The Utilization of Cell-Penetrating Peptides in the Intracellular Delivery of Viral Nanoparticles


Abstract
1. Introduction
2. Cell-penetrating Peptides
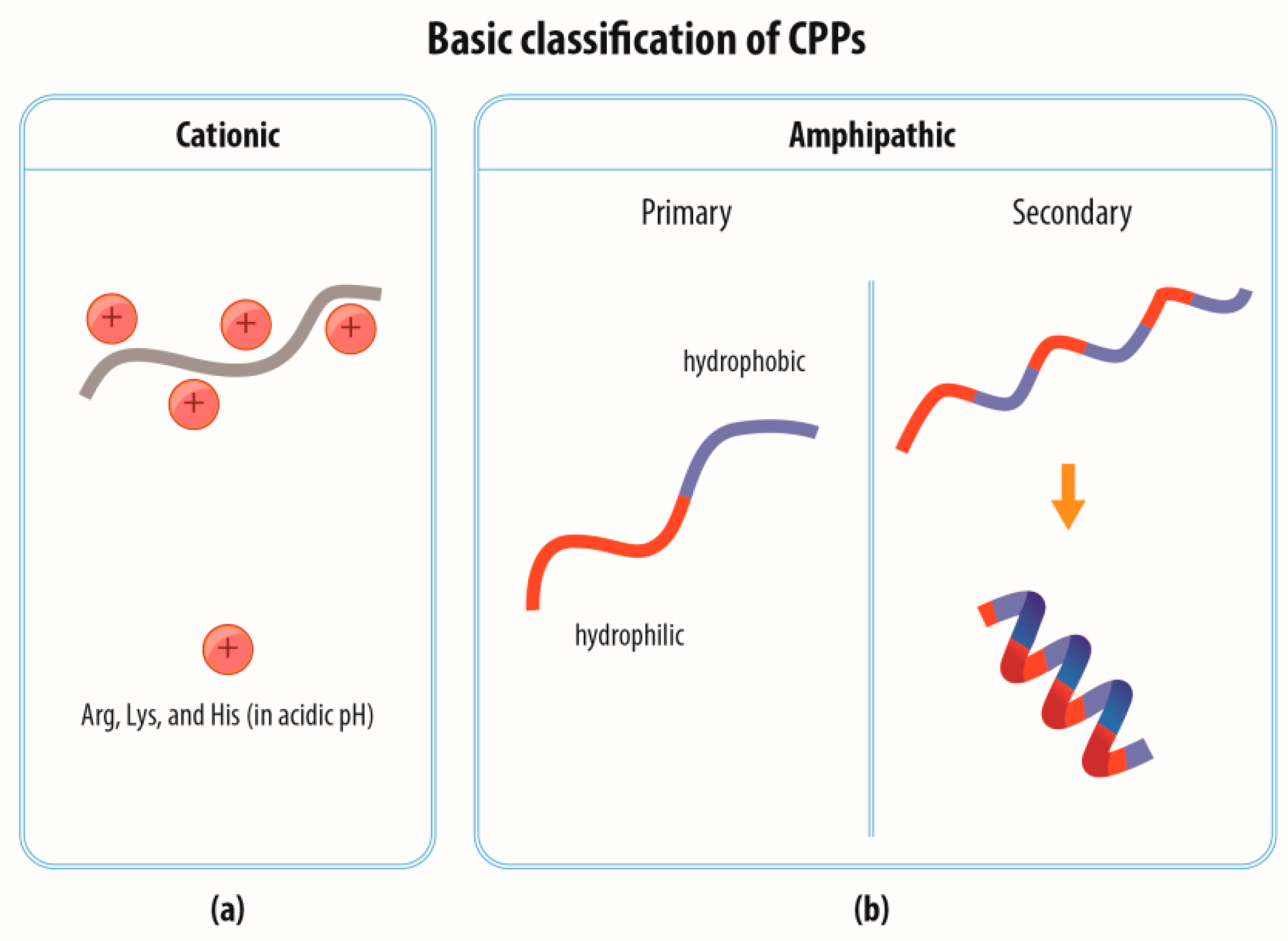
2.1. Classification of CPPs
2.2. Internalization of CPPs
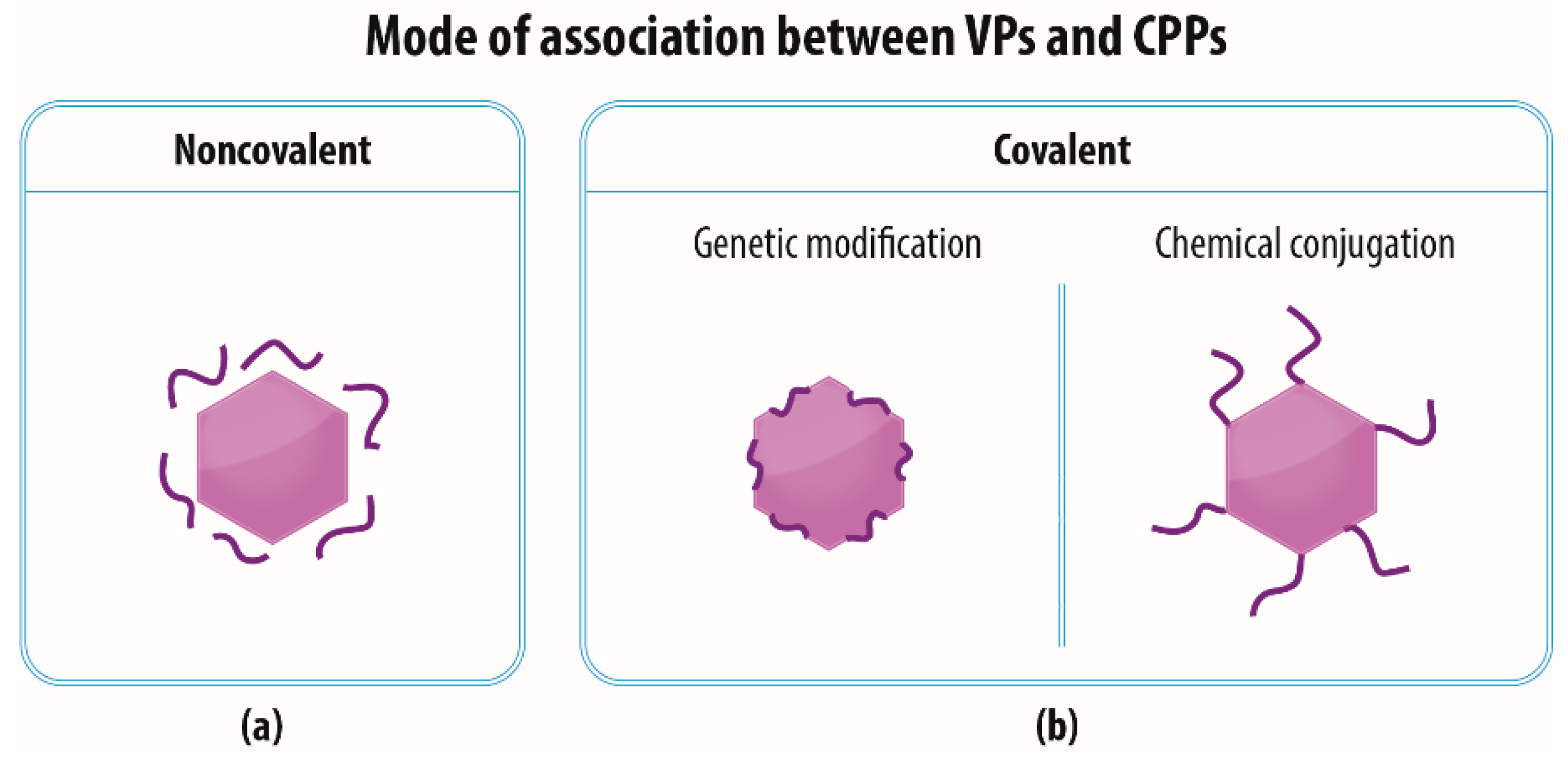
3. The Association of VPs and CPPs
3.1. Noncovalent—Electrostatic Interactions between VPs and CPPs
3.2. Covalent—Genetic Modification of VPs with CPPs
3.3. Covalent—Chemical Conjugation of CPPs to the Surface of VPs
4. Modalities of CPP-functionalized VPs
4.1. Broadening the Spectrum of Cells for VP Entry
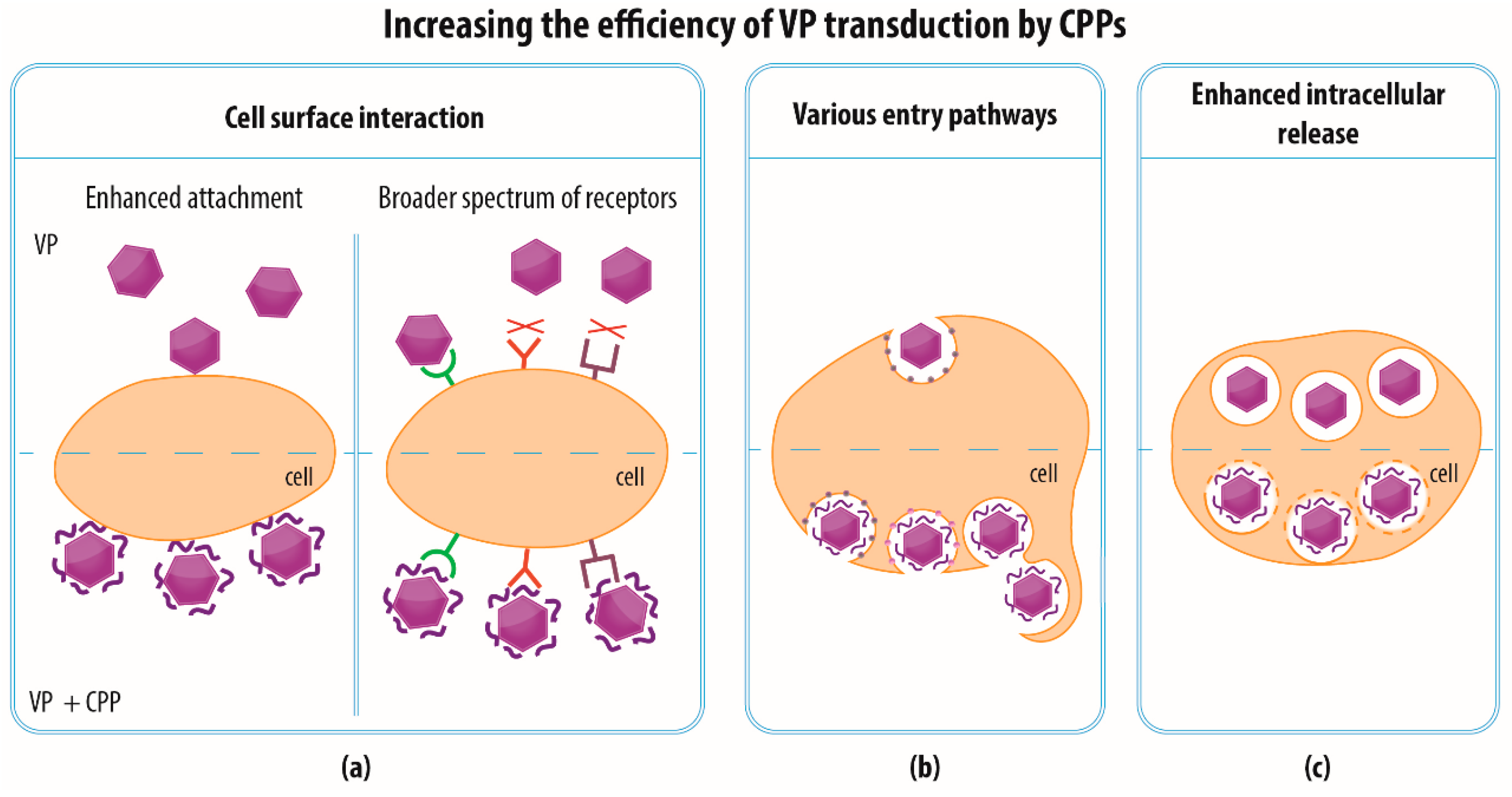
4.2. Increasing the Efficiency of VP Transduction
4.3. Immunomodulation
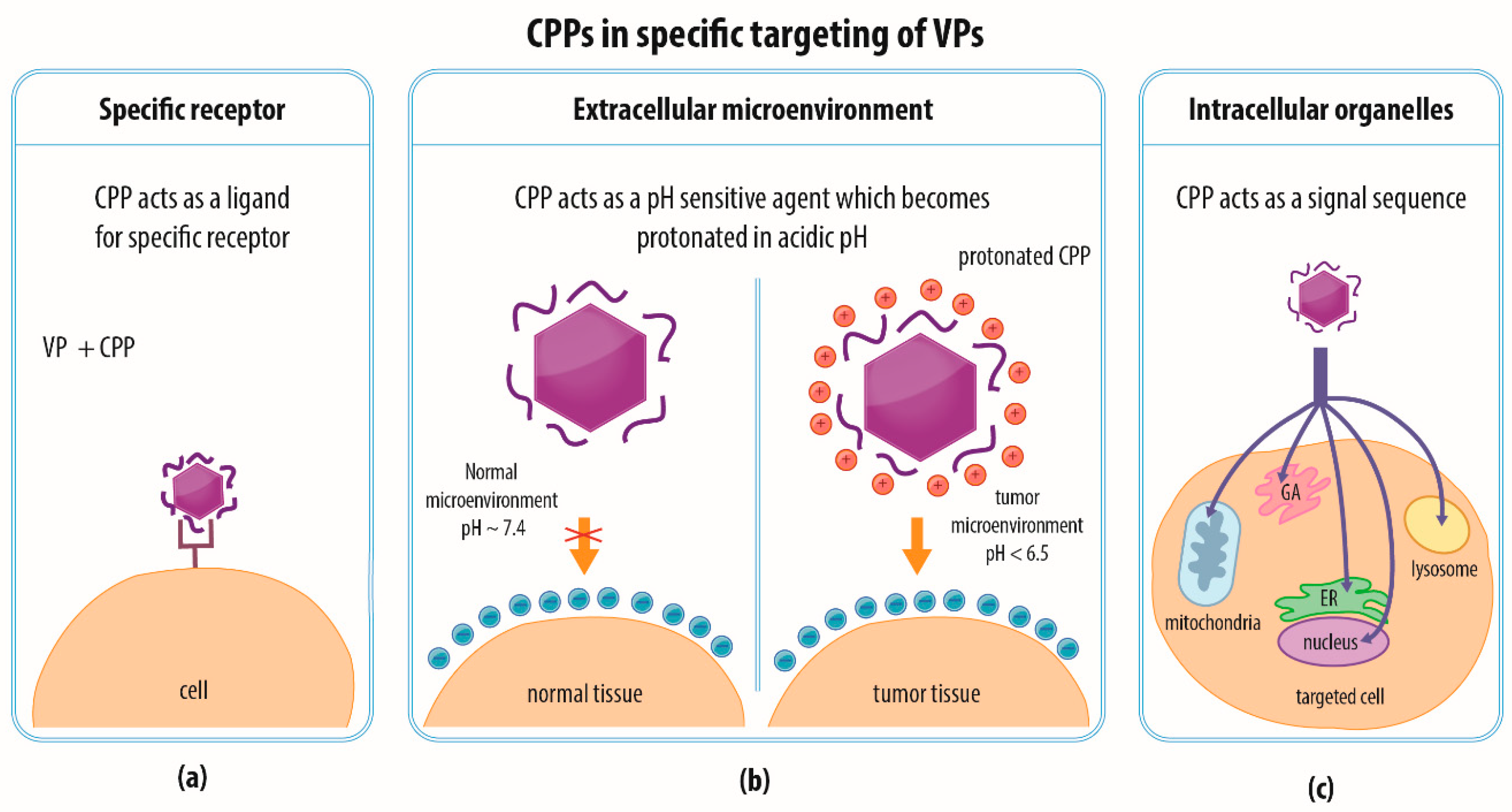
4.4. Combination of Strategies and Specific Targeting
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef]
- Shirbaghaee, Z.; Bolhassani, A. Different applications of virus-like particles in biology and medicine: Vaccination and delivery systems. Biopolymers 2016, 105, 113–132. [Google Scholar] [PubMed]
- Schwarz, B.; Douglas, T. Development of virus-like particles for diagnostic and prophylactic biomedical applications. Wiley Interdiscip. Rev. Nanomed. NanobioTechnol. 2015, 7, 722–735. [Google Scholar] [CrossRef]
- Garcea, R.L.; Gissmann, L. Virus-like particles as vaccines and vessels for the delivery of small molecules. Curr. Opin. Biotechnol. 2004, 15, 513–517. [Google Scholar] [CrossRef]
- Grgacic, E.V.L.; Anderson, D.A. Virus-like particles: Passport to immune recognition. Methods 2006, 40, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Durzyńska, J.; Przysiecka, Ł.; Nawrot, R.; Barylski, J.; Nowicki, G.; Warowicka, A.; Musidlak, O.; Goździcka-Józefiak, A. Viral and other cell-penetrating peptides as vectors of therapeutic agents in medicine. J. Pharmacol. Exp. Ther. 2015, 354, 32–42. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic. Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [PubMed]
- Langel, Ü. CPP, Cell-Penetrating Peptides; Langel, Ü., Ed.; Springer: Singapore, 2019; ISBN 9789811387470. [Google Scholar]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wender, P.A. Role of Membrane Potential and Hydrogen Bonding in the Mechanism of Translocation of Guanidinium-Rich Peptides into Cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Lai, G.H.