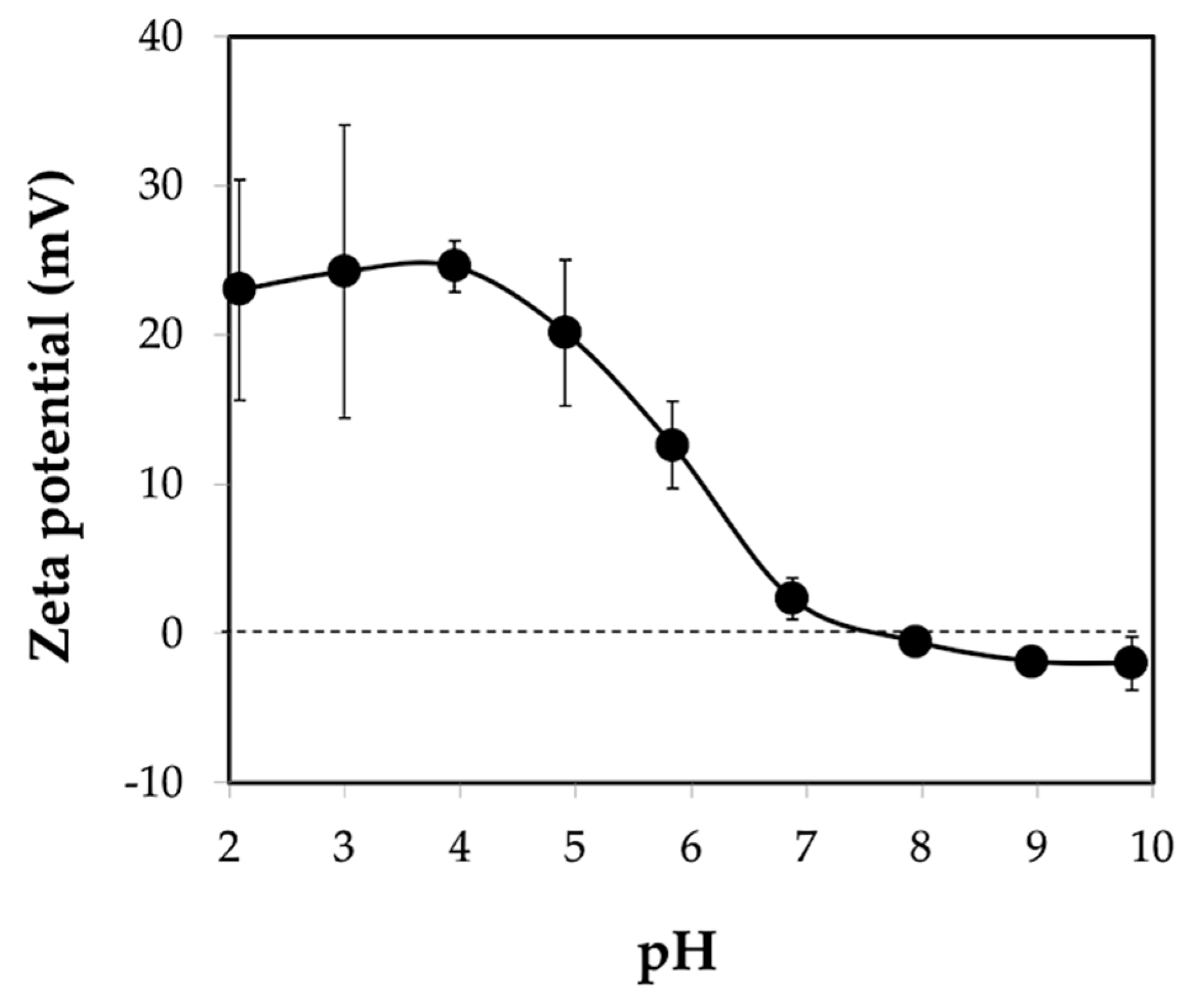
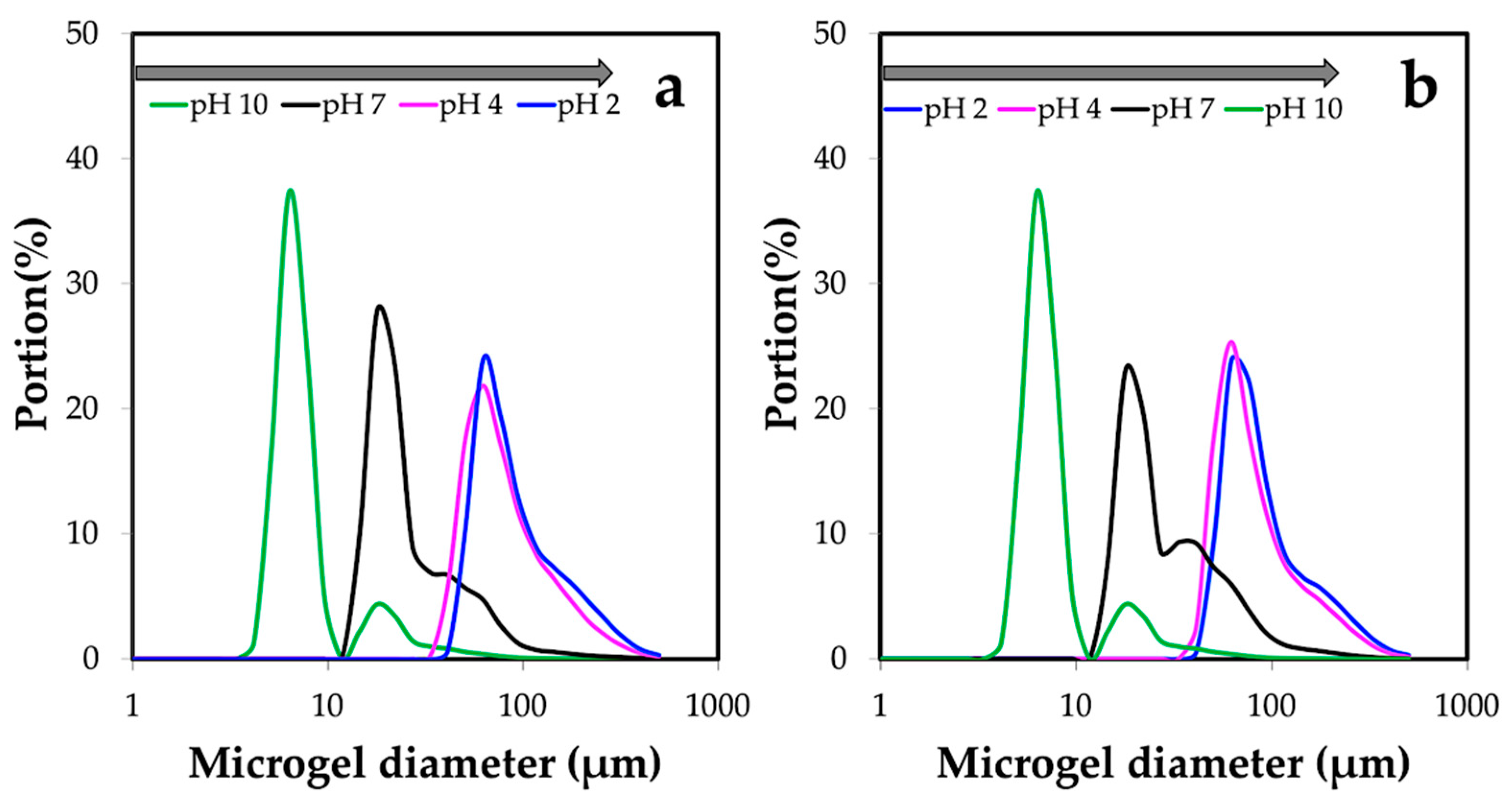
3.3. Properties of Chitin Microgels Prepared by the Reverse Micellar Method
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quantity of Catches of Marine Fisheries by Species. Available online: http://www.stat.go.jp/ english/data/nenkan/66nenkan/1431-08.html (accessed on 27 February 2019).
