Correlations of Equilibrium Properties and Electronic Structure of Pure Metals

Abstract
1. Introduction
2. Methodology
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Trinkle, D.; Woodward, C.F. The chemistry of deformation: How solutes soften pure metals. Science 2005, 310, 1665–1667. [Google Scholar] [CrossRef] [PubMed]
- Leyson, G.P.M.; Curtin, W.A.; Hector, L.G., Jr.; Woodward, C.F. Quantitative prediction of solute strengthening in aluminium alloys. Nat. Mater. 2010, 9, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Varvenne, C.; Luque, A.; Curtin, W.A. Theory of strengthening in fcc high entropy alloys. Acta Mater. 2016, 118, 264–276. [Google Scholar] [CrossRef]
- Aagesen, L.K.; Adams, J.F.; Allison, J.E.; Andrews, W.B.; Araullo-Peters, V.; Berman, T.; Chen, Z.; Daly, S.; Das, S.; DeWitt, S.; et al. Prisms: An integrated, open-source framework for accelerating predictive structural materials science. JOM 2018, 70, 2298–2314. [Google Scholar] [CrossRef]
- Shang, S.L.; Saengdeejing, A.; Mei, Z.G.; Kim, D.E.; Zhang, H.; Ganeshan, S.; Wang, Y.; Liu, Z.K. First-principles calculations of pure elements: Equations of state and elastic stiffness constants. Comput. Mater. Sci. 2010, 48, 813–826. [Google Scholar] [CrossRef]
- Rodney, D.; Ventelon, L.; Clouet, E.; Pizzagalli, L.; Willaime, F. Ab initio modeling of dislocation core properties in metals and semiconductors. Acta Mater. 2017, 124, 633–659. [Google Scholar] [CrossRef]
- Song, Y.; Yang, R.; Li, D.; Wu, W.T.; Guo, Z.X. Calculation of theoretical strengths and bulk moduli of bcc metals. Phys. Rev. B 1999, 59, 14220. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833. [Google Scholar] [CrossRef]
- Foster, J.P.; Wenhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1984. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Soc. 1984, 57, 1259–1281. [Google Scholar]