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Cheng, J.; Deming, T.J.; Kamei, D.T.; et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 16883–16888. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C.; Yaouanc, J.J.; Jaffrès, P.A. Chemical vectors for gene delivery: A current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Kichler, A.; Mason, A.J.; Bechinger, B. Cationic amphipathic histidine-rich peptides for gene delivery. Biochim. Biophys. Acta 2006, 1758, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Galdiero, M.; Galdiero, S. Membranotropic Cell Penetrating Peptides: The Outstanding Journey. Int. J. Mol. Sci. 2015, 16, 25323–25337. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Wikström, S.; Johansson, M. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 2003, 8, 143–150. [Google Scholar] [CrossRef]
- Drin, G.; Cottin, S.; Blanc, E.; Rees, A.R.; Temsamani, J. Studies on the Internalization Mechanism of Cationic Cell-penetrating Peptides. J. Biol. Chem. 2003, 278, 31192–31201. [Google Scholar] [CrossRef]
- Leifert, J.A.; Harkins, S.; Whitton, J.L. Full-length proteins attached to the HIV tat protein transduction domain are neither transduced between cells, nor exhibit enhanced immunogenicity. Gene Ther. 2002, 9, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef] [PubMed]
- LeCher, J.C.; Nowak, S.J.; McMurry, J.L. Breaking in and busting out: Cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.; Marrink, S.J.; Mark, A.E. Alternative Mechanisms for the Interaction of the Cell-Penetrating Peptides Penetratin and the TAT Peptide with Lipid Bilayers. Biophys. J. 2009, 97, 40–49. [Google Scholar] [CrossRef]
- Kosuge, M.; Takeuchi, T.; Nakase, I.; Jones, A.T.; Futaki, S. Cellular Internalization and Distribution of Arginine-Rich Peptides as a Function of Extracellular Peptide Concentration, Serum, and Plasma Membrane Associated Proteoglycans. BioConjug. Chem. 2008, 19, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient Focal Membrane Deformation Induced by Arginine-rich Peptides Leads to Their Direct Penetration into Cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Nakase, I. Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization. Acc. Chem. Res. 2017, 50, 2449–2456. [Google Scholar] [CrossRef]
