Surface Treatment of Carbon Fibers by Oxy-Fluorination





Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Scanning Electron Microscopy
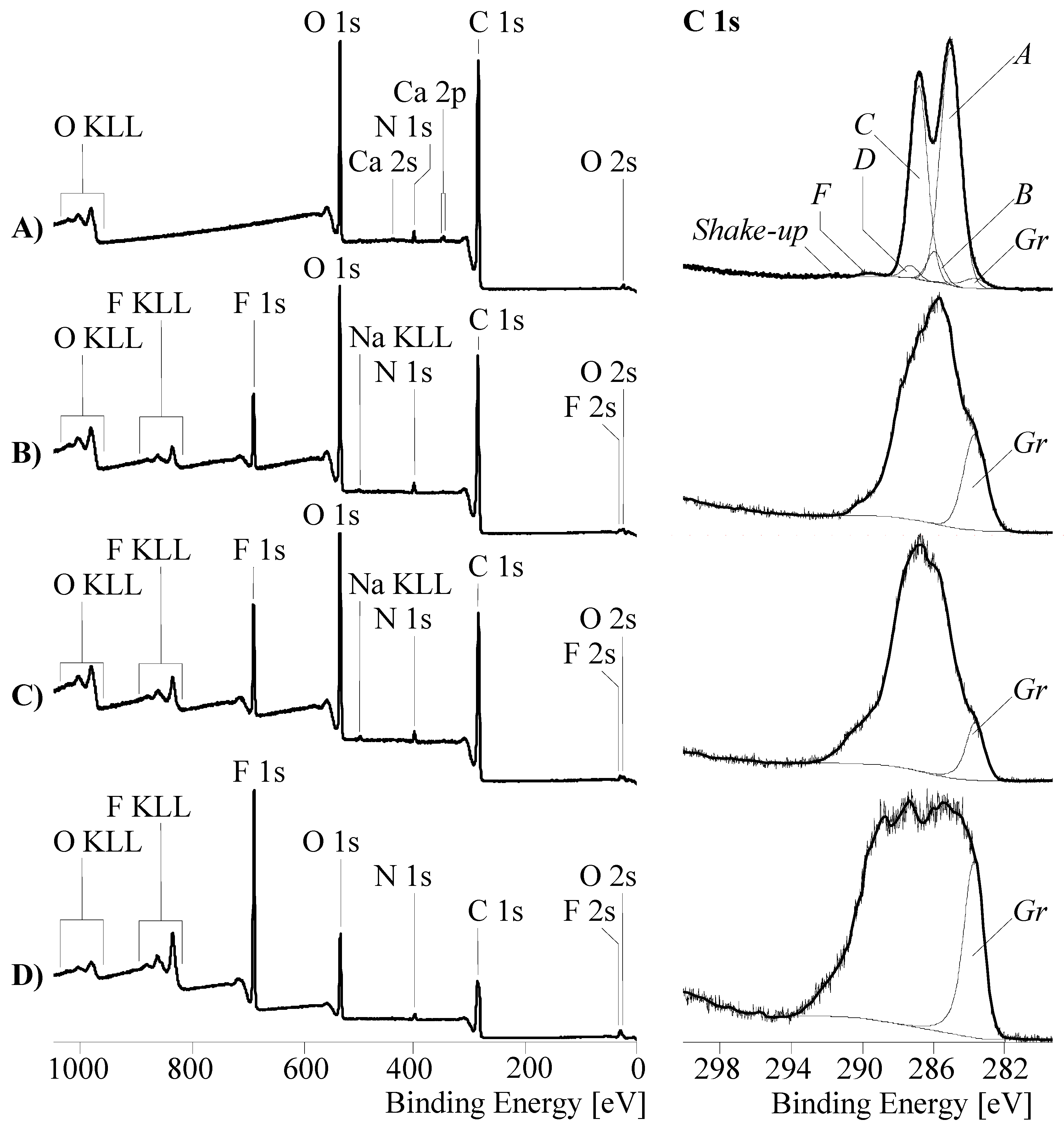
2.3. X-ray Photoelectron Spectroscopy
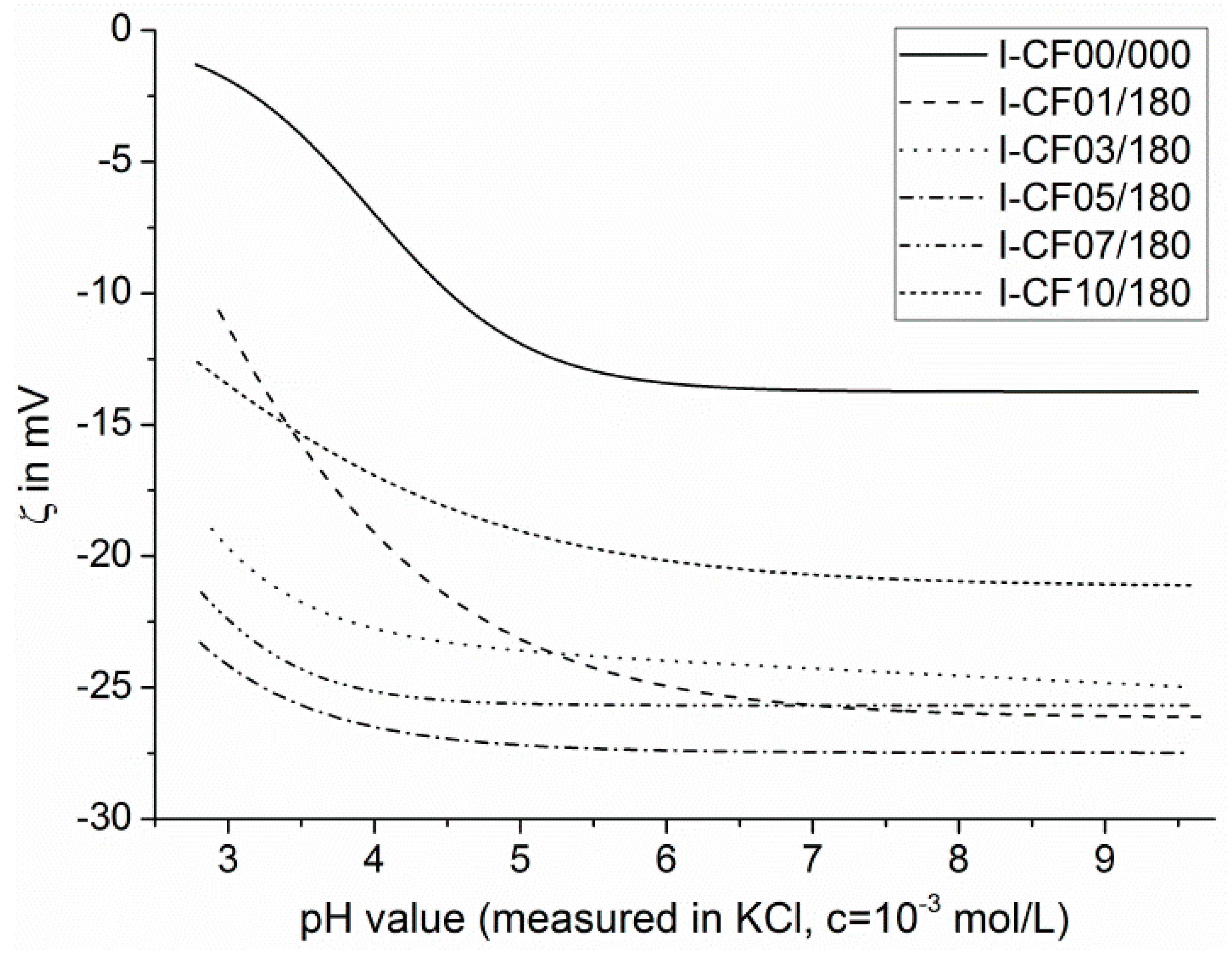
2.4. Electrokinetic Measurements
2.5. Single Fiber Tensile Test
2.6. Determination of the Surface Free Energy
2.7. Determination of Density
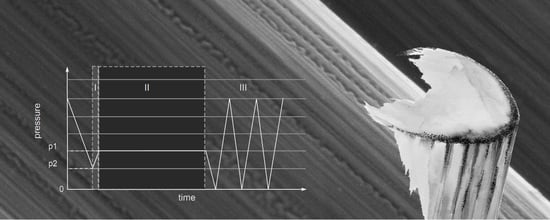
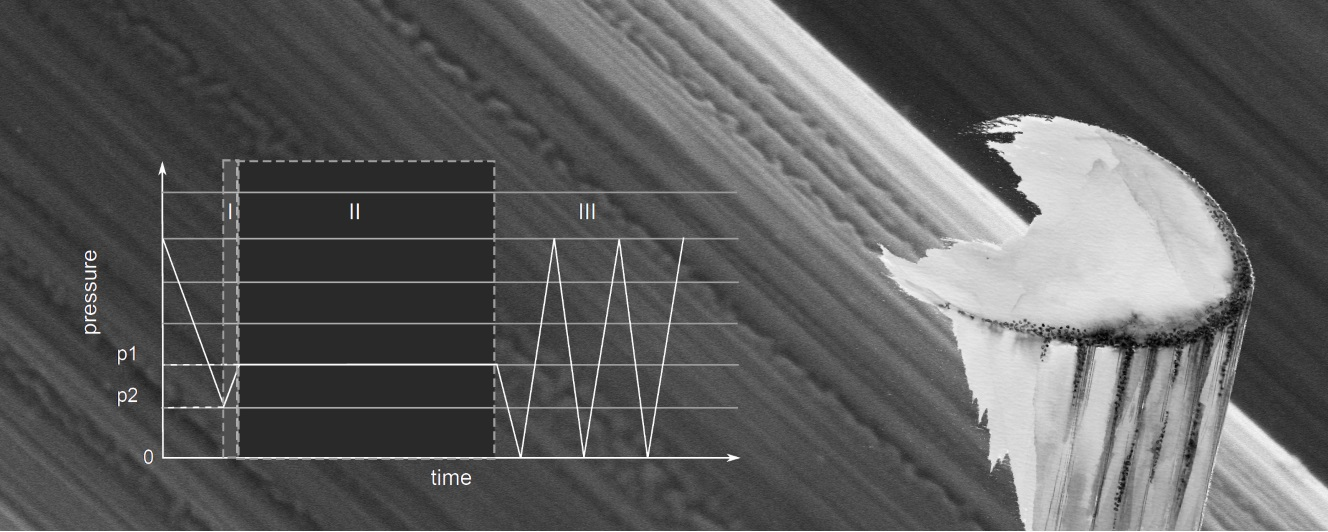
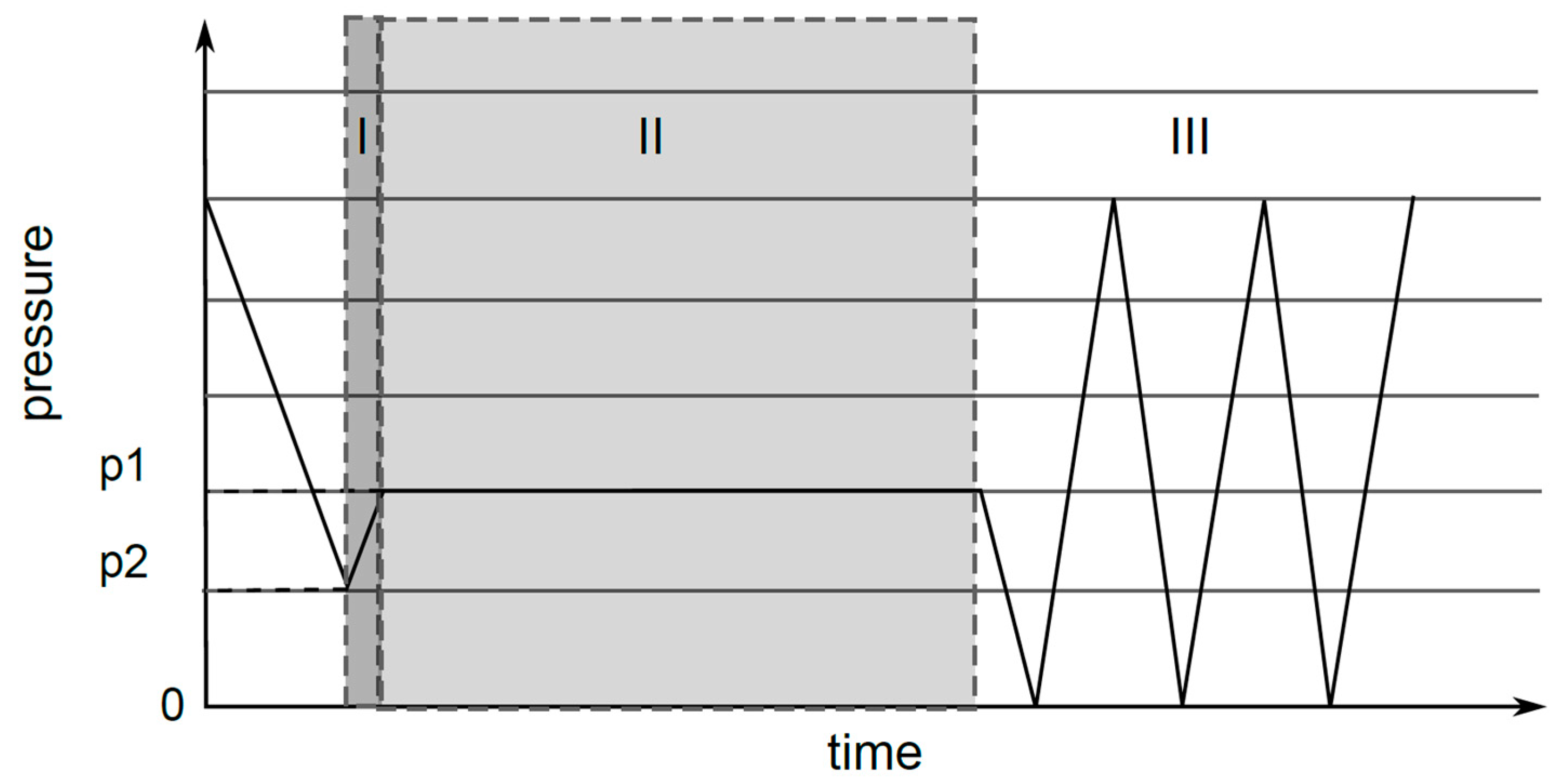
2.8. Oxy-Fluorination Procedure
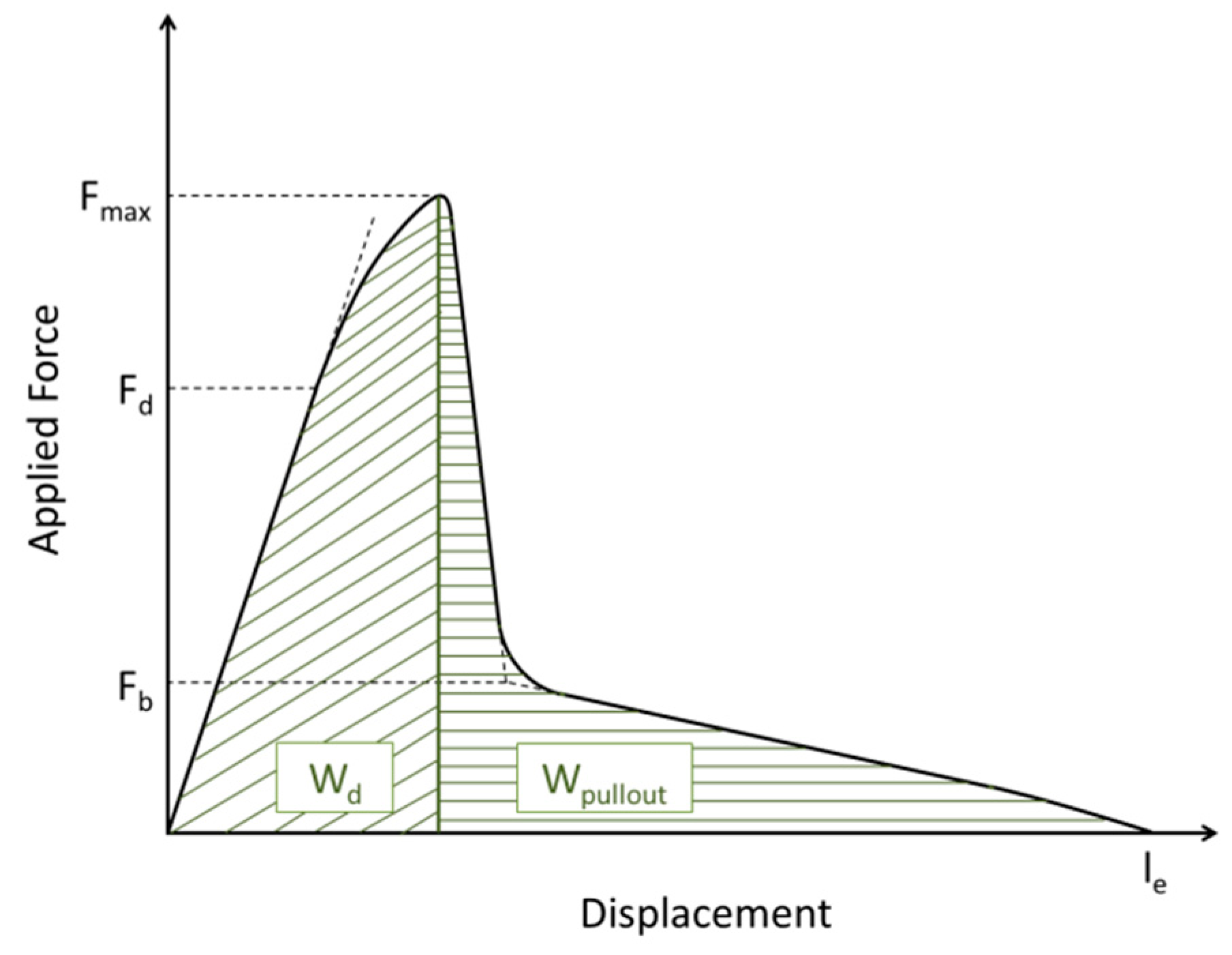
2.9. Single Fiber Pull-Out Test
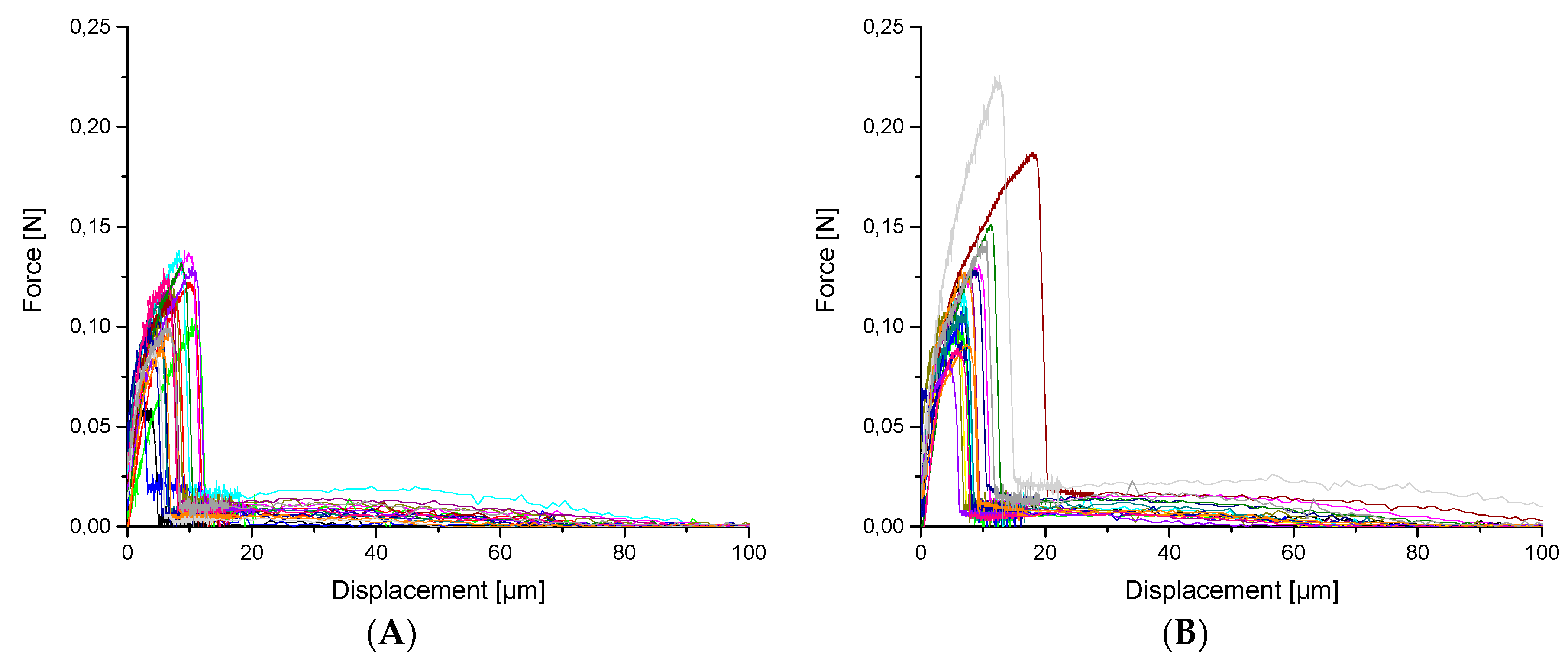
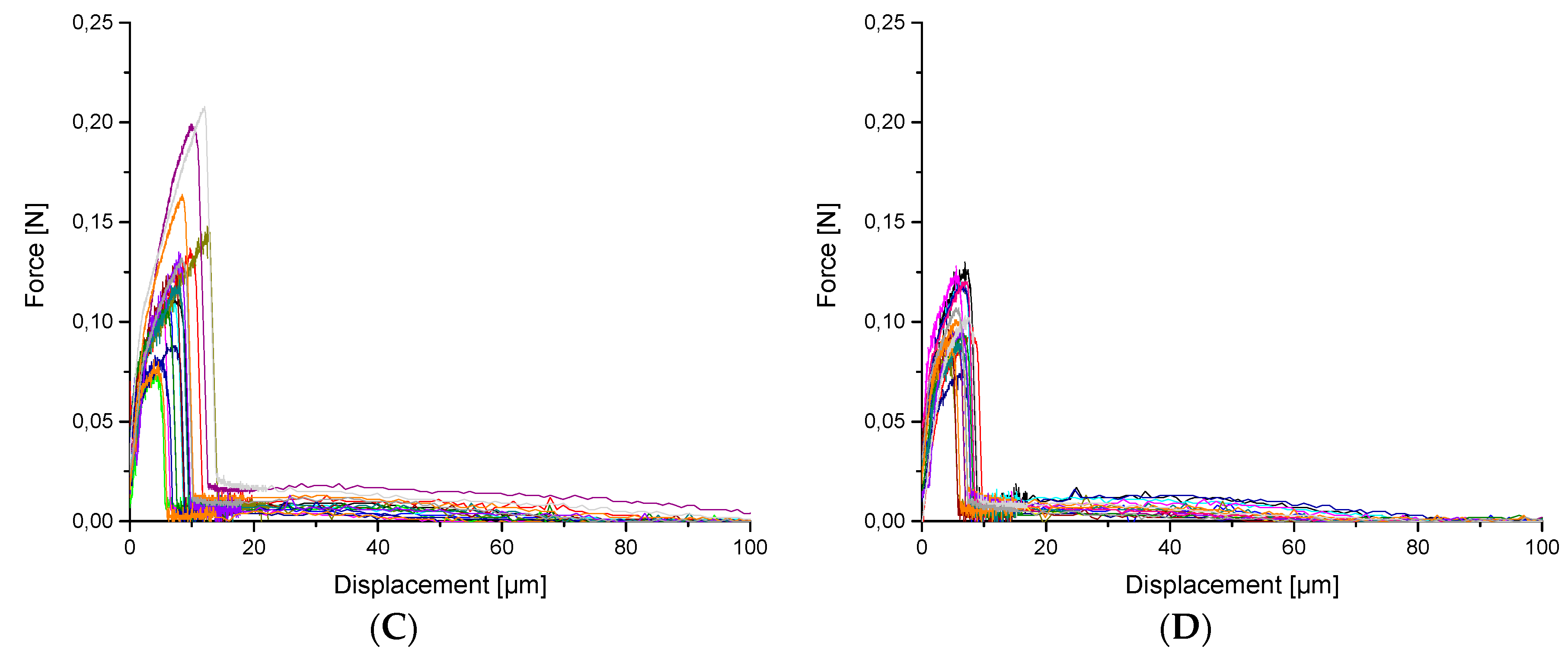
3. Results and Discussion
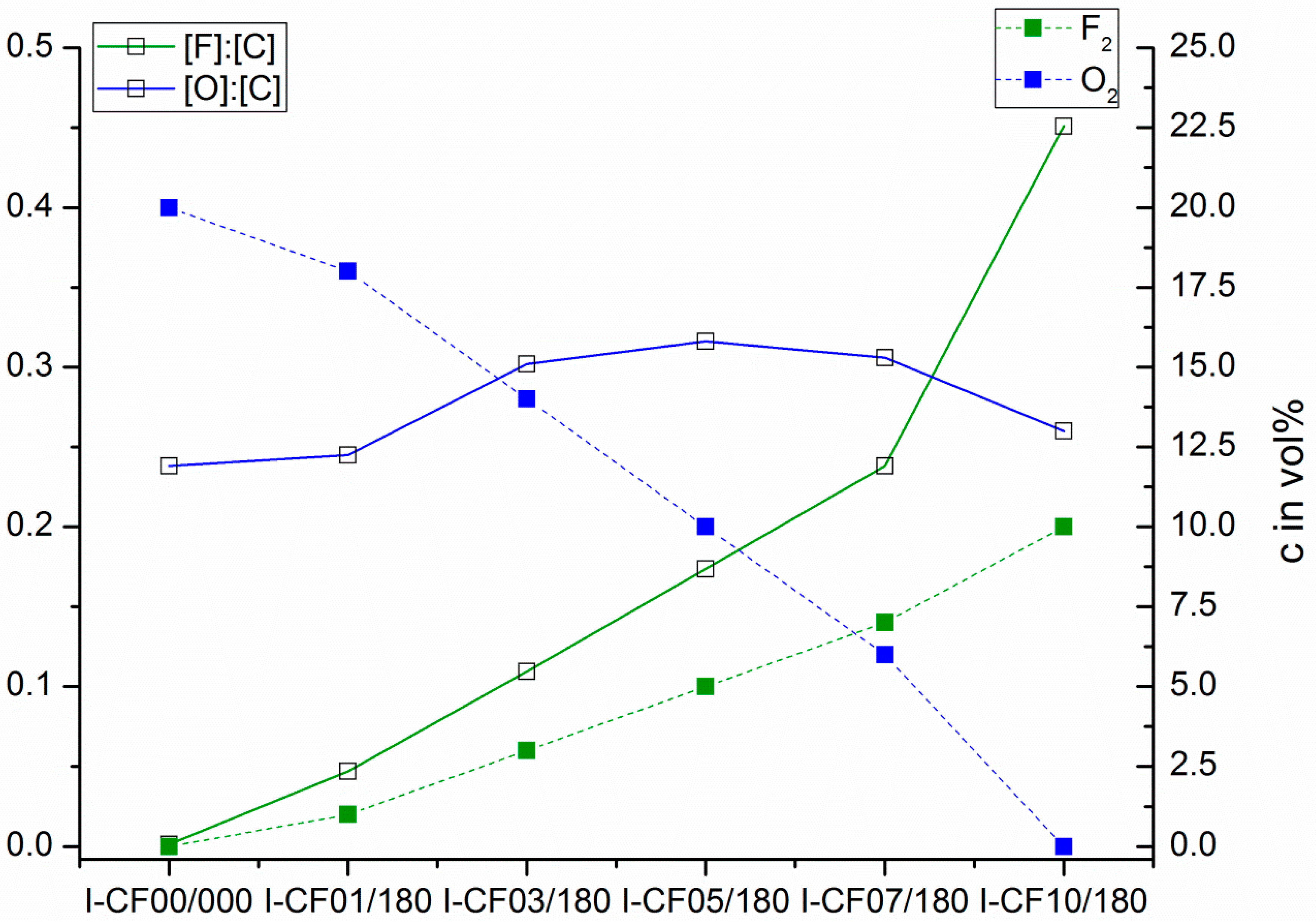
3.1. Surface Characterization and Bulk Properties
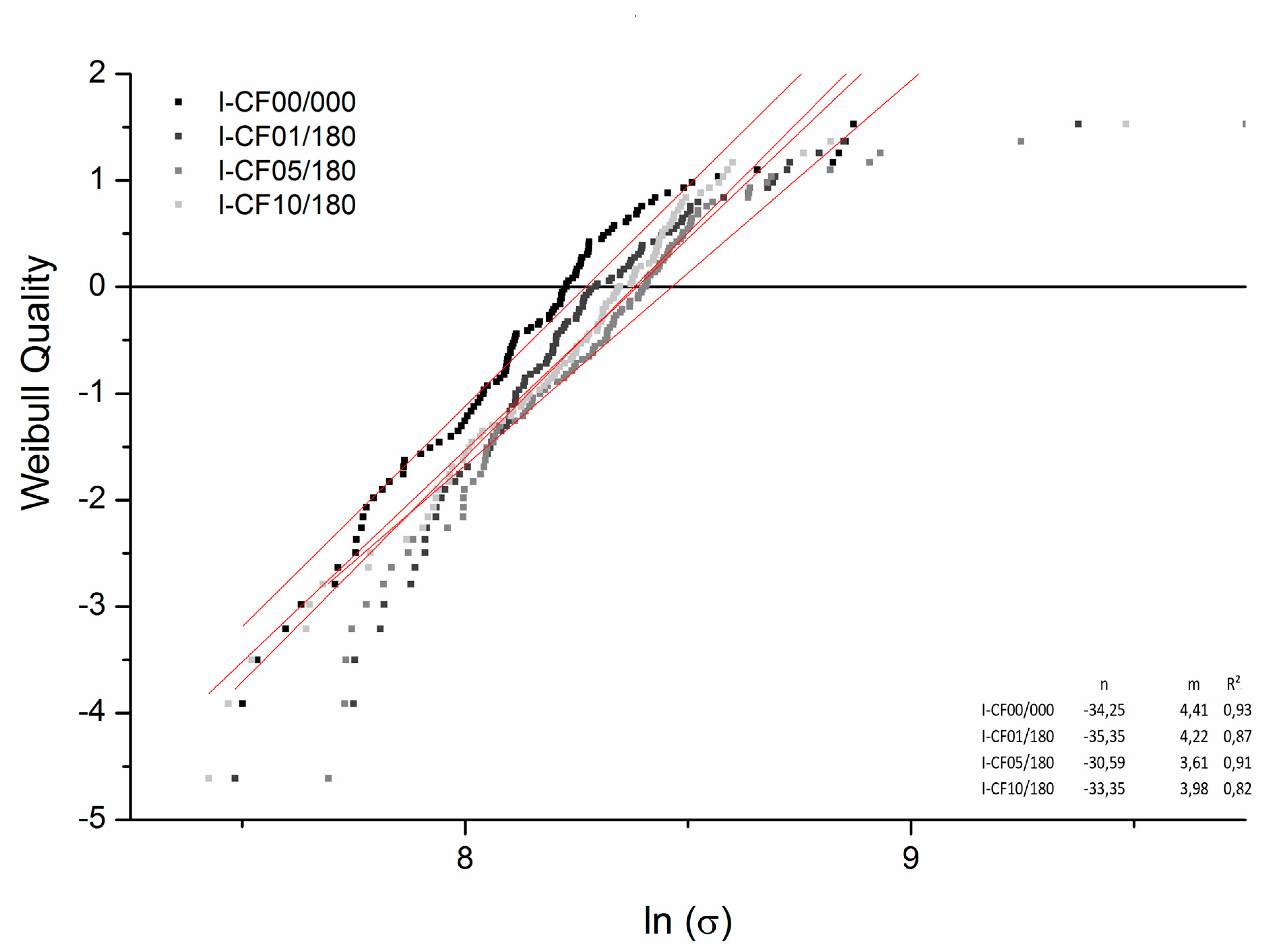
3.2. Mechanical tests
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, S.-J.; Seo, M.-K.; Rhee, K.-Y. Studies on mechnical interfacial properties of oxy-fluorinated carbon fibres-reinforced composites. Mater. Sci. Eng. A Struct. Mater. Prop. Microstr. Process. 2003, 356, 219–226. [Google Scholar] [CrossRef]
- Sharma, M.; Gao, S.; Mäder, E.; Sharma, H.; Wei, L.Y.; Bijwe, J. Carbon fiber surfaces and composite interphases. Compos. Sci. Technol. 2014, 102, 35–50. [Google Scholar] [CrossRef]
- Tang, L.-G.; Kardos, J.L. A review of methods for Improving the interferical Adhesion Between Carbon Fiber and Polymer Matrix. Polym. Compos. 1997, 18, 100–113. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, H.J.; Cho, B.W.; Park, C.R. Effects of sulfuric acid treatment on the microstructure and electrochemical performance of a polyacrylonitrile (PAN)-based carbon anode. Carbon 2005, 43, 163–169. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, S.; Yang, D.; Doelet, J.-P.; Sacher, E. The surface analytical characterization of carbon fibers functionalized by H2SO4/HNO3 treatment. Carbon 2008, 46, 196–205. [Google Scholar] [CrossRef]