- Feth, S.; Gibbs, G.V.; Boisen, M.B.; Hill, F.C. A study of the bonded interactions in nitride molecules in terms of bond critical point properties and relative electronegatives. Phys. Chem. Miner. 1998, 25, 234–241. [Google Scholar] [CrossRef]
- Louit, G.; Hocquet, A.; Ghomi, M.; Meyer, M.; Suehnel, J. An AIM topological analysis of Guanine tetrads interacting with metals. PhysChemComm 2003, 6, 1–5. [Google Scholar] [CrossRef]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population analysis of plane-wave electronic structure calculations of bulk materials. Phys. Rev. B 1996, 54, 16317–16320. [Google Scholar] [CrossRef] [PubMed]
- Al-Douri, Y.; Abid, H.; Aourag, H. Correlation between the bulk modulus and the charge density in semiconductors. Phys. B 2001, 305, 186–190. [Google Scholar] [CrossRef]
- Miedema, A.R.; de Boer, F.R.; de Chatel, P.F. Empirical description of the role of electronegativity in alloy formation. J. Phys. F Met. Phys. 1973, 3, 1558–1576. [Google Scholar] [CrossRef]
- Cheng, D.Y.; Wang, S.Q.; Ye, H.Q. Calculations showing a correlation between electronic density and bulk modulus in fcc and bcc metals. Phys. Rev. B 2001, 64, 024107. [Google Scholar] [CrossRef]
- Li, C.H.; Wu, P. Correlation of Bulk Modulus and the Constituent Element Properties of Binary Intermetallic Compounds. Chem. Mater. 2001, 13, 4642–4648. [Google Scholar] [CrossRef]
- Li, C.H.; Chin, Y.L.; Wu, P. Correlation between bulk modulus of ternary intermetallic compounds and atomic properties of their constituent elements. Intermetallics 2004, 12, 103–109. [Google Scholar] [CrossRef]
- Wills, J.M.; Harrison, W.A. Interionic interactions in transition metals. Phys. Rev. B 1983, 28, 4363. [Google Scholar] [CrossRef]