- Gao, X.; Hong, S.; Liu, Z.; Yue, T.; Dobnikar, J.; Zhang, X. Membrane potential drives direct translocation of cell-penetrating peptides. Nanoscale 2019, 11, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Tünnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006, 20, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Maiolo, J.R.; Ferrer, M.; Ottinger, E.A. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta 2005, 1712, 161–172. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, S.; Järver, P.; Johansson, H.J.; Langel, U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: A comparative study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef]
- Mai, J.C.; Shen, H.; Watkins, S.C.; Cheng, T.; Robbins, P.D. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J. Biol. Chem. 2002, 277, 30208–30218. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Koppelhus, U.; Awasthi, S.K.; Zachar, V.; Holst, H.U.; Ebbesen, P.; Nielsen, P.E. Cell-Dependent Differential Cellular Uptake of PNA, Peptides, and PNA-Peptide Conjugates. Antisense Nucleic Acid Drug Dev. 2002, 12, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.-H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef]
- Birch, D.; Christensen, M.V.; Staerk, D.; Franzyk, H.; Nielsen, H.M. Fluorophore labeling of a cell-penetrating peptide induces differential effects on its cellular distribution and affects cell viability. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2483–2494. [Google Scholar] [CrossRef]
- Fischer, R.; Waizenegger, T.; Köhler, K.; Brock, R. A quantitative validation of fluorophore-labelled cell-permeable peptide conjugates: Fluorophore and cargo dependence of import. Biochim. Biophys. Acta 2002, 1564, 365–374. [Google Scholar] [CrossRef]
- Kristensen, M.; Birch, D.; Mørck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Piovan, C.; Marin, V.; Scavullo, C.; Corna, S.; Giuliani, E.; Bossi, S.; Galy, A.; Fenard, D.; Bordignon, C.; Rizzardi, G.P.; et al. Vectofusin-1 Promotes RD114-TR-Pseudotyped Lentiviral Vector Transduction of Human HSPCs and T Lymphocytes. Mol. Ther. Methods Clin. Dev. 2017, 5, 22–30. [Google Scholar] [CrossRef]
- Youn, J.I.; Park, S.H.; Jin, H.T.; Lee, C.G.; Seo, S.H.; Song, M.Y.; Lee, C.W.; Sung, Y.C. Enhanced delivery efficiency of recombinant adenovirus into tumor and mesenchymal stem cells by a novel PTD. Cancer Gene Ther. 2008, 15, 703–712. [Google Scholar] [CrossRef]
