The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors

 ,
,  ,
, 
Abstract
1. Introduction
2. The Tumor Niche
3. Tumors-On-Chips: A Superior Alternative for Emulating the Tumor Micro-Niche
4. Tumor Spheroids: Fabrication Techniques
5. Tumor-on-Chip Examples
5.1. Spheroids in Microfluidic Chambers
5.2. Spheroids in a Hydrogel Matrix
5.3. Non-Spheroid Models where 3D Cancer Tissues are in Contact with Non-Cancerous Tissues
5.4. Spheroids Surrounded by “Healthy” Tissues
5.5. Vascularized Tumor-On-Chip Systems
6. Applications: Toward Precision Medicine
7. Challenges and Perspectives
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, K.; Zheng, R.; Zeng, H.; Zhang, S.; Xia, C.; Yang, Z.; Li, H.; Zou, X.; He, J. Cancer incidence and mortality in China, 2014. Chin. J. Cancer Res. 2018, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.J.; Rothenberg, M.L.; Pentz, R.; Hricak, H.; Abernethy, A.; Anderson, K.; Gee, A.W.; Harvey, R.D.; Piantadosi, S.; Bertagnolli, M.M.; et al. Accelerating anticancer drug development—Opportunities and trade-offs. Nat. Rev. Clin. Oncol. 2018, 15, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Verjans, E.-T.; Doijen, J.; Luyten, W.; Landuyt, B.; Schoofs, L. Three-dimensional cell culture models for anticancer drug screening: Worth the effort? J. Cell. Physiol. 2018, 233, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Hickman, J.A. Limits to Personalized Cancer Medicine. N. Engl. J. Med. 2016, 375, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T. Finding the tumor copycat: Approximating a human cancer. Nat. Med. 2010, 16, 976–977. [Google Scholar] [CrossRef] [PubMed]
- Heylman, C.; Sobrino, A.; Shirure, V.S.; Hughes, C.C.; George, S.C. A strategy for integrating essential three-dimensional microphysiological systems of human organs for realistic anticancer drug screening. Exp. Biol. Med. (Maywood) 2014, 239, 1240–1254. [Google Scholar] [CrossRef]
- Zheng, F.; Fu, F.; Cheng, Y.; Wang, C.; Zhao, Y.; Gu, Z. Organ-on-a-Chip Systems: Microengineering to Biomimic Living Systems. Small 2016, 12, 2253–2282. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef]
- Trujillo-de Santiago, G.; Lobo-Zegers, M.J.; Montes-Fonseca, S.L.; Zhang, Y.S.; Alvarez, M.M. Gut-microbiota-on-a-chip: An enabling field for physiological research. Microphysiol. Syst. 2018, 2. [Google Scholar] [CrossRef]
- Zhang, B.; Radisic, M. Organ-on-a-chip devices advance to market. Lab Chip 2017, 17, 2395–2420. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-F.; Trubelja, A.; Shen, A.Q.; Bao, G. Tumour-on-a-chip: Microfluidic models of tumour morphology, growth and microenvironment. J. R. Soc. Interface 2017, 14, 20170137. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Luo, Z.; Lee, J.; Kim, H.-J.; Lee, K.; Tebon, P.; Feng, Y.; Dokmeci, M.R.; Sengupta, S.; Khademhosseini, A. Organ-on-a-Chip for Cancer and Immune Organs Modeling. Adv. Healthc. Mater. 2019, 8, 1801363. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Soon, R.H.; Lim, C.T.; Khoo, B.L.; Han, J. Microfluidic modelling of the tumor microenvironment for anti-cancer drug development. Lab Chip 2019, 19, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Caballero, D.; Kaushik, S.; Correlo, V.M.; Oliveira, J.M.; Reis, R.L.; Kundu, S.C. Organ-on-chip models of cancer metastasis for future personalized medicine: From chip to the patient. Biomaterials 2017, 149, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, A.; Mummery, C.L.; Passier, R.; van der Meer, A.D. Personalised organs-on-chips: Functional testing for precision medicine. Lab Chip 2019, 19, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, C.; Park, K.; Konieczny, S.F.; Korc, M. Recapitulation of complex transport and action of drugs at the tumor microenvironment using tumor-microenvironment-on-chip. Cancer Lett. 2016, 380, 319–329. [Google Scholar] [CrossRef]
- Ruppen, J.; Cortes-Dericks, L.; Marconi, E.; Karoubi, G.; Schmid, R.A.; Peng, R.; Marti, T.M.; Guenat, O.T. A microfluidic platform for chemoresistive testing of multicellular pleural cancer spheroids. Lab Chip 2014, 14, 1198–1205. [Google Scholar] [CrossRef]
- Huang, K.; Boerhan, R.; Liu, C.; Jiang, G. Nanoparticles Penetrate into the Multicellular Spheroid-on-Chip: Effect of Surface Charge, Protein Corona, and Exterior Flow. Mol. Pharm. 2017, 14, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Patra, B.; Peng, C.-C.; Liao, W.-H.; Lee, C.-H.; Tung, Y.-C. Drug testing and flow cytometry analysis on a large number of uniform sized tumor spheroids using a microfluidic device. Sci. Rep. 2016, 6, 21061. [Google Scholar] [CrossRef]
- Lim, W.; Park, S.; Lim, W.; Park, S. A Microfluidic Spheroid Culture Device with a Concentration Gradient Generator for High-Throughput Screening of Drug Efficacy. Molecules 2018, 23, 3355. [Google Scholar] [CrossRef] [PubMed]
- Holton, A.B.; Sinatra, F.L.; Kreahling, J.; Conway, A.J.; Landis, D.A.; Altiok, S. Microfluidic Biopsy Trapping Device for the Real-Time Monitoring of Tumor Microenvironment. PLoS ONE 2017, 12, e0169797. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Fluri, D.A.; Marchan, R.; Boonen, K.; Mohanty, S.; Singh, P.; Hammad, S.; Landuyt, B.; Hengstler, J.G.; Kelm, J.M.; et al. 3D spherical microtissues and microfluidic technology for multi-tissue experiments and analysis. J. Biotechnol. 2015, 205, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-T.; Chiang, C.-L.; Chang, C.-H.; Liu, H.-K.; Huang, G.-S.; Yun-Ju Huang, R.; Lee, H.; Huang, C.-S.; Wo, A.M. Modeling of cancer metastasis and drug resistance via biomimetic nano-cilia and microfluidics. Biomaterials 2014, 35, 1562–1571. [Google Scholar] [CrossRef]
- Albanese, A.; Lam, A.K.; Sykes, E.A.; Rocheleau, J.V.; Chan, W.C.W. Tumour-on-a-chip provides an optical window into nanoparticle tissue transport. Nat. Commun. 2013, 4, 2718. [Google Scholar] [CrossRef]
- Kwak, B.; Ozcelikkale, A.; Shin, C.S.; Park, K.; Han, B. Simulation of complex transport of nanoparticles around a tumor using tumor-microenvironment-on-chip. J. Control. Release 2014, 194, 157–167. [Google Scholar] [CrossRef]
- Shin, C.S.; Kwak, B.; Han, B.; Park, K. Development of an in vitro 3D tumor model to study therapeutic efficiency of an anticancer drug. Mol. Pharm. 2013, 10, 2167–2175. [Google Scholar] [CrossRef]
- Piotrowski-Daspit, A.S.; Tien, J.; Nelson, C.M. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and Transition to invasion. Integr. Biol. 2016, 8, 319–331. [Google Scholar] [CrossRef]