- Makino, Y. Empirical determination of bulk moduli of elemental substances by pseudopotential radius. J. Alloys Compd. 1996, 242, 122–128. [Google Scholar] [CrossRef]
- Raju, S.; Mohandas, E.; Raghunathan, V.S. The pressure derivative of bulk modulus of transition metals: An estimation using the method of model potentials and a study of the systematics. J. Phys. Chem. Solids 1997, 58, 1367–1373. [Google Scholar] [CrossRef]
- Goble, R.J.; Scott, S.D. The relationship between mineral hardness and compressibility (or bulk modulus). Can. Mineral. 1985, 23, 273–285. [Google Scholar]
- Singh, N.; Yadav, B.S. The elastic moduli, the volume contribution and the Cauchy ratio ford andf shell metals. Pramana-J. Phys. 1994, 42, 387–394. [Google Scholar] [CrossRef]
- Miracle, D.B.; Senkov, O.N. A critical review of high entropy alloys and related concepts. Acta Mater. 2017, 122, 448–511. [Google Scholar] [CrossRef]
- Tanaka, I.; Rajan, K.; Wolverton, C. Data-centric science for materials innovation. MRS Bull. 2018, 43, 659–663. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Blaha, P.; Schwarz, K.; Luitz, J. Computer Code WIEN2K; Vienna University of Technology: Vienna, Austria, 2000. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics; John Wiley & Sons Inc.: New York, NY, USA, 2005. [Google Scholar]
- Ambrosetti, A.; Silvestrelli, P.L. Cohesive properties of noble metals by van der Waals–corrected density functional theory: Au, Ag, and Cu as case studies. Phys. Rev. B 2016, 94, 045124. [Google Scholar] [CrossRef]
- Janthon, P.; Luo, S.J.; Kozlov, S.M.; Viñes, F.; Limtrakul, J.; Truhlar, D.G.; Illas, F. Bulk properties of transition metals: A challenge for the design of universal density functionals. J. Chem. Theory Comput. 2014, 10, 3832–3839. [Google Scholar] [CrossRef]