- Pittman, C.U., Jr.; Jiang, W.; Yue, Y.Z.; Gradner, S.; Wang, L.; Toghiani, H. Surface properties of electrochemically oxidized carbon fibers. Carbon 1999, 37, 1797–1808. [Google Scholar] [CrossRef]
- Yue, Z.R.; Jiang, W.; Wang, L.; Gardner, S.D.; Pittman, C.U., Jr. Surface characterization of electrochemically oxidized carbon fibers. Carbon 1999, 37, 1785–1796. [Google Scholar] [CrossRef]
- Wu, G.M.; Hung, C.H.; You, J.H.; Liu, S.J. Surface Modification of Reinforcement Fibers for Composites by Acid Treatments. J. Polym. Res. 2004, 11, 31–36. [Google Scholar] [CrossRef]
- Jang, J.; Yang, H. The effect of surface treatment on the performance improvement of carbon fiber/polybenzoxazine composites. J. Mater. Sci. 2000, 35, 2297–2303. [Google Scholar] [CrossRef]
- Pittmann, C.U.; He, G.-R.; Gradner, SD. Chemical Modification of Carbon Fiber Surfaces By Nitric Acid Oxidation Followed By Reaktion With Tetraethylenepentamine. Carbon 1997, 35, 317–331. [Google Scholar] [CrossRef]
- Wu, Z.; Pittman, C.U., Jr.; Grardner, S.D. Nitric Acid Oxidation of Carbon Fibres and the Effects odf Subsequent Treatment in Relfuxing Aqueous NaOH. Carbon 1995, 33, 597–605. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, M.-H. Effect of acidic anode treatment on carbon fibers for increasing fiber-matrix adhesion and its relationship to interlaminar shear. J. Mater. Sci. 2000, 35, 1901–1905. [Google Scholar] [CrossRef]
- Allred, R.E.; Schimpf, W.C. CO2 plasma modification of high-modulus carbon fibers and their adhesion to epoxy resins. J. Adhes. Sci. Technol. 1994, 8, 383–394. [Google Scholar] [CrossRef]