- Yan, N.; Chen, X. Sustainability: Don’t waste seafood waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef]
- Younes, I.; Rinaudo, M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Thorne, J.B.; Vine, G.J.; Snowden, M.J. Microgel applications and commercial considerations. Colloid Polym. Sci. 2011, 289, 625–646. [Google Scholar] [CrossRef]
- Panonnummal, R.; Sabitha, M. Anti-psoriatic and toxicity evaluation of methotrexate loaded chitin nanogel in imiquimod induced mice model. Int. J. Biol. Macromol. 2018, 110, 245–258. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, S.; Gao, C.; Chen, C.; Zhang, X.; Liu, M. Highly stable and degradable multifunctional microgel for self-regulated insulin delivery under physiological conditions. Nanoscale 2013, 5, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Chedly, J.; Soares, S.; Montembault, A.; von Boxberg, Y.; Veron-Ravaille, M.; Mouffle, C.; Benassy, M.-N.; Taxi, J.; David, L.; Nothias, F. Physical chitosan microhydrogels as scaffolds for spinal cord injury restoration and axon regeneration. Biomaterials 2017, 138, 91–107. [Google Scholar] [CrossRef]
- Nishiyama, A.; Shinohara, T.; Pantuso, T.; Tsuji, S.; Yamashita, M.; Shinohara, S.; Myrvik, Q.N.; Henriksen, R.A.; Shibata, Y. Depletion of cellular cholesterol enhances macrophage MAPK activation by chitin microparticles but not by heat-killed Mycobacterium bovis BCG. Am. J. Physiol. Cell Physiol. 2008, 295, 341–349. [Google Scholar] [CrossRef]
- Caldorera-Moore, M.; Guimard, N.; Shi, L.; Roy, K. Designer nanoparticles: Incorporating size, shape, and triggered release into nanoscale drug carriers. Expert Opin. Drug Deliv. 2010, 7, 479–495. [Google Scholar] [CrossRef]
- Vaino, A.R.; Janda, K.D. Solid-phase organic synthesis: A critical understanding of the resin. J. Comb. Chem. 2000, 2, 579–596. [Google Scholar] [CrossRef]
- Seiffert, S. Microgel capsules tailored by droplet-based microfluidics. ChemPhysChem 2013, 14, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Y.; Wan, L.S.C.; Thai, P.Y. Chitosan microspheres prepared by emulsification and ionotropic gelation. Drug Dev. Ind. Pharm. 1997, 23, 981–985. [Google Scholar] [CrossRef]
- Zhou, W.-Q.; Gu, T.-Y.; Su, Z.-G.; Ma, G.-H. Synthesis of macroporous poly(styrene-divinyl benzene) microspheres by surfactant reverse micelles swelling method. Polymer 2007, 48, 1981–1988. [Google Scholar] [CrossRef]
- Spanka, C.; Clapham, B.; Janda, K.D. Preparation of new microgel polymers and their application as supports in organic synthesis. J. Org. Chem. 2002, 67, 3045–3050. [Google Scholar] [CrossRef]
- Guerzoni, L.P.B.; Bohl, J.; Jans, A.; Rose, J.C.; Koehler, J.; Kuehne, A.J.C.; de Laporte, L. Microfluidic fabrication of polyethylene glycol microgel capsules with tailored properties for the delivery of biomolecules. Biomater. Sci. 2017, 5, 1549–1557. [Google Scholar] [CrossRef]
- Uskokovic, V.; Drofenik, M. Sysnthesis of materials within reverse micelles. Surf. Rev. Lett. 2005, 12, 239–277. [Google Scholar] [CrossRef]
- Fulton, J.L.; Smith, R.D. Reverse micelle and microemulsion phases in supercritcal fluids. J. Phys. Chem. 1988, 92, 2903–2907. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, M.; Ma, Z.; Xin, B.; Guo, R.; Xu, X. Sugar Fatty Acid Esters, 1st ed.; Elsevier: Saint Louis, IL, USA, 2015; pp. 215–243. [Google Scholar]
- Rhein, L. Surfactant Action on Skin and Hair: Cleansing and Skin Reactivity Mechanisms; Elsevier Science: Amsterdam, The Netherlands, 2007; pp. 305–369. [Google Scholar]
- Zhang, Y.; Xue, C.; Xue, Y.; Gao, R.; Zhang, X. Determination of the degree of deacetylation of chitin and chitosan by X-ray powder diffraction. Carbohydr. Res. 2005, 340, 1914–1917. [Google Scholar] [CrossRef]
- Kasaai, M.R. A review of several reported procedures to determine the degree of N-acetylation for chitin and chitosan using infrared spectroscopy. Carbohydr. Polym. 2008, 71, 497–508. [Google Scholar] [CrossRef]