- Fenard, D.; Genries, S.; Scherman, D.; Galy, A.; Martin, S.; Kichler, A. Infectivity enhancement of different HIV-1-based lentiviral pseudotypes in presence of the cationic amphipathic peptide LAH4-L1. J. Virol. Methods 2013, 189, 375–378. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gratton, J.P.; Yu, J.; Griffith, J.W.; Babbitt, R.W.; Scotland, R.S.; Hickey, R.; Giordano, F.J.; Sessa, W.C. Cell-permeable peptides improve cellular uptake and therapeutic gene delivery of replication-deficient viruses in cells and in vivo. Nat. Med. 2003, 9, 357–362. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, Y.J.; Ji, M.; Fang, J.; Siriwon, N.; Zhang, L.; Wang, P. Enhancing gene delivery of adeno-associated viruses by cell-permeable peptides. Mol. Ther. Methods Clin. Dev. 2014, 1, 12. [Google Scholar] [CrossRef]
- Park, S.H.; Doh, J.; Park, S.; Lim, J.Y.; Kim, S.M.; Youn, J.I.; Jin, H.T.; Seo, S.H.; Song, M.Y.; Sung, S.Y.; et al. Branched oligomerization of cell-permeable peptides markedly enhances the transduction efficiency of adenovirus into mesenchymal stem cells. Gene Ther. 2010, 17, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Jamali, A.; Kapitza, L.; Schaser, T.; Johnston, I.C.; Buchholz, C.J.; Hartmann, J. Highly Efficient and Selective CAR-Gene Transfer Using CD4 and CD8-Targeted Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2019, 13, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Kühnel, F.; Schulte, B.; Wirth, T.; Woller, N.; Schäfers, S.; Zender, L.; Manns, M.; Kubicka, S. Protein Transduction Domains Fused to Virus Receptors Improve Cellular Virus Uptake and Enhance Oncolysis by Tumor-Specific Replicating Vectors. J. Virol. 2004, 78, 13743–13754. [Google Scholar] [CrossRef]
- Fenard, D.; Ingrao, D.; Seye, A.; Buisset, J.; Genries, S.; Martin, S.; Kichler, A.; Galy, A. Vectofusin-1, a New Viral Entry Enhancer, Strongly Promotes Lentiviral Transduction of Human Hematopoietic Stem Cells. Mol. Ther. Nucleic Acids 2013, 2, e90. [Google Scholar] [CrossRef]
- Lehmusvaara, S.; Rautsi, O.; Hakkarainen, T.; Wahlfors, J. Utility of cell-permeable peptides for enhancement of virus-mediated gene transfer to human tumor cells. BioTechniques 2006, 40, 573–576. [Google Scholar] [CrossRef]