- Bogoeva-Gaceva, G.; Mäder, E.; Häussler, L.; Dekanski, A. Characterization of the surface and interphase of plasma-treated HM carbon fibres. Compos. A. 1997, 28, 445–452. [Google Scholar] [CrossRef]
- Farrow, G.J.; Jones, C. The Effect of Low Power Nitrogen Plasma Treatment of Carbon Fibres on the Interfacial Shear Strength of Carbon Fibre/Epoxy Composites. J. Adhes. 1994, 45, 29–42. [Google Scholar] [CrossRef]
- Han, S.H.; Oh, H.J.; Kim, S.S. Evaluation of fiber surface treatment on the interfacial behavior of carbon fiber-reinforced polypropylene composites. Compos. B. 2014, 60, 98–105. [Google Scholar] [CrossRef]
- Käppler, I.; Hund, R.D.; Cherif, C. Surface modification of carbon fibres using plasma technique. AUTEX Res. J. 2014, 14, 34–38. [Google Scholar] [CrossRef]
- Park, S.-J.; Chang, Y.-H.; Moon, C.-W.; Suh, D.-H.; Im, S.-S.; Kim, Y.-C. A Study of Atmospheric Plasma Treatment on Surface Energetics of Carbon Fibers. Bull. Korean Chem. Soc. 2010, 31, 335–338. [Google Scholar] [CrossRef]
- Scheffler, C.; Wörfel, E.; Förster, T.; Poitzsch, C.; Klotte, L.; Mäder, G. Influence of microwave plasma treatment on the surface properties of carbon fibers and their adhesion in a polypropylene matrix. IOP Conf. Ser. Mater. Sci. Eng. 2016, 139, 012046. [Google Scholar] [CrossRef]
- Tiwari, S.; Sharma, M.; Panier, S.; Mutel, B.; Mitschang, P.; Bijwe, J. Influence of cold remote nitrogen oxygen plasma treatment on carbon fabric and its composites with specialty polymers. J. Mater. Sci. 2011, 46, 964–974. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Ban, K.; Shibata, K.; Okazaki, S.; Kogoma, M. Wettability and Adhesion Characteristics of Plasma-Treated Carbon Fibers. J. Adhes. 1996, 57, 45–53. [Google Scholar] [CrossRef]