- Bender, B.F.; Aijian, A.P.; Garrell, R.L. Digital microfluidics for spheroid-based invasion assays. Lab Chip 2016, 16, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Lanz, H.L.; Saleh, A.; Kramer, B.; Cairns, J.; Ng, C.P.; Yu, J.; Trietsch, S.J.; Hankemeier, T.; Joore, J.; Vulto, P.; et al. Therapy response testing of breast cancer in a 3D high-throughput perfused microfluidic platform. BMC Cancer 2017, 17, 709. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.E.; Yang, N.; Pehlke, C.; Keely, P.J.; Eliceiri, K.W.; Friedl, A.; Beebe, D.J. Transition to invasion in breast cancer: A microfluidic in vitro model enables examination of spatial and temporal effects. Integr. Biol. (Camb.) 2011, 3, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Hyun, E.; Seo, J.; Blundell, C.; Kim, H.C.; Lee, E.; Lee, S.H.; Moon, A.; Moon, W.K.; Huh, D. A microengineered pathophysiological model of early-stage breast cancer. Lab Chip 2015, 15, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Gioiella, F.; Urciuolo, F.; Imparato, G.; Brancato, V.; Netti, P.A. An Engineered Breast Cancer Model on a Chip to Replicate ECM-Activation In vitro during Tumor Progression. Adv. Healthc. Mater. 2016, 5, 3074–3084. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, J.M.; Virumbrales-Muñoz, M.; Lacueva, A.; Lanuza, P.M.; Checa-Chavarria, E.; Botella, P.; Fernández, E.; Doblare, M.; Allison, S.J.; Phillips, R.M.; et al. Development and characterization of a microfluidic model of the tumour microenvironment. Sci. Rep. 2016, 6, 36086. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gao, Y.; Hao, Y.; Li, E.; Wang, Y.; Zhang, J.; Wang, W.; Gao, Z.; Wang, Q. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials 2013, 34, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Regier, M.C.; Maccoux, L.J.; Weinberger, E.M.; Regehr, K.J.; Berry, S.M.; Beebe, D.J.; Alarid, E.T. Transitions from mono- to co- to tri-culture uniquely affect gene expression in breast cancer, stromal, and immune compartments. Biomed. Microdevices 2016, 18, 70. [Google Scholar] [CrossRef]
- Hassell, B.A.; Goyal, G.; Lee, E.; Sontheimer-Phelps, A.; Levy, O.; Chen, C.S.; Ingber, D.E. Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In vitro. Cell Rep. 2017, 21, 508–516. [Google Scholar] [CrossRef]
- Buchanan, C.F.; Verbridge, S.S.; Vlachos, P.P.; Rylander, M.N. Flow shear stress regulates endothelial barrier function and expression of angiogenic factors in a 3D microfluidic tumor vascular model. Cell Adh. Migr. 2014, 8, 517–524. [Google Scholar] [CrossRef]
- Trietsch, S.J.; Israëls, G.D.; Joore, J.; Hankemeier, T.; Vulto, P. Microfluidic titer plate for stratified 3D cell culture. Lab Chip 2013, 13, 3548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shiratsuchi, H.; Lin, J.; Chen, G.; Reddy, R.M.; Azizi, E.; Fouladdel, S.; Chang, A.C.; Lin, L.; Jiang, H.; et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget 2014, 5, 12383–12397. [Google Scholar] [CrossRef] [PubMed]
- Menon, N.V.; Chuah, Y.J.; Cao, B.; Lim, M.; Kang, Y. A microfluidic co-culture system to monitor tumor-stromal interactions on a chip. Biomicrofluidics 2014, 8, 064118. [Google Scholar] [CrossRef] [PubMed]
- Businaro, L.; De Ninno, A.; Schiavoni, G.; Lucarini, V.; Ciasca, G.; Gerardino, A.; Belardelli, F.; Gabriele, L.; Mattei, F. Cross talk between cancer and immune cells: Exploring complex dynamics in a microfluidic environment. Lab Chip 2013, 13, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cao, Y.; Zhang, S.; Zhao, Y.; Liu, X.; Shi, H.; Hu, K.; Zhu, G.; Ma, B.; Niu, H. A bladder cancer microenvironment simulation system based on a microfluidic co-culture model. Oncotarget 2015, 6, 37695–37705. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Brewer, B.M.; Yang, L.; Franco Coronel, O.E.; Hayward, S.W.; Webb, D.J.; Li, D. Stretching Fibroblasts Remodels Fibronectin and Alters Cancer Cell Migration. Sci. Rep. 2015, 5, 8334. [Google Scholar] [CrossRef]
- Jarvis, M.; Arnold, M.; Ott, J.; Pant, K.; Prabhakarpandian, B.; Mitragotri, S. Microfluidic co-culture devices to assess penetration of nanoparticles into cancer cell mass. Bioeng. Transl. Med. 2017, 2, 268–277. [Google Scholar] [CrossRef]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef]
- Bruce, A.; Evans, R.; Mezan, R.; Shi, L.; Moses, B.S.; Martin, K.H.; Gibson, L.F.; Yang, Y. Three-Dimensional Microfluidic Tri-Culture Model of the Bone Marrow Microenvironment for Study of Acute Lymphoblastic Leukemia. PLoS ONE 2015, 10, e0140506; [Google Scholar] [CrossRef]
- Aung, A.; Theprungsirikul, J.; Lim, H.L.; Varghese, S. Chemotaxis-driven assembly of endothelial barrier in a tumor-on-a-chip platform. Lab Chip 2016, 16, 1886–1898. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef] [PubMed]
- Montanez-Sauri, S.I.; Sung, K.E.; Berthier, E.; Beebe, D.J. Enabling screening in 3D microenvironments: Probing matrix and stromal effects on the morphology and proliferation of T47D breast carcinoma cells. Integr. Biol. 2013, 5, 631. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, S.-K.; Khawar, I.A.; Jeong, S.-Y.; Chung, S.; Kuh, H.-J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 4. [Google Scholar] [CrossRef] [PubMed]
- Mannino, R.G.; Santiago-Miranda, A.N.; Pradhan, P.; Qiu, Y.; Mejias, J.C.; Neelapu, S.S.; Roy, K.; Lam, W.A. 3D microvascular model recapitulates the diffuse large B-cell lymphoma tumor microenvironment in vitro. Lab Chip 2017, 17, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, A.; Ghousifam, N.; Hoopes, P.J.; Yankeelov, T.E.; Rylander, M.N. In vitro vascularized liver and tumor tissue microenvironments on a chip for dynamic determination of nanoparticle transport and toxicity. Biotechnol. Bioeng. 2019, 116, 1201–1219. [Google Scholar] [CrossRef] [PubMed]
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018, 18, 3687–3702. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Wang, H.; Sun, M.; Xu, J.; Zhao, S.; Liu, Z.; Gooch, K.J.; Zhao, Y.; Lu, X.; He, X. Microfluidics Enabled Bottom-Up Engineering of 3D Vascularized Tumor for Drug Discovery. ACS Nano 2017, 11, 6691–6702. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, A.; Phan, D.T.T.; Datta, R.; Wang, X.; Hachey, S.J.; Romero-López, M.; Gratton, E.; Lee, A.P.; George, S.C.; Hughes, C.C.W. 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 2016, 6, 31589. [Google Scholar] [CrossRef] [PubMed]
- Dereli-Korkut, Z.; Akaydin, H.D.; Ahmed, A.H.R.; Jiang, X.; Wang, S. Three Dimensional Microfluidic Cell Arrays for ex Vivo Drug Screening with Mimicked Vascular Flow. Anal. Chem. 2014, 86, 2997–3004. [Google Scholar] [CrossRef]
- Pradhan, S.; Smith, A.M.; Garson, C.J.; Hassani, I.; Seeto, W.J.; Pant, K.; Arnold, R.D.; Prabhakarpandian, B.; Lipke, E.A. A Microvascularized Tumor-mimetic Platform for Assessing Anti-cancer Drug Efficacy. Sci. Rep. 2018, 8, 3171. [Google Scholar] [CrossRef]
- Chen, M.B.; Whisler, J.A.; Jeon, J.S.; Kamm, R.D. Mechanisms of tumor cell extravasation in an in vitro microvascular network platform. Integr. Biol. 2013, 5, 1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; Whisler, J.A.; Fröse, J.; Yu, C.; Shin, Y.; Kamm, R.D. On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 2017, 12, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Tu, T.-Y.; Kim, C.; Thiery, J.P.; Kamm, R.D. Identification of drugs as single agents or in combination to prevent carcinoma dissemination in a microfluidic 3D environment. Oncotarget 2015, 6, 36603–36614. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Soroush, F.; Sheffield, J.B.; Wang, B.; Prabhakarpandian, B.; Kiani, M.F. A Biomimetic Microfluidic Tumor Microenvironment Platform Mimicking the EPR Effect for Rapid Screening of Drug Delivery Systems. Sci. Rep. 2017, 7, 9359. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, W.; Ryu, H.; Jeon, N.L. A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation. Biomicrofluidics 2014, 8, 054102. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [PubMed]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Boussommier-Calleja, A.; Atiyas, Y.; Haase, K.; Headley, M.; Lewis, C.; Kamm, R.D. The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model. Biomaterials 2019, 198, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Tsuchida, C.; Zheng, Y.; Himmelfarb, J.; Akilesh, S. A 3D Human Renal Cell Carcinoma-on-a-Chip for the Study of Tumor Angiogenesis. Neoplasia 2018, 20, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.S.; Bersini, S.; Gilardi, M.; Dubini, G.; Charest, J.L.; Moretti, M.; Kamm, R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA 2015, 112, 214–219. [Google Scholar] [CrossRef]