- Posey, N.D.; Tew, G.N. Associative and Dissociative Processes in Non-Covalent Polymer-Mediated Intracellular Protein Delivery. Chem. Asian J. 2018, 13, 3351–3365. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef]
- Han, T.; Tang, Y.; Ugai, H.; Perry, L.; Siegal, G.P.; Contreras, J.L.; Wu, H. Genetic incorporation of the protein transduction domain of Tat into Ad5 fiber enhances gene transfer efficacy. Virol. J. 2007, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, S.; Tashiro, K.; Sakurai, F.; Sakurai, H.; Kawabata, K.; Yayama, K.; Okamoto, H.; Nakagawa, S.; Mizuguchi, H. Fiber-modified adenovirus vectors containing the TAT peptide derived from HIV-1 in the fiber knob have efficient gene transfer activity. Gene Ther. 2007, 14, 1160–1165. [Google Scholar] [CrossRef]
- Liu, S.; Mao, Q.; Zhang, W.; Zheng, X.; Bian, Y.; Wang, D.; Li, H.; Chai, L.; Zhao, J.; Xia, H. Genetically modified adenoviral vector with the protein transduction domain of Tat improves gene transfer to CAR-deficient cells. Biosci. Rep. 2009, 29, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Jin, C.; Leja, J.; Majdalani, N.; Nilsson, B.; Eriksson, F.; Essand, M. Adenovirus with Hexon Tat-Protein Transduction Domain Modification Exhibits Increased Therapeutic Effect in Experimental Neuroblastoma and Neuroendocrine Tumors. J. Virol. 2011, 85, 13114–13123. [Google Scholar] [CrossRef]
- Chen, H.Z.; Wu, C.P.; Chao, Y.C.; Liu, C.Y.Y. Membrane penetrating peptides greatly enhance baculovirus transduction efficiency into mammalian cells. Biochem. Biophys. Res. Commun. 2011, 405, 297–302. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, Y.; Zhao, R. Establishment of MicroRNA delivery system by PP7 bacteriophage-like particles carrying cell-penetrating peptide. J. Biosci. Bioeng. 2017, 124, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, Y.; Zhao, R.; Gao, K. Intracellular delivery of messenger RNA by recombinant PP7 virus-like particles carrying low molecular weight protamine. BMC Biotechnol. 2016, 16, 3020. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jia, T.; Xu, X.; Chang, L.; Zhang, R.; Fu, Y.; Li, Y.; Yang, X.; Zhang, K.; Lin, G.; et al. Novel miR-122 delivery system based on MS2 virus like particle surface displaying cell-penetrating peptide TAT for hepatocellular carcinoma. Oncotarget 2016, 7, 59402–59416. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Akuta, T.; Okuyama, H.; Senda, T.; Yokoi, H.; Inokuchi, H.; Fujita, S.; Hayakawa, T.; Takeda, K.; Hasegawa, M.; et al. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J. Biol. Chem. 2001, 276, 26204–26210. [Google Scholar] [CrossRef] [PubMed]