- Yuan, L.Y.; Shyu, S.S.; Lai, J.Y. Plasma Surface Treatments on Carbon Fibers. II. Mechanical Property and Interfacial Shear Strength. J. Appl. Polym. Sci. 1991, 42, 2525–2534. [Google Scholar] [CrossRef]
- Mathur, R.B.; Gupta, V.; Bahl, O.P.; Tressaud, A.; Flandrois, S. Improvement in the mechanical properties of polyacrylonitrile (PAN)–based carbon fibers after fluorination. Synth. Metals 2000, 114, 197–200. [Google Scholar] [CrossRef]
- Tressaud, A.; Durand, E.; Labrugere, C.; Kharitonov, A.P.; Kharitonova, L.N. Modification of surface properties of carbon-based and polymeric materials through fluorination routes: From fundamental research to industrial applications. J. Fluor. Chem. 2007, 128, 378–391. [Google Scholar] [CrossRef]
- Tressaud, A.; Guimon, C.; Gupta, V.; Mouget, F. Fluorine-intercalated carbon fibers: II: An X-ray photoelectron spectroscopy study. Mater. Sci. Engin B. 1995, 30, 62–68. [Google Scholar] [CrossRef]
- Kharitonov, A.P. Direct fluorination of polymers–From fundamental research to industrial applications. Prog. Organ. Coat. 2008, 61, 192–204. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Kharitonova, L.N. Surface modification of polymers by direct fluorination: A convenient approach to improve commercial properties of polymeric articles. Pure Appl. Chem. 2009, 81, 451–471. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Simbirtseva, G.V.; Bouznik, V.M.; Chebzubov, M.G.; Dubois, M.; Guérin, K. Modification of Ultra-High-Molecular Weight Polyethylene by Various Fluorinating Routes. J. Polym. Sci. A Polym. Chem. 2011, 49, 3559–3573. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Taege, R.; Ferrier, G.; Piven, N.P. The kinetics and mechanism of the direct fluorination of polyethylenes. Surf. Coat. Int. B Coat. Trans. 