- Dolocan, V.; Dolocan, A.; Dolocan, V.O. Relation of inter-atomic forces in solids to bulk modulus, cohesive energy and thermal expansion. Mod. Phys. Lett. B 2008, 22, 2481–2492. [Google Scholar] [CrossRef]
- Tal, Y. Cohesive properties of metals as determined from atomic charge densities. Can. J. Chem. 1996, 74, 870–874. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
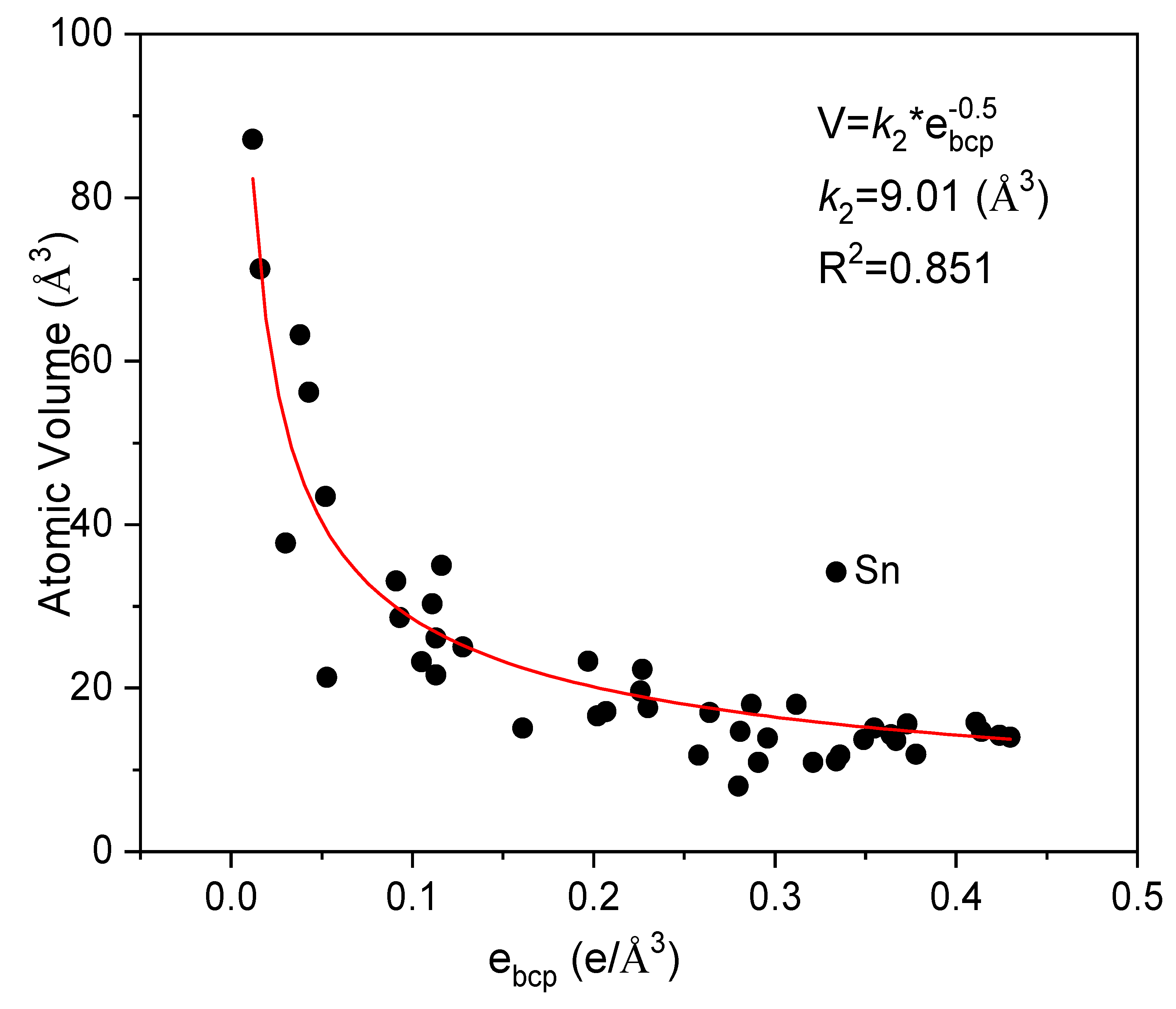{kind=link}
{kind=link}
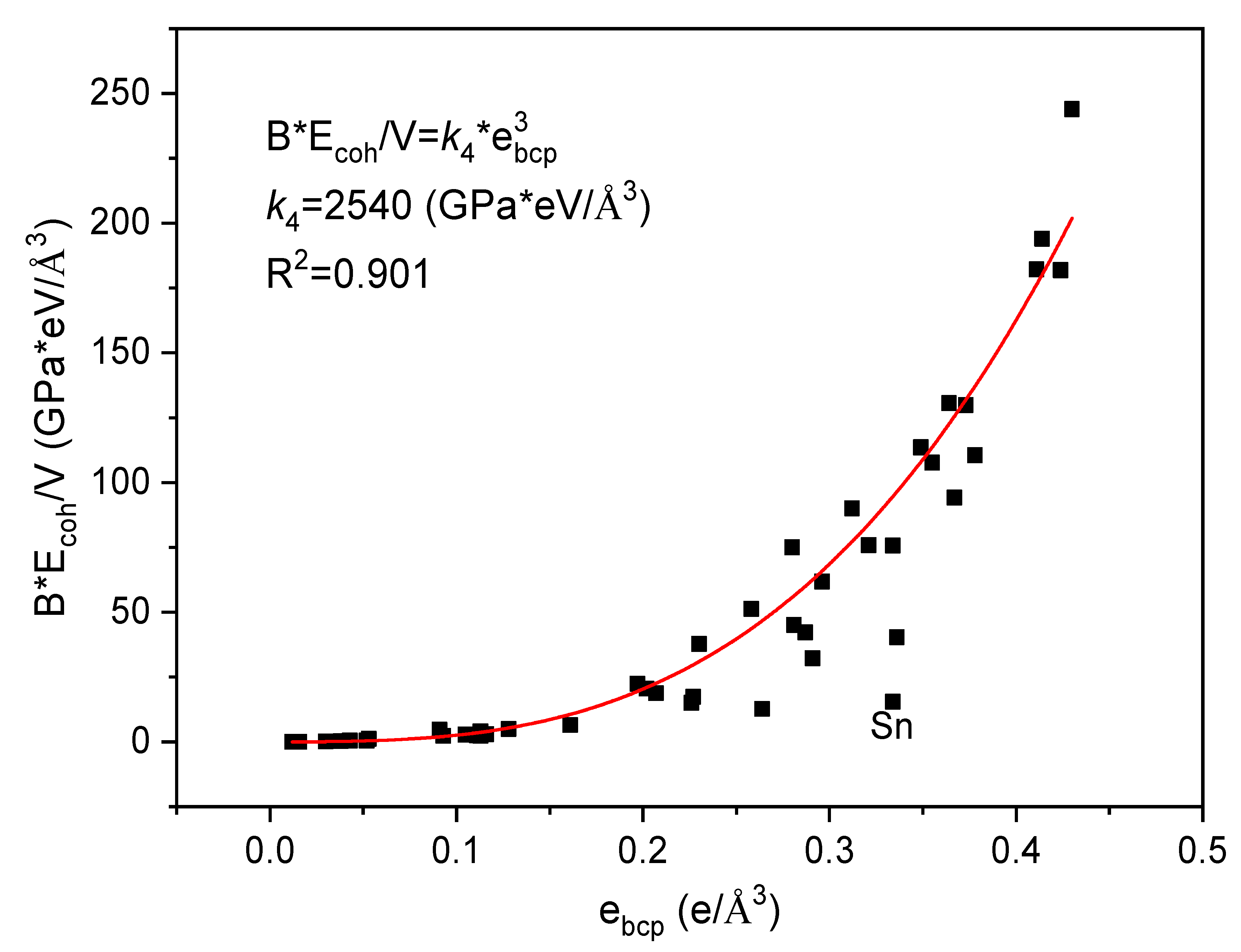{kind=link}
| Metals | Z | Ecoh (eV/atom) | V (Å3/atom) | ebcp (e/Å3) | B (GPa) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Present | Exp ** | Present | Shang * | Exp ** | Present | Shang * | Exp ** | ||
| Lib | 3 | 1.59 | 1.63 | 20.4 | 20.3 | 21.3 | 0.053 | 13.9 | 11.6 |
| Nab | 9 | 1.05 | 1.113 | 36.9 | 37.1 | 37.7 | 0.030 | 7.9 | 6.8 |
| Kb | 9 | 0.795 | 0.934 | 73.5 | 73.7 | 71.3 | 0.016 | 3.5 | 3.2 |
| Rbb | 9 | 0.682 | 0.852 | 91.2 | 90.8 | 87.1 | 0.012 | 2.7 | 3.1 |
| Beh | 2 | 3.69 | 3.32 | 7.9 | 7.9 | 8.01 | 0.280 | 121.1 | 100.3 |
| Mgh | 10 | 1.47 | 1.51 | 22.9 | 22.9 | 23.2 | 0.105 | 35.7 | 35.4 |
| Caf | 10 | 1.86 | 1.84 | 42.6 | 41.8 | 43.4 | 0.052 | 17.4 | 15.2 |
| Srf | 10 | 1.55 | 1.72 | 53.9 | 53.9 | 56.2 | 0.043 | 11.8 | 11.6 |
| Bab | 10 | 1.79 | 1.90 | 62.3 | 62.5 | 63.2 | 0.038 | 9.0 | 10.3 |
| Sch | 11 | 4.26 | 3.90 | 24.0 | 24.5 | 25.0 | 0.128 | 54.9 | 43.5 |
| Tih | 12 | 5.56 | 4.85 | 17.1 | 17.3 | 17.6 | 0.230 | 112.8 | 105.1 |
| Vb | 13 | 5.30 | 5.31 | 13.2 | 13.5 | 13.9 | 0.290 | 182.9 | 161.9 |
| Crb | 14 | 4.03 | 4.10 | 11.4 | 11.6 | 11.9 | 0.378 | 257.7 | 190.1 |
| Mnc | 13 | 3.91 | 2.92 | 10.7 | -- | 10.9 | 0.291 | 120.0 | 120.0 |
| Feb | 14 | 4.49 | 4.28 | 10.5 | 11.4 | 11.8 | 0.336 | 189.3 | 168.3 |
| Coh | 15 | 4.92 | 4.39 | 10.3 | 10.9 | 11.1 | 0.334 | 212.5 | 191.4 |
| Nif | 16 | 4.77 | 4.44 | 10.7 | 10.9 | 10.9 | 0.321 | 195.6 | 186.0 |