- Jeon, J.S.; Zervantonakis, I.K.; Chung, S.; Kamm, R.D.; Charest, J.L. In vitro Model of Tumor Cell Extravasation. PLoS ONE 2013, 8, e56910. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.D.; Searson, P.C. Tumor and Stem Cell Biology Live-Cell Imaging of Invasion and Intravasation in an Artificial Microvessel Platform. Cancer Res. 2014, 74, 4937–4945. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Vleminckx, K.; Vakaet, L.; Mareel, M.; Fiers, W.; Van Roy, F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991, 66, 107–119. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; Latorre, D.; Cascione, F.; Leporatti, S.; Trerotola, M.; Giudetti, A.M.; Capobianco, L.; Lunetti, P.; Rizzello, A.; et al. Proteomics analysis of E-cadherin knockdown in epithelial breast cancer cells. J. Biotechnol. 2015, 202, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef]
- Lan, J.; Sun, L.; Xu, F.; Liu, L.; Hu, F.; Song, D.; Hou, Z.; Wu, W.; Luo, X.; Wang, J.; et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019, 79, 146–158. [Google Scholar] [CrossRef]
- Bai, J.; Adriani, G.; Dang, T.-M.; Tu, T.-Y.; Penny, H.-X.L.; Wong, S.-C.; Kamm, R.D.; Thiery, J.-P. Contact-dependent carcinoma aggregate dispersion by M2a macrophages via ICAM-1 and β2 integrin interactions. Oncotarget 2015, 6, 25295–25307. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Kim, P. Time series assessment of the effects of hypoxic stress on glioma tumorsphere development within engineered microscale niches. Biomaterials 2019, 194, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl. N. Z.) 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Miermont, A.; Lim, C.T.; Kamm, R.D. A 3D microvascular network model to study the impact of hypoxia on the extravasation potential of breast cell lines. Sci. Rep. 2018, 8, 17949. [Google Scholar] [CrossRef] [PubMed]
- Soltani, M.; Chen, P. Effect of tumor shape and size on drug delivery to solid tumors. J. Biol. Eng. 2012, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Sefidgar, M.; Soltani, M.; Raahemifar, K.; Bazmara, H.; Nayinian, S.M.; Bazargan, M. Effect of tumor shape, size, and tissue transport properties on drug delivery to solid tumors. J. Biol. Eng. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Kashaninejad, N.; Nikmaneshi, M.; Moghadas, H.; Kiyoumarsi Oskouei, A.; Rismanian, M.; Barisam, M.; Saidi, M.; Firoozabadi, B.; Kashaninejad, N.; Nikmaneshi, M.R.; et al. Organ-Tumor-on-a-Chip for Chemosensitivity Assay: A Critical Review. Micromachines 2016, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, W.; Kuss, M.; Mirza, S.; Qi, D.; Krasnoslobodtsev, A.; Zeng, J.; Band, H.; Band, V.; Duan, B. 3D Bioprinting of Breast Cancer Models for Drug Resistance Study. ACS Biomater. Sci. Eng. 2018, 4, 4401–4411. [Google Scholar] [CrossRef]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Joffe, A.R.; Bara, M.; Anton, N.; Nobis, N. The ethics of animal research: A survey of the public and scientists in North America. BMC Med. Eth. 2016, 17, 17. [Google Scholar] [CrossRef]
- Masterton, M.; Renberg, T.; Kälvemark Sporrong, S. Patients’ attitudes towards animal testing: “To conduct research on animals is, I suppose, a necessary evil. ” Biosocieties 2014, 9, 24–41. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Ellis, L.M.; Fidler, I.J. Finding the tumor copycat: Therapy fails, patients don’t. Nat. Med. 2010, 16, 974–975. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models-Advances and prospects. Acta Biomater. 2018, 75, 11–34. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, D.M.; Hattersley, S.D.; Stafford, N.J.; Haswell, S.; Greenman, J. Development of Microfluidic-based Analytical Methodology for Studying the Effects of Chemotherapy Agents on Cancer Tissue. Curr. Anal. Chem. 2013, 9, 2–8. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Charest, J.L.; Kamm, R.D. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, L.; He, J.; Guo, Z.; Ying, L.; Xu, Z.; Zhang, J.; Lu, J.; Wang, Q. Functional Investigation of NCI-H460-Inducible Myofibroblasts on the Chemoresistance to VP-16 with a Microfluidic 3D Co-Culture Device. PLoS ONE 2013, 8, e61754. [Google Scholar] [CrossRef]
- Wang, S.; Li, E.; Gao, Y.; Wang, Y.; Guo, Z.; He, J.; Zhang, J.; Gao, Z.; Wang, Q. Study on Invadopodia Formation for Lung Carcinoma Invasion with a Microfluidic 3D Culture Device. PLoS ONE 2013, 8, e56448. [Google Scholar] [CrossRef]
- Neoh, K.H.; Hassan, A.A.; Chen, A.; Sun, Y.; Liu, P.; Xu, K.-F.; Wong, A.S.T.; Han, R.P.S. Rethinking liquid biopsy: Microfluidic assays for mobile tumor cells in human body fluids. Biomaterials 2018, 150, 112–124. [Google Scholar] [CrossRef]
- Um, E.; Oh, J.M.; Granick, S.; Cho, Y.-K. Cell migration in microengineered tumor environments. Lab Chip 2017, 17, 4171–4185. [Google Scholar] [CrossRef]
- Portillo-Lara, R.; Annabi, N. Microengineered cancer-on-a-chip platforms to study the metastatic microenvironment. Lab Chip 2016, 16, 4063–4081. [Google Scholar] [CrossRef]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Futur. Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [PubMed]
- Ozcelikkale, A.; Moon, H.; Linnes, M.; Han, B. In vitro microfluidic models of tumor microenvironment to screen transport of drugs and nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Zhang, Y.-N.; Zhang, W. Cancer-on-a-chip systems at the frontier of nanomedicine. Drug Discov. Today 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.C.; de Melo-Diogo, D.; Moreira, A.F.; Carvalho, M.P.; Correia, I.J. Spheroids Formation on Non-Adhesive Surfaces by Liquid Overlay Technique: Considerations and Practical Approaches. Biotechnol. J. 2018, 13, 1700417. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Hoffmann, O.I.; Ilmberger, C.; Magosch, S.; Joka, M.; Jauch, K.-W.; Mayer, B. Impact of the spheroid model complexity on drug response. J. Biotechnol. 2015, 205, 14–23. [Google Scholar] [CrossRef]
- Lazzari, G.; Nicolas, V.; Matsusaki, M.; Akashi, M.; Couvreur, P.; Mura, S. Multicellular spheroid based on a triple co-culture: A novel 3D model to mimic pancreatic tumor complexity. Acta Biomater. 2018, 78, 296–307. [Google Scholar] [CrossRef]