2005, 38, 201–212. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Taege, R.; Ferrier, G.; Teplyakov, V.V.; Syrtsova, D.A.; Koops, G.-H. Direct fluorination–Useful tool to enhance commercial properties of polymer articles. J. Fluor. Chem. 2005, 126, 251–263. [Google Scholar] [CrossRef]
- Käppler, I.; Matthäi, P.; Cherif, C. Adhesion Problematic for Novel Non-Crimp Fabric and Surface Modification of Carbon-Fibres Using Oxy-Fluorination. Int. J. Chem. Nucl. Mater. Metall. Eng. 2014, 8, 1390–1395. [Google Scholar]
- Kruppke, I.; Bartusch, M.; Hickmann, R.; Hund, R.-D.; Cherif, C. Effects of (Oxy-) Fluorination on Various High-Performance Yarns. Molecules 2016, 21, 1127. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Tanaka, F.; Okuda, H.; Okabe, T. Tensile strength distribution of carbon fibers at short gauge lengths. Adv. Compos. Mater. 2014, 25, 535–550. [Google Scholar] [CrossRef]
- Kaelble, D.H.; Uy, K.C. A Reinterpretation of organic liquid-polytetrafluoroethylene surface interactions. J. Adhes. 1970, 2, 50–60. [Google Scholar] [CrossRef]
- Owens, D.K.; Wendt, R.C. Estimation of the Surface Free Energy of Polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- Rabel, W. Liquid interfaces in theory and applied technology. Phys. Blätter 1977, 33, 151–156. [Google Scholar] [CrossRef]
- Mäder, E.; Grundke, K.; Jacobasch, H.J.; Wachinger, G. Surface, interphase and composite property relation in fibre-reinforced polymers. Composites 1994, 25, 739–744. [Google Scholar] [CrossRef]
- Pisanova, E.V.; Zhandarov, S.F.; Dovgyalo, V.A. Interfacial adhesion and failure modes in single filament thermoplastic composites. Polym. Compos. 1994, 15, 147–155. [Google Scholar] [CrossRef]