| Cuf | 17 | 3.47 | 3.49 | 12.0 | 12.0 | 11.8 | 0.258 | 137.5 | 137.0 |
| Znh | 12 | 1.12 | 1.35 | 15.1 | 15.4 | 15.1 | 0.161 | 51.8 | 59.8 |
| Yh | 11 | 4.24 | 4.37 | 32.6 | 32.7 | 33.1 | 0.091 | 40.8 | 36.6 |
| Zrh | 12 | 6.39 | 6.25 | 23.3 | 23.4 | 23.3 | 0.197 | 95.3 | 83.3 |
| Nbb | 13 | 6.86 | 7.57 | 18.3 | 18.3 | 18.0 | 0.287 | 172.3 | 170.2 |
| Mob | 14 | 6.33 | 6.82 | 15.7 | 16.0 | 15.6 | 0.373 | 260.4 | 272.5 |
| Tch | 15 | 6.97 | 6.85 | 14.5 | 14.6 | 14.3 | 0.364 | 296.1 | 297.0 |
| Ruh | 16 | 7.06 | 6.74 | 13.9 | 13.9 | 13.6 | 0.367 | 309.4 | 320.8 |
| Rhf | 15 | 5.96 | 5.75 | 14.2 | 14.2 | 13.7 | 0.349 | 253.4 | 270.4 |
| 16 | 3.72 | 3.89 | 15.4 | 15.5 | 14.7 | 0.281 | 163.7 | 180.8 | |
| Agf | 17 | 2.52 | 2.95 | 17.9 | 18.0 | 17.1 | 0.207 | 91.3 | 100.7 |
| Cdh | 18 | 0.764 | 1.16 | 22.4 | 23.0 | 21.6 | 0.113 | 35.8 | 46.7 |
| Hfh | 26 | 6.56 | 6.44 | 22.2 | 22.4 | 22.3 | 0.227 | 109.1 | 109.0 |
| Tab | 27 | 8.28 | 8.10 | 18.1 | 18.3 | 18.0 | 0.312 | 195.3 | 200.0 |
| Wb | 26 | 8.41 | 8.90 | 16.0 | 16.2 | 15.8 | 0.411 | 302.2 | 323.2 |
| Reh | 27 | 7.82 | 8.03 | 15.0 | 15.0 | 14.7 | 0.414 | 366.8 | 372.0 |
| Osh | 28 | 8.45 | 8.17 | 14.4 | 14.4 | 14.0 | 0.430 | 395.5 | 418.0 |
| Irf | 29 | 7.55 | 6.94 | 14.6 | 14.6 | 14.2 | 0.424 | 342.8 | 355.0 |
| Ptf | 30 | 5.48 | 5.84 | 15.8 | 15.8 | 15.1 | 0.355 | 243.4 | 278.3 |
| Auf | 17 | 3.03 | 3.81 | 18.2 | 18.2 | 17.0 | 0.264 | 137.6 | 173.2 |
| Alf | 9 | 3.43 | 3.39 | 16.6 | 16.6 | 16.6 | 0.202 | 74.3 | 72.2 |
| Gac | 13 | 2.68 | 2.81 | 20.4 | -- | 19.6 | 0.226 | 50.3 | 56.9 |
| Inc | 13 | 2.36 | 2.52 | 27.6 | -- | 26.1 | 0.113 | 37.4 | 41.1 |
| Tlh | 13 | 2.08 | 1.88 | 30.9 | 31.3 | 28.6 | 0.093 | 27.2 | 35.9 |
| Snc | 14 | 3.16 | 3.14 | 36.7 | -- | 34.2 | 0.334 | 111.0 | 111.0 |