- Rohovie, M.J.; Nagasawa, M.; Swartz, J.R. Virus-like particles: Next-generation nanoparticles for targeted therapeutic delivery. Bioeng. Transl. Med. 2017, 2, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Asavatanabodee, R.; Eto, Y.; Watanabe, H.; Morishige, T.; Yao, X.; Kida, S.; Maeda, M.; Mukai, Y.; Mizuguchi, H.; et al. Tat conjugation of adenovirus vector broadens tropism and enhances transduction efficiency. Life Sci. 2008, 83, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Eto, Y.; Yoshioka, Y.; Asavatanabodee, R.; Kida, S.; Maeda, M.; Mukai, Y.; Mizuguchi, H.; Kawasaki, K.; Okada, N.; Nakagawa, S. Transduction of adenovirus vectors modified with cell-penetrating peptides. Peptides 2009, 30, 1548–1552. [Google Scholar] [CrossRef]
- Nigatu, A.S.; Vupputuri, S.; Flynn, N.; Ramsey, J.D. Effects of cell-penetrating peptides on transduction efficiency of PEGylated adenovirus. Biomed. Pharmacother. 2015, 71, 153–160. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, K.; Yildiz, I.; Dirksen, A.; Fischer, R.; Dawson, P.E.; Steinmetz, N.F. Development of viral nanoparticles for efficient intracellular delivery. Nanoscale 2012, 4, 3567–3576. [Google Scholar] [CrossRef]
- Gan, B.K.; Yong, C.Y.; Ho, K.L.; Omar, A.R.; Alitheen, N.B.; Tan, W.S. Targeted Delivery of Cell Penetrating Peptide Virus-like Nanoparticles to Skin Cancer Cells. Sci. Rep. 2018, 8, 8499. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, Y.; Jia, T.; Zhang, K.; Li, J.; Wang, L. Development of a microRNA delivery system based on bacteriophage MS2 virus-like particles. FEBS J. 2012, 279, 1198–1208. [Google Scholar] [CrossRef]
- Wei, B.; Wei, Y.; Zhang, K.; Wang, J.; Xu, R.; Zhan, S.; Lin, G.; Wang, W.; Liu, M.; Wang, L.; et al. Development of an antisense RNA delivery system using conjugates of the MS2 bacteriophage capsids and HIV-1 TAT cell-penetrating peptide. Biomed. Pharmacother. 2009, 63, 313–318. [Google Scholar] [CrossRef]