- Miller, B.; Muri, P.; Rebenfeld, L. A microbond method for determination of the shear strength of a fiber–resin interface. Compos. Sci Technol. 1987, 28, 17–32. [Google Scholar] [CrossRef]
- Zhandarov, S.; Maeder, E. Characterization of fiber/matrix interface strength: Applicability of different tests, approaches and parameters. Compos. Sci. Technol. 2005, 65, 149–160. [Google Scholar] [CrossRef]
- Zhandarov, S.F.; Maeder, E.; Yurkevich, O.R. Indirect estimation of fiber/polymer bond strength and interfacial friction from maximum load values recorded in the microbond and pull-out tests. Part I: Local bond strength. J. Adhes. Sci. Technol. 2002, 16, 1171–1200. [Google Scholar] [CrossRef]
- Krekel, G.; Huettinger, K.J.; Hoffmann, W.P.; Silver, D.S. The relevance of the surface structure and surface chemistry of carbon fibres in their adhesion to high-temperature thermoplastics. J. Mater. Sci. 1994, 29, 2968–2980. [Google Scholar] [CrossRef]
- Nanse, G.; Papirer, E.; Fious, P.; Moguet, F.; Tressaud, A. Fluorination of Carbon Blacks: An X-ray Photoelectron Spectroscopy Study: I. A Literature Review of XPS Studies of Fluorinated Carbons. XPS Investigation of Some Reference Compounds. Carbon 1997, 35, 175–194. [Google Scholar] [CrossRef]
- Mishra, A.K.; Chattopadhyay, D.K.; Sreedhar, B.; Raju, K.V.S.N. FT-IR and XPS studies of polyurethane-urea-imide coatings. Progr. Org. Coat. 2006, 55, 231–243. [Google Scholar] [CrossRef]
- Sherwood, P.M.A. Surface analysis of carbon and carbon fibers for composites. J. Electr. Spectrosc. Relat. Phenom. 1996, 81, 319–342. [Google Scholar] [CrossRef]
- Bismarck, A.; Tahhan, R.; Springer, J.; Schulz, A.; Klapötke, T.M.; Zell, H. Influence of fluorination on the properties of carbon fibres. J. Fluor. Chem. 1997, 84, 127–134. [Google Scholar] [CrossRef]