| Pbf | 14 | 2.93 | 2.03 | 31.7 | 31.9 | 30.3 | 0.111 | 40.6 | 43.0 |
| Bic | 15 | 2.58 | 2.18 | 36.8 | -- | 35.0 | 0.116 | 34.0 | 31.5 |
| Metal. | ebcp (e/Å3) | B * (GPa) | ||||
|---|---|---|---|---|---|---|
| hcp | bcc | fcc | hcp | bcc | fcc | |
| Li | 0.0539 | 0.0535 | 0.0534 | 13.5 | 13.9 | 13.5 |
| Na | 0.0304 | 0.0306 | 0.0301 | 7.6 | 7.9 | 7.5 |
| K | 0.0163 | 0.0165 | 0.0163 | 3.5 | 3.5 | 3.5 |
| Rb | 0.0138 | 0.0122 | 0.0136 | 2.7 | 2.7 | 2.7 |
| Be | 0.2809 | 0.2558 | 0.2714 | 121.1 | 122.9 | 118.5 |
| Mg | 0.1059 | 0.0892 | 0.0979 | 35.7 | 34.9 | 34.7 |
| Ca | 0.0518 | 0.0520 | 0.0523 | 17.7 | 16.0 | 17.4 |
| Sr | 0.0435 | 0.0424 | 0.0431 | 11.4 | 12.2 | 11.8 |
| Ba | 0.0401 | 0.0388 | 0.0390 | 8.4 | 9.0 | 8.3 |
| Sc | 0.1282 | 0.1244 | 0.1232 | 54.9 | 53.2 | 51.8 |
| Ti | 0.2300 | 0.1874 | 0.2037 | 112.8 | 107.3 | 109.0 |
| V | 0.3232 | 0.2900 | 0.2755 | 173.2 | 182.9 | 176.0 |
| Cr | 0.3782 | 0.3788 | 0.3337 | 233.5 | 257.7 | 236.7 |
| Mn | 0.2998 | 0.2998 | 0.2998 | 279.7 | 241.8 | 278.9 |
| Fe | 0.3810 | 0.3362 | 0.4084 | 288.3 | 189.3 | 194.6 |
| Co | 0.3343 | 0.3770 | 0.3641 | 212.5 | 203.6 | 210.2 |
| Ni | 0.3194 | 0.3399 | 0.3214 | 193.8 | 189.1 | 195.6 |
| Cu | 0.2580 | 0.1939 | 0.2584 | 136.1 | 134.6 | 137.5 |
| Zn | 0.1616 | 0.1456 | 0.2004 | 51.8 | 63.3 | 68.4 |
| Y | 0.0915 | 0.0981 | 0.0876 | 40.8 | 39.3 | 39.7 |
| Zr | 0.1972 | 0.1546 | 0.1781 | 95.3 | 89.7 | 92.9 |
| Nb | 0.2992 | 0.2871 | 0.2529 | 162.8 | 172.3 | 165.7 |
| Mo | 0.3713 | 0.3738 | 0.3342 | 233.8 | 260.4 | 239.4 |
| Tc | 0.3648 | 0.4048 | 0.3740 | 296.1 | 290.6 | 294.7 |
| Ru | 0.3670 | 0.4015 | 0.3809 | 309.4 | 279.1 | 304.2 |
| Rh | 0.3431 | 0.3581 | 0.3493 | 251.1 | 225.9 | 253.4 |
| Pd | 0.2912 | 0.3016 | 0.2813 | 163.6 | 163.5 | 163.7 |
| Ag | 0.2067 | 0.1289 | 0.2078 | 91.1 | 88.9 | 91.3 |
| Cd | 0.1139 | 0.0920 | 0.1395 | 35.8 | 35.8 | 42.6 |