- Sun, Q.; Tan, S.H.; Chen, Q.; Ran, R.; Hui, Y.; Chen, D.; Zhao, C.-X. Microfluidic Formation of Coculture Tumor Spheroids with Stromal Cells as a Novel 3D Tumor Model for Drug Testing. ACS Biomater. Sci. Eng. 2018, 4, 4425–4433. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Durymanov, M.; Kroll, C.; Permyakova, A.; O’Neill, E.; Sulaiman, R.; Person, M.; Reineke, J. Subcutaneous Inoculation of 3D Pancreatic Cancer Spheroids Results in Development of Reproducible Stroma-Rich Tumors. Transl. Oncol. 2019, 12, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Q.; Kuo, J.-C.; Wei, M.-T.; Chen, Y.-C.; Yang, M.-H.; Chiou, A. Early stage mechanical remodeling of collagen surrounding head and neck squamous cell carcinoma spheroids correlates strongly with their invasion capability. Acta Biomater. 2019, 84, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Manuel Iglesias, J.; Beloqui, I.; Garcia-Garcia, F.; Leis, O.; Vazquez-Martin, A.; Eguiara, A.; Cufi, S.; Pavon, A.; Menendez, J.A.; Dopazo, J.; et al. Mammosphere Formation in Breast Carcinoma Cell Lines Depends upon Expression of E-cadherin. PLoS ONE 2013, 8, e77281. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Phung, Y.; Feng, M.; Nagashima, K.; Zhang, J.; Broaddus, V.C.; Hassan, R.; FitzGerald, D.; Ho, M. The Development and Characterization of a Human Mesothelioma In vitro 3D Model to Investigate Immunotoxin Therapy. PLoS ONE 2011, 6, e14640. [Google Scholar] [CrossRef] [PubMed]
- Sant, S.; Johnston, P.A. The production of 3D tumor spheroids for cancer drug discovery. Drug Discov. Today Technol. 2017, 23, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-Z.; Chou, L.-F.; Chien, C.-C.M.; Chang, H.-Y. Dynamic analysis of hepatoma spheroid formation: Roles of E-cadherin and β1-integrin. Cell Tissue Res. 2006, 324, 411–422. [Google Scholar] [CrossRef]
- Lee, W.G.; Ortmann, D.; Hancock, M.J.; Bae, H.; Khademhosseini, A. A Hollow Sphere Soft Lithography Approach for Long-Term Hanging Drop Methods. Tissue Eng. Part C Methods 2010, 16, 249–259. [Google Scholar] [CrossRef]
- Ho, V.H.B.; Müller, K.H.; Barcza, A.; Chen, R.; Slater, N.K.H. Generation and manipulation of magnetic multicellular spheroids. Biomaterials 2010, 31, 3095–3102. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, J.; Chen, Y. Agarose multi-wells for tumour spheroid formation and anti-cancer drug test. Microelectron. Eng. 2016, 158, 41–45. [Google Scholar] [CrossRef]
- Fu, J.J.; Zhou, Y.; Shi, X.X.; Kang, Y.J.; Lu, Z.S.; Li, Y.; Li, C.M.; Yu, L. Spontaneous formation of tumor spheroid on a hydrophilic filter paper for cancer stem cell enrichment. Coll. Surf. B Biointerfaces 2019, 174, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.M.; Loh, X.J.; Tan, E.Y.; Loo, J.S.C.; Ho, V.H.B. Development of a Magnetic 3D Spheroid Platform with Potential Application for High-Throughput Drug Screening. Mol. Pharm. 2014, 11, 2182–2189. [Google Scholar] [CrossRef] [PubMed]
- Şükrüoğlu Erdoğan, Ö.; Kılıç Erciyas, S.; Bilir, A.; Buğra Tunçer, Ş.; Akdeniz Ödemiş, D.; Kurul, S.; Karanlık, H.; Cabıoğlu, N.; Yazıcı, H. Methylation Changes of Primary Tumors, Monolayer, and Spheroid Tissue Culture Environments in Malignant Melanoma and Breast Carcinoma. BioMed Res. Int. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lewin Mejia, D.; Chiang, B.; Luker, K.E.; Luker, G.D. Hybrid collagen alginate hydrogel as a platform for 3D tumor spheroid invasion. Acta Biomater. 2018, 75, 213–225. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Melchels, F.P.W.; Holzapfel, B.M.; Meckel, T.; Hutmacher, D.W.; Loessner, D. Gelatine methacrylamide-based hydrogels: An alternative three-dimensional cancer cell culture system. Acta Biomater. 2014, 10, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Hainline, K.M.; Gu, F.; Handley, J.F.; Tian, Y.F.; Wu, Y.; de Wet, L.; Vander Griend, D.J.; Collier, J.H. Self-Assembling Peptide Gels for 3D Prostate Cancer Spheroid Culture. Macromol. Biosci. 2019, 19, 1800249. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Camões, S.P.; Filipe, E.; Cipriano, M.; Barcia, R.N.; Filipe, M.; Teixeira, M.; Simões, S.; Gaspar, M.; Mosqueira, D.; et al. Three-dimensional spheroid cell culture of umbilical cord tissue-derived mesenchymal stromal cells leads to enhanced paracrine induction of wound healing. Stem Cell Res. Ther. 2015, 6, 90. [Google Scholar] [CrossRef]
- Lammel, T.; Tsoukatou, G.; Jellinek, J.; Sturve, J. Development of three-dimensional (3D) spheroid cultures of the continuous rainbow trout liver cell line RTL-W1. Ecotoxicol. Environ. Saf. 2019, 167, 250–258. [Google Scholar] [CrossRef]
- Sebastian, A.; Buckle, A.-M.; Markx, G.H. Formation of multilayer aggregates of mammalian cells by dielectrophoresis. J. Micromech. Microeng. 2006, 16, 1769–1777. [Google Scholar] [CrossRef]
- Liu, J.; Kuznetsova, L.A.; Edwards, G.O.; Xu, J.; Ma, M.; Purcell, W.M.; Jackson, S.K.; Coakley, W.T. Functional three-dimensional HepG2 aggregate cultures generated from an ultrasound trap: Comparison with HepG2 spheroids. J. Cell. Biochem. 2007, 102, 1180–1189. [Google Scholar] [CrossRef]
- Chen, K.; Wu, M.; Guo, F.; Li, P.; Chan, C.Y.; Mao, Z.; Li, S.; Ren, L.; Zhang, R.; Huang, T.J. Rapid formation of size-controllable multicellular spheroids via 3D acoustic tweezers. Lab Chip 2016, 16, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Hamid, Q.; Sun, W.; Clyne, A.M. Bioprinting of 3D breast epithelial spheroids for human cancer models. Biofabrication 2019, 11, 025003. [Google Scholar] [CrossRef] [PubMed]
- Dubiak-Szepietowska, M.; Karczmarczyk, A.; Jönsson-Niedziółka, M.; Winckler, T.; Feller, K.-H. Development of complex-shaped liver multicellular spheroids as a human-based model for nanoparticle toxicity assessment in vitro. Toxicol. Appl. Pharmacol. 2016, 294, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Huang, G.; Liu, J.; Zhang, X.; Ma, Y.; Lu, T.; Xu, F. Bioprinting-Based High-Throughput Fabrication of Three-Dimensional MCF-7 Human Breast Cancer Cellular Spheroids. Engineering 2015, 1, 269–274. [Google Scholar] [CrossRef]
- Sabhachandani, P.; Sarkar, S.; Mckenney, S.; Ravi, D.; Evens, A.M.; Konry, T. Microfluidic assembly of hydrogel-based immunogenic tumor spheroids for evaluation of anticancer therapies and biomarker release. J. Control. Release 2019, 295, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Siltanen, C.; Yaghoobi, M.; Haque, A.; You, J.; Lowen, J.; Soleimani, M.; Revzin, A. Microfluidic fabrication of bioactive microgels for rapid formation and enhanced differentiation of stem cell spheroids. Acta Biomater. 2016, 34, 125–132. [Google Scholar] [CrossRef]
- Watanabe, T.; Motohiro, I.; Ono, T. Microfluidic Formation of Hydrogel Microcapsules with a Single Aqueous Core by Spontaneous Cross-Linking in Aqueous Two-Phase System Droplets. Langmuir 2019, 35, 2358–2367. [Google Scholar] [CrossRef]
- Chen, B.; Wu, Y.; Ao, Z.; Cai, H.; Nunez, A.; Liu, Y.; Foley, J.; Nephew, K.; Lu, X.; Guo, F. High-throughput acoustofluidic fabrication of tumor spheroids. Lab Chip 2019, 19, 1755–1763. [Google Scholar] [CrossRef]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef]
- Frey, O.; Misun, P.M.; Fluri, D.A.; Hengstler, J.G.; Hierlemann, A. Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis. Nat. Commun. 2014, 5, 4250. [Google Scholar] [CrossRef]