- Cárdenas, G.; Cabrera, G.; Taboada, E.; Miranda, S.P. Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J. Appl. Polym. Sci. 2004, 93, 1876–1885. [Google Scholar] [CrossRef]
- Pereira, A.G.B.; Muniz, E.C.; Hsieh, Y.-L. 1H NMR and 1H–13C HSQC surface characterization of chitosan-chitin sheath-core nanowhiskers. Carbohydr. Polym. 2015, 123, 46–52. [Google Scholar] [CrossRef]
- Wu, Y.; Sasaki, T.; Irie, S.; Sakurai, K. A novel biomass-ionic liquid platform for the utilization of native chitin. Polymer 2008, 49, 2321–2327. [Google Scholar] [CrossRef]
- Jang, M.-K.; Kong, B.-G.; Jeong, Y.-I.; Lee, C.H.; Nah, J.-W. Physicochemical characterization of α-chitin, β-chitin, and γ-chitin separated from natural resources. J. Polym. Sci. A 2004, 42, 3423–3432. [Google Scholar] [CrossRef]
- Guinesi, L.S.; Cavalheiro, É.T.G. The use of DSC curves to determine the acetylation degree of chitin/chitosan samples. Thermochim. Acta 2006, 444, 128–133. [Google Scholar] [CrossRef]
- Alothman, A.Z. A Review: Fundamental aspects of silicate mesoporous materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Pereira, A.G.B.; Muniz, E.C.; Hsieh, Y.-L. Chitosan-sheath and chitin-core nanowhiskers. Carbohydr. Polym. 2014, 107, 158–166. [Google Scholar] [CrossRef]
- Alabaraoye, E.; Achilonu, M.; Hester, R. Biopolymer (Chitin) from various marine seashell wastes: Isolation and Characterization. J. Polym. Environ. 2017, 26, 1–12. [Google Scholar] [CrossRef]
- Kumirska, J.; Weinhold, M.X.; Thöming, J.; Stepnowski, P. Biomedical activity of chitin/chitosan based materials—influence of physicochemical properties apart from molecular weight and degree of n-acetylation. Polymers 2011, 3, 1875–1901. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Concentration of chitin solution (% w/w) | O:W volume ratio | HLB of surfactant | Concentration of Span 80 (% w/w) | Concentration of HCl (M) | Volume of HCl (μl) |
|---|---|---|---|---|---|---|
| Experiment 1 | 1 2 3 | 15:1 | 4.3 | 5 | 1.0 | 800 |
| Experiment 2 | 3 | 3:1 7:1 15:1 | 4.3 | 5 | 1.0 | 800 |
| Experiment 3 | 3 | 15:1 | 4.3 5 6 | 5 | 1.0 | 800 |
| Experiment 4 | 3 | 15:1 | 4.3 | 3 5 7 | 1.0 | 800 |
| Experiment 5 | 3 | 15:1 | 4.3 | 5 | 0.05 0.1 1.0 | 800 |
| Experiment 6 | 3 | 15:1 | 4.3 | 5 | 1.0 | 400 800 1200 |
| Elements | Crab Shell (mass%) | Extracted Chitin (mass%) | Commercial Chitin (mass%) | Commercial Chitosan (mass%) |
|---|---|---|---|---|
| C | 29.2 | 52.6 | 52.2 | 50.8 |
| O | 46.3 | 47.2 | 47.6 | 49.1 |
| Na | 0.968 | trace | trace | trace |
| Mg | 1.19 | trace | trace | 0.0135 |
| P | 2.93 | 0.0293 | 0.0022 | 0.0074 |
| S | 0.344 | 0.0158 | 0.0123 | 0.0044 |
| Cl | 0.971 | 0.0167 | 0.112 | trace |
| Ca | 17.2 | 0.0782 | 0.0072 | 0.0457 |
| Fe | 0.016 | 0.0104 | 0.0053 | 0.0084 |
| Samples | %CrI | %DA from XRD | %DA from FTIR | Peak Temperature (°C) |
|---|---|---|---|---|
| Extracted chitin | 70.3 | 55.3 | 60.9 | 330 |
| Commercial chitin | 74.7 | 61.1 | 62.6 | 340 |
| Commercial chitosan | 44.7 | 21.2 | 45.7 | 295 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taokaew, S.; Ofuchi, M.; Kobayashi, T. Size Distribution and Characteristics of Chitin Microgels Prepared via Emulsified Reverse-Micelles. Materials 2019, 12, 1160. https://doi.org/10.3390/ma12071160
Taokaew S, Ofuchi M, Kobayashi T. Size Distribution and Characteristics of Chitin Microgels Prepared via Emulsified Reverse-Micelles. Materials. 2019; 12(7):1160. https://doi.org/10.3390/ma12071160
Chicago/Turabian StyleTaokaew, Siriporn, Mitsumasa Ofuchi, and Takaomi Kobayashi. 2019. "Size Distribution and Characteristics of Chitin Microgels Prepared via Emulsified Reverse-Micelles" Materials 12, no. 7: 1160. https://doi.org/10.3390/ma12071160
APA StyleTaokaew, S., Ofuchi, M., & Kobayashi, T. (2019). Size Distribution and Characteristics of Chitin Microgels Prepared via Emulsified Reverse-Micelles. Materials, 12(7), 1160. https://doi.org/10.3390/ma12071160