- Anand, P.; O’Neil, A.; Lin, E.; Douglas, T.; Holford, M. Tailored delivery of analgesic ziconotide across a blood brain barrier model using viral nanocontainers. Sci. Rep. 2015, 5, 12497. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.H.; Chen, P.Y.; Wei, K.C.; Huang, C.-W.; Shiue, Y.L.; Huang, C.Y.; Yang, H.W. Convection-Enhanced Delivery of a Virus-Like Nanotherapeutic Agent with Dual-Modal Imaging for Besiegement and Eradication of Brain Tumors. Theranostics 2019, 9, 1752–1763. [Google Scholar] [CrossRef]
- Kim, D.; Lee, Y.; Dreher, T.W.; Cho, T.J. Empty Turnip yellow mosaic virus capsids as delivery vehicles to mammalian cells. Virus Res. 2018, 252, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Salerno, J.C.; Ngwa, V.M.; Nowak, S.J.; Chrestensen, C.A.; Healey, A.N.; McMurry, J.L. Novel cell-penetrating peptide-adaptors effect intracellular delivery and endosomal escape of protein cargos. J. Cell Sci. 2016, 129, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Koudelka, K.J.; Destito, G.; Plummer, E.M.; Trauger, S.A.; Siuzdak, G.; Manchester, M. Endothelial Targeting of Cowpea Mosaic Virus (CPMV) via Surface Vimentin. PLoS Pathog. 2009, 5, e1000417. [Google Scholar] [CrossRef]
- Mansouri, M.; Berger, P. Baculovirus for gene delivery to mammalian cells: Past, present and future. Plasmid 2018, 98, 1–7. [Google Scholar] [CrossRef]
- Reeh, M.; Bockhorn, M.; Görgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the Coxsackievirus and Adenovirus Receptor (CAR) in human neoplasms: A multitumour array analysis. Br. J. Cancer 2013, 109, 1848–1858. [Google Scholar] [CrossRef]
- Wirth, T.; Zender, L.; Schulte, B.; Mundt, B.; Plentz, R.; Rudolph, K.L.; Manns, M.; Kubicka, S.; Kühnel, F. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res. 2003, 63, 3181–3188. [Google Scholar]
- Rebetz, J.; Na, M.; Su, C.; Holmqvist, B.; Edqvist, A.; Nyberg, C.; Widegren, B.; Salford, L.G.; Sjögren, H.O.; Arnberg, N.; et al. Fiber Mediated Receptor Masking in Non-Infected Bystander Cells Restricts Adenovirus Cell Killing Effect but Promotes Adenovirus Host Co-Existence. PLoS ONE 2009, 4, e8484. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; et al. Adenovirus Serotype 5 Hexon Mediates Liver Gene Transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, L.S.; Hamon, L.; Schirer, A.; Schoup, M.; Cosette, J.; Majdoul, S.; Pastré, D.; Stockholm, D.; Holic, N.; Hellwig, P.; et al. Vectofusin-1, a potent peptidic enhancer of viral gene transfer forms pH-dependent α-helical nanofibrils, concentrating viral particles. Acta Biomater. 2017, 64, 259–268. [Google Scholar] [CrossRef]