- Rismani Yazdi, S.; Shadmani, A.; Bürgel, S.C.; Misun, P.M.; Hierlemann, A.; Frey, O. Adding the ‘heart’ to hanging drop networks for microphysiological multi-tissue experiments. Lab Chip 2015, 15, 4138–4147. [Google Scholar] [CrossRef]
- Costa, E.C.; Gaspar, V.M.; Coutinho, P.; Correia, I.J. Optimization of liquid overlay technique to formulate heterogenic 3D co-cultures models. Biotechnol. Bioeng. 2014, 111, 1672–1685. [Google Scholar] [CrossRef]
- Sarisozen, C.; Abouzeid, A.H.; Torchilin, V.P. The effect of co-delivery of paclitaxel and curcumin by transferrin-targeted PEG-PE-based mixed micelles on resistant ovarian cancer in 3-D spheroids and in vivo tumors. Eur. J. Pharm. Biopharm. 2014, 88, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Mirab, F.; Kang, Y.J.; Majd, S. Preparation and characterization of size-controlled glioma spheroids using agarose hydrogel microwells. PLoS ONE 2019, 14, e0211078. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lee, S.J.; Lim, J.; Kim, B.C.; Han, S.J.; Kim, D.S. Versatile Fabrication of Size- and Shape-Controllable Nanofibrous Concave Microwells for Cell Spheroid Formation. ACS Appl. Mater. Interfaces 2018, 10, 37878–37885. [Google Scholar] [CrossRef]
- Portillo-Lara, R.; Alvarez, M.M. Enrichment of the Cancer Stem Phenotype in Sphere Cultures of Prostate Cancer Cell Lines Occurs through Activation of Developmental Pathways Mediated by the Transcriptional Regulator ΔNp63α. PLoS ONE 2015, 10, e0130118. [Google Scholar] [CrossRef]
- Zanoni, M.; Pignatta, S.; Arienti, C.; Bonafè, M.; Tesei, A. Anticancer drug discovery using multicellular tumor spheroid models. Expert Opin. Drug Discov. 2019, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Choi, J.-H.; Kim, M.; Rhee, W.J.; Son, B.; Jung, H.-K.; Park, T.H. High-throughput generation of spheroids using magnetic nanoparticles for three-dimensional cell culture. Biomaterials 2013, 34, 8555–8563. [Google Scholar] [CrossRef]
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, eaas8998. [Google Scholar] [CrossRef]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in multicellular spheroids formation. J. R. Soc. Interface 2017, 14, 20160877. [Google Scholar] [CrossRef]
- Asghar, W.; El Assal, R.; Shafiee, H.; Pitteri, S.; Paulmurugan, R.; Demirci, U. Engineering cancer microenvironments for in vitro 3-D tumor models. Mater. Today 2015, 18, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Gao, D.; Li, S.; Jiang, Y. Co-culture of tumor spheroids and monocytes in a collagen matrix-embedded microfluidic device to study the migration of breast cancer cells. Chin. Chem. Lett. 2019, 30, 331–336. [Google Scholar] [CrossRef]
- Carvalho, M.P.; Costa, E.C.; Miguel, S.P.; Correia, I.J. Tumor spheroid assembly on hyaluronic acid-based structures: A review. Carbohydr. Polym. 2016, 150, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Tan, S.-J.; Yang, Z.; Tayag, C.; Han, B. Tumor Bioengineering Using a Transglutaminase Crosslinked Hydrogel. PLoS ONE 2014, 9, e105616. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kim, M.H.; Lee, J.H.; Seliktar, D.; Cho, N.-J.; Tan, L.P. Modulation of Huh7.5 Spheroid Formation and Functionality Using Modified PEG-Based Hydrogels of Different Stiffness. PLoS ONE 2015, 10, e0118123. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Trujillo-de Santiago, G.; Alvarez, M.M.; Tamayol, A.; Annabi, N.; Khademhosseini, A. Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels. Biomaterials 2015, 73, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Shrike Zhang, Y.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8, 014101. [Google Scholar] [CrossRef]
- Leonard, F.; Godin, B. 3D in vitro Model for Breast Cancer Research Using Magnetic Levitation and Bioprinting Method; Humana Press: New York, NY, USA, 2016; pp. 239–251. [Google Scholar]
- Kwak, B.; Lee, Y.; Lee, J.; Lee, S.; Lim, J. Mass fabrication of uniform sized 3D tumor spheroid using high-throughput microfluidic system. J. Control. Release 2018, 275, 201–207. [Google Scholar] [CrossRef]
- Chan, H.F.; Zhang, Y.; Ho, Y.-P.; Chiu, Y.-L.; Jung, Y.; Leong, K.W. Rapid formation of multicellular spheroids in double-emulsion droplets with controllable microenvironment. Sci. Rep. 2013, 3, 3462. [Google Scholar] [CrossRef]
- Chen, M.C.W.; Gupta, M.; Cheung, K.C. Alginate-based microfluidic system for tumor spheroid formation and anticancer agent screening. Biomed. Microdevices 2010, 12, 647–654. [Google Scholar] [CrossRef]
- Ota, H.; Miki, N. Microfluidic experimental platform for producing size-controlled three-dimensional spheroids. Sens. Actuators A Phys. 2011, 169, 266–273. [Google Scholar] [CrossRef]
- Fu, C.-Y.; Tseng, S.-Y.; Yang, S.-M.; Hsu, L.; Liu, C.-H.; Chang, H.-Y. A microfluidic chip with a U-shaped microstructure array for multicellular spheroid formation, culturing and analysis. Biofabrication 2014, 6, 015009. [Google Scholar] [CrossRef] [PubMed]
- Ziółkowska, K.; Kwapiszewski, R.; Stelmachowska, A.; Chudy, M.; Dybko, A.; Brzózka, Z. Development of a three-dimensional microfluidic system for long-term tumor spheroid culture. Sens. Actuators B Chem. 2012, 173, 908–913. [Google Scholar] [CrossRef]
- Sabhachandani, P.; Motwani, V.; Cohen, N.; Sarkar, S.; Torchilin, V.; Konry, T. Generation and functional assessment of 3D multicellular spheroids in droplet based microfluidics platform. Lab Chip 2016, 16, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Moshksayan, K.; Kashaninejad, N.; Warkiani, M.E.; Lock, J.G.; Moghadas, H.; Firoozabadi, B.; Saidi, M.S.; Nguyen, N.-T. Spheroids-on-a-chip: Recent advances and design considerations in microfluidic platforms for spheroid formation and culture. Sens. Actuators B Chem. 2018, 263, 151–176. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.; Wang, Y.; Lin, S.; Jiang, Y. A novel 3D breast-cancer-on-chip platform for therapeutic evaluation of drug delivery systems. Anal. Chim. Acta 2018, 1036, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, T.; McAllister, M.; Patek, S.; Flint, D.; Underwood, M.; Sim, A.; Edwards, J.; Zagnoni, M. Drug screening of biopsy-derived spheroids using a self-generated microfluidic concentration gradient. Sci. Rep. 2018, 8, 14672. [Google Scholar] [CrossRef] [PubMed]
- Charoen, K.M.; Fallica, B.; Colson, Y.L.; Zaman, M.H.; Grinstaff, M.W. Embedded multicellular spheroids as a biomimetic 3D cancer model for evaluating drug and drug-device combinations. Biomaterials 2014, 35, 2264–2271. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Osaki, T.; Sivathanu, V.; Kamm, R.D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr. Opin. Biotechnol. 2018, 52, 116–123. [Google Scholar] [CrossRef]
- Moya, M.L.; Hsu, Y.-H.; Lee, A.P.; Hughes, C.C.W.; George, S.C. In vitro perfused human capillary networks. Tissue Eng. Part C. Methods 2013, 19, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, H.; Chung, M.; Jeon, N.L. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 2013, 13, 1489. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Mukundan, S.; Jaramillo, M.; Oesterreich, S.; Sant, S. Three-Dimensional Breast Cancer Models Mimic Hallmarks of Size-Induced Tumor Progression. Cancer Res. 2016, 76, 3732–3743. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.M.; Aizenberg, J.; Analoui, M.; Andrews, A.M.; Bisker, G.; Boyden, E.S.; Kamm, R.D.; Karp, J.M.; Mooney, D.J.; Oklu, R.; et al. Emerging Trends in Micro- and Nanoscale Technologies in Medicine: From Basic Discoveries to Translation. ACS Nano 2017, 11, 5195–5214. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-A.; King, A.D.; Shih, H.-C.; Peng, C.-C.; Wu, C.-Y.; Liao, W.-H.; Tung, Y.-C. Generation of oxygen gradients in microfluidic devices for cell culture using spatially confined chemical reactions. Lab Chip 2011, 11, 3626. [Google Scholar] [CrossRef]