| Hf | 0.2274 | 0.1709 | 0.1974 | 109.1 | 101.8 | 103.6 |
| Ta | 0.3216 | 0.3122 | 0.2815 | 188.0 | 195.3 | 191.2 |
| W | 0.4057 | 0.4118 | 0.3641 | 274.3 | 302.2 | 281.9 |
| Re | 0.4146 | 0.4619 | 0.4205 | 366.8 | 357.2 | 365.2 |
| Os | 0.4305 | 0.4717 | 0.4435 | 395.5 | 353.0 | 388.8 |
| Ir | 0.4193 | 0.4351 | 0.4247 | 339.0 | 298.6 | 342.8 |
| Pt | 0.3832 | 0.3583 | 0.3550 | 235.2 | 233.8 | 243.4 |
| Au | 0.2691 | 0.1508 | 0.2646 | 135.0 | 134.9 | 137.6 |
| Al | 0.2014 | 0.1518 | 0.2024 | 70.8 | 65.2 | 74.3 |
| Ga | 0.1773 | 0.1070 | 0.1664 | 45.9 | 47.4 | 48.0 |
| In | 0.1231 | 0.0745 | 0.1206 | 34.4 | 34.7 | 36.2 |
| Tl | 0.0931 | 0.0603 | 0.1057 | 27.2 | 27.9 | 28.4 |
| Sn | 0.1448 | 0.1590 | 0.1355 | 47.6 | 47.5 | 47.2 |
| Pb | 0.1173 | 0.0682 | 0.1118 | 40.2 | 39.8 | 40.6 |
| Bi | 0.1286 | 0.0730 | 0.1269 | 52.0 | 52.6 | 51.9 |
| ebcp | V | B | Ecoh/V | |
|---|---|---|---|---|
| ebcp | -- | (k2/V)2 | B0.5/k1 | Ecoh/(Vk3) + 0.03 |
| V | -- | |||
| B | -- | |||
| Ecoh/V | k3(ebcp − 0.03) | -- |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, J.; He, D.; Song, Y. Correlations of Equilibrium Properties and Electronic Structure of Pure Metals. Materials 2019, 12, 2932. https://doi.org/10.3390/ma12182932
Dai J, He D, Song Y. Correlations of Equilibrium Properties and Electronic Structure of Pure Metals. Materials. 2019; 12(18):2932. https://doi.org/10.3390/ma12182932
Chicago/Turabian StyleDai, Jianhong, Dongye He, and Yan Song. 2019. "Correlations of Equilibrium Properties and Electronic Structure of Pure Metals" Materials 12, no. 18: 2932. https://doi.org/10.3390/ma12182932
APA StyleDai, J., He, D., & Song, Y. (2019). Correlations of Equilibrium Properties and Electronic Structure of Pure Metals. Materials, 12(18), 2932. https://doi.org/10.3390/ma12182932