- Nordstroem, Y.; Joffe, R.; Sjoeholm, E. Mechanical Characterization and Application of Weibull Statistics to the Strength of Softwood Lignin-Based Carbon Fibers. J. Appl. Polym. Sci. 2013, 130, 3689–3697. [Google Scholar] [CrossRef]
- Beetz, C.P., Jr. A self-consistant Weibull analysis of carbon fibre strength distribution. Fibre Sci. Technol. 1982, 16, 81–94. [Google Scholar] [CrossRef]
- Williams, W.S.; Steffens, D.A.; Bacon, R. Bending Behavior and Tensile Strength of Carbon Fibers. J. Appl. Phys. 1970, 41, 4893–4901. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen | F2/O2 Mixture in N2 | Conditions |
|---|---|---|
| I-CF00/000 | No treatment | |
| I-CF01/180 | 1 vol%/18 vol% | 25 °C/180 s |
| I-CF03/180 | 3 vol%/14 vol% | 25 °C/180 s |
| I-CF05/180 | 5 vol%/10 vol% | 25 °C/180 s |
| I-CF07/180 | 7 vol%/6 vol% | 25 °C/180 s |
| I-CF10/180 | 10 vol%/0 vol% | 25 °C/180 s |
| Specimen | I-CF00/000 | I-CF01/180 | I-CF03/180 | I-CF05/180 | I-CF07/180 | I-CF10/180 |
|---|---|---|---|---|---|---|
| Density (g/cm3) | 1.719 ± 0.034 | 1.753 ± 0.019 | 1.768 ± 0.044 | 1.756 ± 0.024 | 1.786 ± 0.092 | 1.803 ± 0.045 |
| Sample | σtotal (mN·m−1) | σP (mN·m−1) | σD (mN·m−1) | θwater (°) | θCH2I2 (°) | [O]:[C] | [F]:[C] |
|---|---|---|---|---|---|---|---|
| I-CF00/000 | 60.60 ± 0.42 | 16.90 ± 0.25 | 43.71 ± 0.33 | 49.05 ± 3.30 | 30,66 ± 6.69 | 0.238 | 0.013 |
| I-CF01/180 | 49.13 ± 0.81 | 14.27 ± 0.51 | 34.87 ± 0.64 | 59.98 ± 5.39 | 48.33 ± 9.41 | 0.245 | 0.047 |
| I-CF03/180 | 50.68 ± 1.01 | 14.16 ± 0.58 | 36.52 ± 0.83 | 58.89 ± 6.99 | 45.49 ± 7.40 | 0.302 | 0.109 |
| I-CF05/180 | 52.11 ± 0.79 | 15.42 ± 0.47 | 36.68 ± 0.63 | 56.66 ± 5.57 | 45.18 ± 7.62 | 0.319 | 0.173 |
| I-CF07/180 | 48.84 ± 0.82 | 15.66 ± 0.47 | 33.18 ± 0.68 | 58.83 ± 5.80 | 51.83 ± 4.67 | 0.306 | 0.238 |