- Huang, J.-W.; Pan, H.-J.; Yao, W.-Y.; Tsao, Y.-W.; Liao, W.-Y.; Wu, C.-W.; Tung, Y.-C.; Lee, C.-H. Interaction between lung cancer cell and myofibroblast influenced by cyclic tensile strain. Lab Chip 2013, 13, 1114. [Google Scholar] [CrossRef] [PubMed]
- Campillo, N.; Falcones, B.; Otero, J.; Colina, R.; Gozal, D.; Navajas, D.; Farré, R.; Almendros, I. Differential Oxygenation in Tumor Microenvironment Modulates Macrophage and Cancer Cell Crosstalk: Novel Experimental Setting and Proof of Concept. Front. Oncol. 2019, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, S.; Truong, D.; Mouneimne, G.; Nikkhah, M. Microfluidic Tumor-Vascular Model to Study Breast Cancer Cell Invasion and Intravasation. Adv. Healthc. Mater. 2018, 7, 1701257. [Google Scholar] [CrossRef]
- Riahi, R.; Yang, Y.L.; Kim, H.; Jiang, L.; Wong, P.K.; Zohar, Y. A microfluidic model for organ-specific extravasation of circulating tumor cells. Biomicrofluidics 2014, 8, 024103. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Pei, Y.; Xie, M.; Jin, Z.-H.; Xiao, Y.-S.; Wang, Y.; Zhang, L.-N.; Li, Y.; Huang, W.-H. An artificial blood vessel implanted three-dimensional microsystem for modeling transvascular migration of tumor cells. Lab Chip 2015, 15, 1178–1187. [Google Scholar] [CrossRef]
- Xu, Z.; Li, E.; Guo, Z.; Yu, R.; Hao, H.; Xu, Y.; Sun, Z.; Li, X.; Lyu, J.; Wang, Q. Design and Construction of a Multi-Organ Microfluidic Chip Mimicking the in vivo Microenvironment of Lung Cancer Metastasis. ACS Appl. Mater. Interfaces 2016, 8, 25840–25847. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Ta, H.P.; Yen, D.P.; Lee, S.-S.; Raola, S.; Shen, K. A Microdevice Platform Recapitulating Hypoxic Tumor Microenvironments. Sci. Rep. 2017, 7, 15233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-P.; Ma, Y.; Lou, Q.; Zhu, H.; Yang, B.; Fang, Q. Three-Dimensional Cell Culture and Drug Testing in a Microfluidic Sidewall-Attached Droplet Array. Anal. Chem. 2017, 89, 10153–10157. [Google Scholar] [CrossRef] [PubMed]
- Khoo, B.L.; Grenci, G.; Jing, T.; Lim, Y.B.; Lee, S.C.; Thiery, J.P.; Han, J.; Lim, C.T. Liquid biopsy and therapeutic response: Circulating tumor cell cultures for evaluation of anticancer treatment. Sci. Adv. 2016, 2, e1600274. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Snow, C.J.; Simmers, M.B.; Hoang, S.A.; Figler, R.A.; Allende, J.A.; Roller, D.G.; Parsons, J.T.; Wulfkuhle, J.D.; Petricoin, E.F.; et al. Development of a multicellular pancreatic tumor microenvironment system using patient-derived tumor cells. Lab Chip 2019, 19, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Bower, R.; Green, V.L.; Kuvshinova, E.; Kuvshinov, D.; Karsai, L.; Crank, S.T.; Stafford, N.D.; Greenman, J. Maintenance of head and neck tumor on-chip: Gateway to personalized treatment? Future Sci. OA 2017, 3, FSO174. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, M.; Péant, B.; Lateef, M.A.; Rousset, N.; Kendall-Dupont, J.; Carmona, E.; Monet, F.; Saad, F.; Provencher, D.; Mes-Masson, A.-M.; et al. Micro-dissected tumor tissues on chip: An ex vivo method for drug testing and personalized therapy. Lab Chip 2016, 16, 312–325. [Google Scholar] [CrossRef]
- Mierke, C.T. Cancer cells regulate biomechanical properties of human microvascular endothelial cells. J. Biol. Chem. 2011, 286, 40025–40037. [Google Scholar] [CrossRef]
- Carr, S.D.; Green, V.L.; Stafford, N.D.; Greenman, J. Analysis of Radiation-Induced Cell Death in Head and Neck Squamous Cell Carcinoma and Rat Liver Maintained in Microfluidic Devices. Otolaryngol. Neck Surg. 2014, 150, 73–80. [Google Scholar] [CrossRef]
- Junaid, A.; Mashaghi, A.; Hankemeier, T.; Vulto, P. An end-user perspective on Organ-on-a-Chip: Assays and usability aspects. Curr. Opin. Biomed. Eng. 2017, 1, 15–22. [Google Scholar] [CrossRef]
- Ho, C.M.B.; Ng, S.H.; Li, K.H.H.; Yoon, Y.-J. 3D printed microfluidics for biological applications. Lab Chip 2015, 15, 3627–3637. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; He, J.; Li, X.; Zhang, B.; Lei, Q.; Liu, Y.; Li, D.; Mao, M.; He, J.; Li, X.; et al. The Emerging Frontiers and Applications of High-Resolution 3D Printing. Micromachines 2017, 8, 113. [Google Scholar] [CrossRef]
- Ong, L.J.Y.; Islam, A.; DasGupta, R.; Iyer, N.G.; Leo, H.L.; Toh, Y.-C. A 3D printed microfluidic perfusion device for multicellular spheroid cultures. Biofabrication 2017, 9, 045005. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cho, D.-W. One-step fabrication of an organ-on-a-chip with spatial heterogeneity using a 3D bioprinting technology. Lab Chip 2016, 16, 2618–2625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Duchamp, M.; Oklu, R.; Ellisen, L.W.; Langer, R.; Khademhosseini, A. Bioprinting the Cancer Microenvironment. ACS Biomater. Sci. Eng. 2016, 2, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Clyne, A.M.; Swaminathan, S.; Lantada, A.D. Biofabrication Biofabrication strategies for creating microvascular complexity Biofabrication strategies for creating microvascular complexity. Biofabrication 2019, 11, 32001. [Google Scholar] [CrossRef] [PubMed]
- Miri, A.K.; Khalilpour, A.; Cecen, B.; Maharjan, S.; Shin, S.R.; Khademhosseini, A. Multiscale bioprinting of vascularized models. Biomaterials 2018, 198, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.K.; Dai, G.; Zou, H.; Yoo, S.S. Generation of 3-D glioblastoma-vascular niche using 3-D bioprinting. In Proceedings of the 2015 41st Annual Northeast Biomedical Engineering Conference (NEBC), Troy, NY, USA, 17–19 April 2015; IEEE: Piscataway, NJ, USA, 2015; pp. 1–2. [Google Scholar]
- Trujillo-de Santiago, G.; Alvarez, M.M.; Samandari, M.; Prakash, G.; Chandrabhatla, G.; Rellstab-Sánchez, P.I.; Byambaa, B.; Pour Shahid Saeed Abadi, P.; Mandla, S.; Avery, R.K.; et al. Chaotic printing: Using chaos to fabricate densely packed micro- and nanostructures at high resolution and speed. Mater. Horizons 2018, 5, 813–822. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.S.; Heinrich, M.A.; De Ferrari, F.; Jang, H.L.; Bakht, S.M.; Alvarez, M.M.; Yang, J.; Li, Y.-C.; Trujillo-de Santiago, G.; et al. Rapid Continuous Multimaterial Extrusion Bioprinting. Adv. Mater. 2017, 29, 1604630. [Google Scholar] [CrossRef]
- Knowlton, S.; Onal, S.; Yu, C.H.; Zhao, J.J.; Tasoglu, S. Bioprinting for cancer research. Trends Biotechnol. 2015, 33, 504–513. [Google Scholar] [CrossRef]
- Samavedi, S.; Joy, N. 3D printing for the development of in vitro cancer models. Curr. Opin. Biomed. Eng. 2017, 2, 35–42. [Google Scholar] [CrossRef]
- Dai, X.; Ma, C.; Lan, Q.; Xu, T. 3D bioprinted glioma stem cells for brain tumor model and applications of drug susceptibility. Biofabrication 2016, 8, 045005. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Liu, L.; Ouyang, J.; Li, X.; Zhang, X.; Lan, Q.; Xu, T. Coaxial 3D bioprinting of self-assembled multicellular heterogeneous tumor fibers. Sci. Rep. 2017, 7, 1457. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Meyer, C.M.; Joung, D.; Vallera, D.A.; McAlpine, M.C.; Panoskaltsis-Mortari, A. 3D Bioprinted In vitro Metastatic Models via Reconstruction of Tumor Microenvironments. Adv. Mater. 2019, 31, 1806899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yao, R.; Ouyang, L.; Ding, H.; Zhang, T.; Zhang, K.; Cheng, S.; Sun, W. Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication 2014, 6, 035001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhu, W.; Nowicki, M.; Miao, S.; Cui, H.; Holmes, B.; Glazer, R.I.; Zhang, L.G. 3D Bioprinting a Cell-Laden Bone Matrix for Breast Cancer Metastasis Study. ACS Appl. Mater. Interfaces 2016, 8, 30017–30026. [Google Scholar] [CrossRef]
- Duchamp, M.; Liu, T.; Genderen, A.M.; Kappings, V.; Oklu, R.; Ellisen, L.W.; Zhang, Y.S. Sacrificial Bioprinting of a Mammary Ductal Carcinoma Model. Biotechnol. J. 2019, 9, 1700703. [Google Scholar] [CrossRef]
- Steuperaert, M.; Debbaut, C.; Carlier, C.; De Wever, O.; Descamps, B.; Vanhove, C.; Ceelen, W.; Segers, P. A 3D CFD model of the interstitial fluid pressure and drug distribution in heterogeneous tumor nodules during intraperitoneal chemotherapy. Drug Deliv. 2019, 26, 404–415. [Google Scholar] [CrossRef]
- Garza-García, L.D.; García-López, E.; Camacho-León, S.; del Refugio Rocha-Pizaña, M.; López-Pacheco, F.; López-Meza, J.; Araiz-Hernández, D.; Tapia-Mejía, E.J.; Trujillo-de Santiago, G.; Rodríguez-González, C.A.; et al. Continuous flow micro-bioreactors for the production of biopharmaceuticals: The effect of geometry, surface texture, and flow rate. Lab Chip 2014, 14, 1320–1329. [Google Scholar] [CrossRef]
- Munaz, A.; Vadivelu, R.K.; John, J.A.S.; Nguyen, N.-T. A lab-on-a-chip device for investigating the fusion process of olfactory ensheathing cell spheroids. Lab Chip 2016, 16, 2946–2954. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kankala, R.; Wang, S.-B.; Chen, A.-Z.; Zhao, Y.; Kankala, R.K.; Wang, S.-B.; Chen, A.-Z. Multi-Organs-on-Chips: Towards Long-Term Biomedical Investigations. Molecules 2019, 24, 675. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description/Flow Conditions | Cancer Type/Cell Line | Materials (Microfluidic Device) | Biomaterial (ECM) | Application | Reference |
|---|---|---|---|---|---|
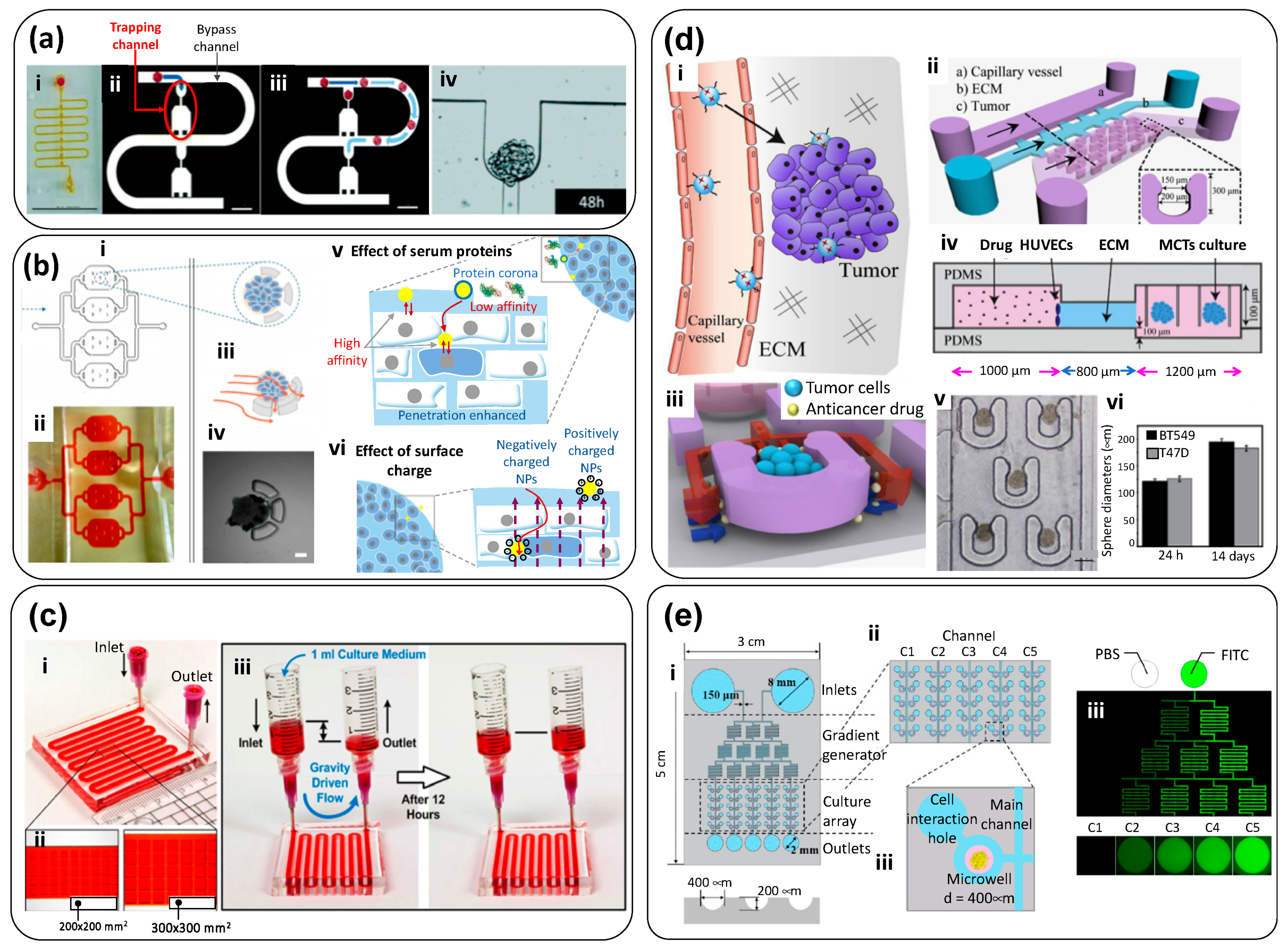
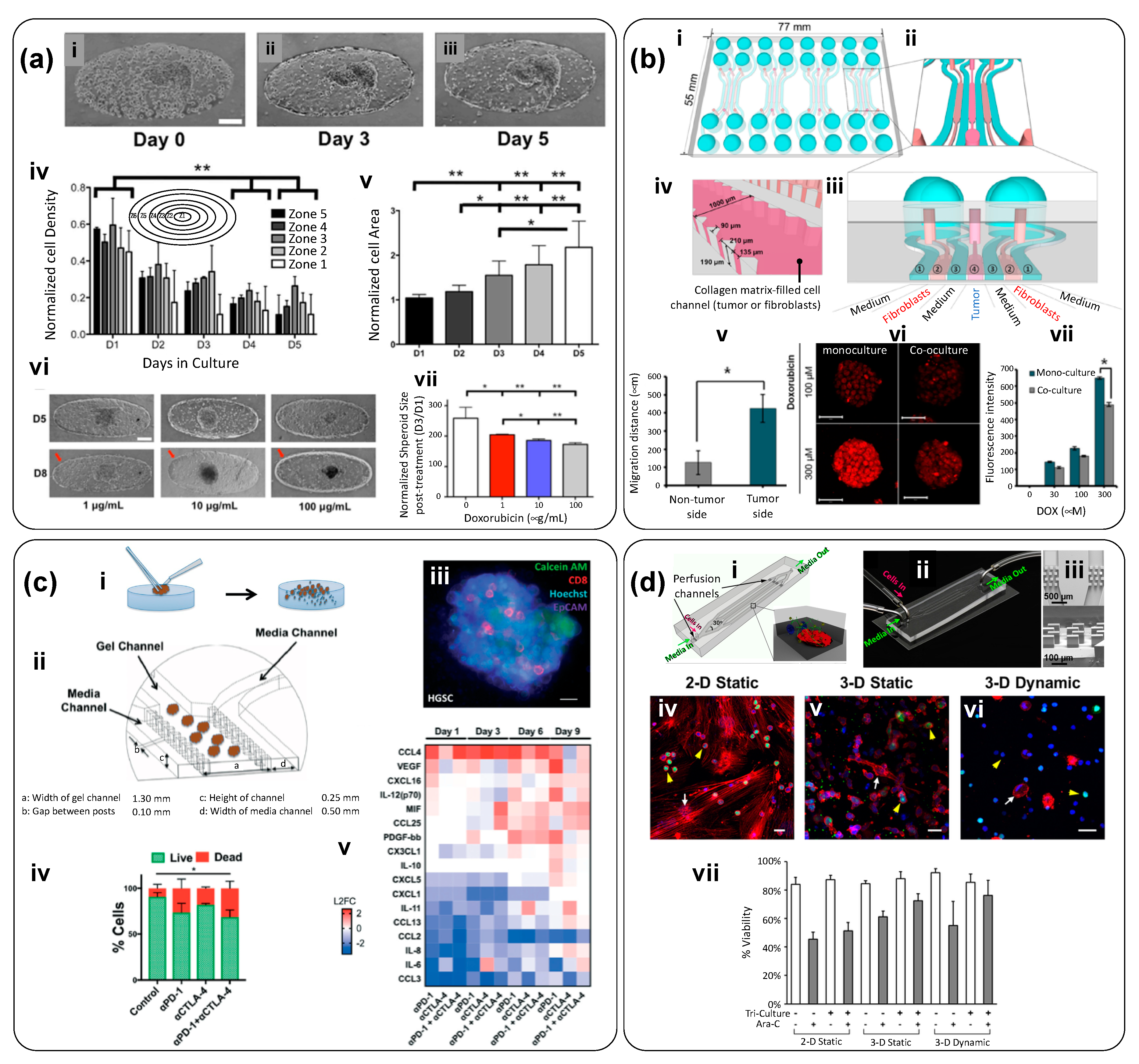
| Microfluidic channel with multiple micro traps for individual spheroids (Figure 2). Nutrients diffuse from the main channel to trapped spheroids. Flow rate (main channel): 100 µL/h | malignant pleural mesothelioma (MPM) cells | PDMS | No extracellular matrix (ECM) was used | To compare the therapeutic efficiency of cisplatin, a drug commonly prescribed for MPM patients, and compare chemoresistance of cancer spheroids exposed to cisplatin in static (ex-device) versus dynamic environments. | [20]; Figure 3a |
| Triple layer microfluidic conduit in which spheroids are captured in semicircular traps for growth/maturation. | Human Hepatic cancer; HepG2 | PDMS (top layer); glass (middle layer); PVC (bottom layer) | Non-embedded spheroids | To study nanoparticle penetration into tumor spheroids: The effects of protein corona, protein size, and charge were analyzed. | [21]; Figure 3b |
| Simple, bi-layered, and pump-independent microfluidic system devised for continuous formation and perfusion of cancer spheroids contained within rectangular cavities located on the floor of the microfluidic circuit. Flow is driven simply by a pressure head induced by a difference in the height of the column of liquid (culture medium) between the inlet and the outlet reservoir. | Human hepatic cancer; (HepG2) | PDMS | Non-embedded spheroids | To study the process of formation of hepatic cancer spheroids under continuous perfusion. Spheroids are formed within cavities in the bottom floor of the device, and cavities of two different sizes are tested. To test the anti-cancer effects of three compounds (tirapazamine, cisplatin, and resveratrol) on hepatic cancer spheroids. | [22]; Figure 3c |
| Microfluidic chamber (Figure 3) composed of a central compartment and two side channels: the central channel was filled with cancer spheroids embedded in gelatin (cross-linked using glutaraldehyde); the two side channels were used for continuous feeding of liquid streams at 30 µL/h (0.5–30 µL/min). (The linear speed was ~278 µm/s). Breast cancer spheroids, prepared ex-device by an overflow method, were embedded in the gelatin-filled central compartment. | Breast Cancer; MCF7 cells | PDMS | Gelatin cross-linked using glutaraldehyde | To evaluate therapeutic efficiency of doxorubicin (DOX), an anthracycline antibiotic that intercalates DNA, in multi-cellular tumor spheroids (MTS) fabricated ex-device. | [29] |
| Microfluidic system composed of an inlet channel connected to a visualization chamber, where a tumor spheroid is physically trapped by slight compression against a glass coverslip. | Human breast cancer; MDA-MB-435 cells | PDMS and glass coverslip | Non-embedded spheroids | To study the transport, penetration, and accumulation of nanoparticles in cancer spheroids in real time (under a microscope) | [27]; Figure 4a |
| Microfluidic device formed by two stacked layers of microchannels with a porous membrane sandwiched between the layers. The top layer has a channel simulating the capillary of the tumor vasculature. The endothelium of the capillary is mimicked by culturing MVECs on the porous membrane. The bottom layer has three channels, which are partitioned with periodically placed posts. The center channel simulates the tumor surroundings where spheroids dispersed in collagen are placed, and the two side channels simulate the lymphatics. In the tumor channel, cancer cells grow within a 3D collagen matrix, while the interstitial fluid flows through the matrix and creates an elevated interstitial fluid pressure. Nanoparticles are transported through this 3D tissue structure and reach the cancer cells. | Breast Cancer; (MCF7 cells) and endothelial cells (MVECs) | PDMS (layers); polycarbonate membrane | Collagen I and Matrigel (for the membrane coating) | To simulate the complex transport of nanoparticles around a tumor spheroid in a TOC system, where well-defined pressure gradients can be established. The authors studied the effect of size, concentration, and dynamic conditions in targeted delivery of anti-cancer compounds encapsulated in nanoparticles | [28]; Figure 4b |
| Y-shape device with two microchannel lines that enable the co-culture (sharing an interface) of mammary epithelial cells (MCF-DCIS) and non-cancerous human mammary fibroblasts (HMFs) (Figure 5). Sample loading and fluid changes are performed using a surface-tension driven pump. | Breast cancer; Mammary epithelial ductal carcinoma in situ cells (MCF-DCIS) | PDMS | Hydrogel mix: 1:1 matrigel and collagen I (0.8 mg/mL) | To evaluate the progression of breast cancer cells from ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) | [33]; Figure 5a |