| I-CF10/180 | 44.62 ± 0.38 | 15.87 ± 0.22 | 28.75 ± 0.30 | 61.98 ± 2.52 | 59.66 ± 2.98 | 0.260 | 0.451 |
| Specimen | I-CF00/000 | I-CF01/180 | I-CF05/180 | I-CF10/180 |
|---|---|---|---|---|
| Fmax(mean) (cN) | 14.17 ± 2.75 | 15.08 ± 2.97 | 15.60 ± 2.87 | 15.11 ± 2.81 |
| Fmax(median) (cN) | 14.33 | 15.08 | 15.50 | 15.39 |
| σmax(mean) (MPa) | 3538.94 | 3964.36 | 4281.16 | 3975.53 |
| Specimen | I-CF00/000 | I-CF01/180 | I-CF05/180 | I-CF10/180 |
|---|---|---|---|---|
| τapp (N/mm2) | 65 ± 10 | 65 ± 5 | 68 ± 15 | 64 ± 6 |
| τd (N/mm2) | 66 ± 11 | 66 ± 7 | 63 ± 11 | 62 ± 5 |
| τf (N/mm2) | 5.5 ± 3.4 | 5.5 ± 2.1 | 4.0 ± 2.0 | 4.7 ± 2.0 |
| Wd (mN/mm) | 0.71 ± 0.30 | 0.97 ± 0.66 | 0.94 ± 0.48 | 0.60 ± 0.16 |
| Wpullou (mN/mm) | 0.70 ± 0.26 | 0.91 ± 0.46 | 0.73 ± 0.30 | 0.61 ± 0.19 |
| Wtotal (mN/mm) | 1.42 ± 0.51 | 1.88 ± 1.09 | 1.70 ± 0.69 | 1.21 ± 0.31 |
| le (µm) | 77 ± 17 | 86 ± 21 | 76 ± 17 | 71 ± 11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruppke, I.; Scheffler, C.; Simon, F.; Hund, R.-D.; Cherif, C. Surface Treatment of Carbon Fibers by Oxy-Fluorination. Materials 2019, 12, 565. https://doi.org/10.3390/ma12040565
Kruppke I, Scheffler C, Simon F, Hund R-D, Cherif C. Surface Treatment of Carbon Fibers by Oxy-Fluorination. Materials. 2019; 12(4):565. https://doi.org/10.3390/ma12040565
Chicago/Turabian StyleKruppke, Iris, Christina Scheffler, Frank Simon, Rolf-Dieter Hund, and Chokri Cherif. 2019. "Surface Treatment of Carbon Fibers by Oxy-Fluorination" Materials 12, no. 4: 565. https://doi.org/10.3390/ma12040565
APA StyleKruppke, I., Scheffler, C., Simon, F., Hund, R.-D., & Cherif, C. (2019). Surface Treatment of Carbon Fibers by Oxy-Fluorination. Materials, 12(4), 565. https://doi.org/10.3390/ma12040565