| Microfluidic chip composed of two compartments for micro-tissue (3D-µTP) accommodation: the inner chamber is for the tumor and the outer one for the stromal compartment. The chambers are separated by an interface that allows physical contact. The two chambers have a dedicated channel for cell culture loading, while the other two side channels allowed the flow of culture medium at a nominal flow rate of 3.0 µL/min. | Breast cancer; Normal mammary Fibroblasts (NF) and Cancer Associated Fibroblasts (CAF), Human breast adenocarcinoma cells (MCF7) | PDMS | ECM produced by micro-tissues | To replicate in vitro the stromal activation that occurs during tumor epithelial invasion | [35]; Figure 5b |
| Microfluidic device to study a tumor microenvironment in real time. This tumor-on-chip consisted of a central microchamber flanked by two lateral microchannels. Tumor cells were embedded within a collagen matrix in the central microchamber (Figure 5d-i,ii), while the lateral microchannels were used to perfuse medium, oxygen, and anticancer drugs. The configuration of the central chamber enables the spontaneous generation of normoxic, hypoxic, and necrotic regions within the device. | Two cancer cell lines in independent experiments—glioblastoma U-251 MG cells and HCT-116 cells from colon carcinoma | polystyrene | Collagen | To establish clear oxygen gradients and hypoxic conditions in a microfluidic device and to study the effect of anticancer drugs (DOX for colon carcinoma and TMZ for glioblastoma) in cell populations under controlled oxygen gradients, without resorting to the use of spheroids. | [36]; Figure 5d |
| Four-unit microfluidic chip. Each unit consists of three cell-loading channels (to fill with cells in collagen) and four medium channels (Figure 6-i–iv). Channel width was 1000 μm and channel depth was approximately 190 μm; material/gas exchange was accommodated between the channels. | Normal colon fibroblasts (CCD-18Co) and human colorectal cancer cells (HT-29 cells) | PDMS | Collagen Type 1 | To study the crosstalk and mutual activation between fibroblasts and cancer cells. To evaluate the effect of different doses of DOX in monoculture and co-culture conditions within this TOC system. | [51]; Figure 6b |
| Commercial device: ‘3-D cell culture chip’ (DAX-1) from AIM BIOTECH: https://www.aimbiotech.com/. Each single layer slide format chip (75 mm × 25 mm), consisting of 3 microfluidic chambers, each with a central gel channel (width 1.3 mm) flanked by two medium channels (width 0.5 mm). The height of the microfluidic chambers is 0.25 mm. (Figure 3a–c). | Patient-derived spheroids from real tumor samples: Samples contained cancerous cells, stromal cells and immune cells. | cyclic olefin polymer (COP) | Collagen Type 1 (rat tail) | Patient-derived spheroids (PDOSTS) were cultured in contact with immune system cells to recreate the tumor immune microenvironment and screen the response of tumors to immunotherapy (therapies based on antibodies) | [48]; Figure 6c |
| 3D microfluidic device that allows co-culture. The device comprises four perfusion microchannels with only one inlet and one outlet. The cells are loaded and culture medium is fed using two ports (“cell in” and “media in”) placed at the inlet side of the device. The velocity rate within the microchannels was 0.27 ± 0.18 μm/s. | Acute lymphoblastic leukemia (SUP-B15 cells), and bone marrow mesenchymal stem cells (BMSC). | PDMS | Collagen Type 1 | To elucidate cell–cell and cell–matrix interactions on leukemia progression and to test therapeutic agents in a co-culture. | [49]; Figure 6d |
| Dual compartment human-on-chip system composed of a liver compartment (healthy or tumorous), and a breast cancer compartment. Each compartment (volume ~0.5 cm3) contains a microtissue formed by a mixture of cells (liver or breast cancer cells) in collagen. Compartments are connected by a vascular (endothelialized) channel that crosses through each microtissue (Figure 7b-i–ii). | Liver or breast cancer; Human breast cancer cells (MDA-MB-231); healthy liver cells (THLE-3); carcinoma liver cells (C3Asub28); and telomerase immortalized microvascular endothelial (TIME) cells. | PDMS and glass cover | Collagen Type I: at 7 mg/mL (for cancerous tissues) and at 4 mg/mL (for healthy tissues) | To recapitulate an interactive liver–tumor tissue microenvironment on a chip for the investigation of nanoparticle transport and toxicity. | [55]; Figure 7b |
| Microfluidic platform consisting of a series of three rhombic tissue chambers (Figure 7e-i–iii). These are connected to two adjacent channels by two capillary burst valves that retain a mixture of cells and ECM inside the chambers. At the two ends of the tissue chambers are two gel-loading ports for introduction of introduced the cell–ECM suspension. Four media reservoirs are attached to the inlets and outlets of the microfluidic channels. | Three colorectal cancer cell lines (HCT116, SW620, and SW480); two breast cancer lines (MCF-7 and MDA-MB-231), and a melanoma cell line (MNT-1). Endothelial cells. | PDMS and glass cover | A mix of fibrinogen in PBS with Ca2+/Mg2+ to a final concentration of 10 mg/mL; and thrombin (30 U/mL) and laminin (1 mg/mL) | To develop a vascularized TOC and recreate a vascularized environment relevant to the progression of cancerous tissue and the testing of anti-cancer agents. To demonstrate that vascular-targeting agents with different mechanisms of action (i.e., VEGF blockers, vascularization inhibitors, etc.) can be distinguished in this TOC. For example, to show in vitro that drugs targeting only VEGFRs (i.e., apatinib and vandetanib) are not effective, whereas drugs that target VEGFRs, PDGFR, and Tie2 (i.e., linifanib and cabozantinib) do regress the vasculature. | [58]; Figure 7e |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trujillo-de Santiago, G.; Flores-Garza, B.G.; Tavares-Negrete, J.A.; Lara-Mayorga, I.M.; González-Gamboa, I.; Zhang, Y.S.; Rojas-Martínez, A.; Ortiz-López, R.; Álvarez, M.M. The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials 2019, 12, 2945. https://doi.org/10.3390/ma12182945
Trujillo-de Santiago G, Flores-Garza BG, Tavares-Negrete JA, Lara-Mayorga IM, González-Gamboa I, Zhang YS, Rojas-Martínez A, Ortiz-López R, Álvarez MM. The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials. 2019; 12(18):2945. https://doi.org/10.3390/ma12182945
Chicago/Turabian StyleTrujillo-de Santiago, Grissel, Brenda Giselle Flores-Garza, Jorge Alfonso Tavares-Negrete, Itzel Montserrat Lara-Mayorga, Ivonne González-Gamboa, Yu Shrike Zhang, Augusto Rojas-Martínez, Rocío Ortiz-López, and Mario Moisés Álvarez. 2019. "The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors" Materials 12, no. 18: 2945. https://doi.org/10.3390/ma12182945
APA StyleTrujillo-de Santiago, G., Flores-Garza, B. G., Tavares-Negrete, J. A., Lara-Mayorga, I. M., González-Gamboa, I., Zhang, Y. S., Rojas-Martínez, A., Ortiz-López, R., & Álvarez, M. M. (2019). The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials, 12(18), 2945. https://doi.org/10.3390/ma12182945