- Kim, P.H.; Kim, T.I.; Yockman, J.W.; Kim, S.W.; Yun, C.O. The effect of surface modification of adenovirus with an arginine-grafted bioreducible polymer on transduction efficiency and immunogenicity in cancer gene therapy. Biomaterials 2010, 31, 1865–1874. [Google Scholar] [CrossRef]
- Rubsamen, R.; Herst, C.; Lloyd, P.; Heckerman, D. Eliciting cytotoxic T-lymphocyte responses from synthetic vectors containing one or two epitopes in a C57BL/6 mouse model using peptide-containing biodegradable microspheres and adjuvants. Vaccine 2014, 32, 4111–4116. [Google Scholar] [CrossRef]
- Grau, M.; Walker, P.R.; Derouazi, M. Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell. Mol. Life Sci. 2018, 75, 2887–2896. [Google Scholar] [CrossRef]
- Schumacher, T.; Ruehland, C.; Schultheiss, C.; Brinkman, M.; Roedel, F.; Reiser, C.O.A.; Hess, J.; Reichel, C. Advanced Antigen Delivery of Murine Survivin: Chimeric Virus-Like Particles in Cancer Vaccine Research. Int. J. Biomed. Sci. IJBS 2007, 3, 199–205. [Google Scholar] [PubMed]
- Zhang, T.T.; Kang, T.H.; Ma, B.; Xu, Y.; Hung, C.F.; Wu, T.C. LAH4 enhances CD8+ T cell immunity of protein/peptide-based vaccines. Vaccine 2012, 30, 784–793. [Google Scholar] [CrossRef]
- Wang, H.Y.; Wang, R.F. Enhancing cancer immunotherapy by intracellular delivery of cell-penetrating peptides and stimulation of pattern-recognition receptor signaling. Adv. Immunol. 2012, 114, 151–176. [Google Scholar]
- Brooks, N.A.; Pouniotis, D.S.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G.A. Cell-penetrating peptides: Application in vaccine delivery. Biochim. Biophys. Acta 2010, 1805, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Sahdev, P.; Ochyl, L.J.; Moon, J.J. Biomaterials for nanoparticle vaccine delivery systems. Pharm. Res. 2014, 31, 2563–2582. [Google Scholar] [CrossRef]
- Nakamura, T.; Moriguchi, R.; Kogure, K.; Shastri, N.; Harashima, H. Efficient MHC class I presentation by controlled intracellular trafficking of antigens in octaarginine-modified liposomes. Mol. Ther. 2008, 16, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Akhras, S.; Toda, M.; Boller, K.; Himmelsbach, K.; Elgner, F.; Biehl, M.; Scheurer, S.; Gratz, M.; Vieths, S.; Hildt, E. Cell-permeable capsids as universal antigen carrier for the induction of an antigen-specific CD8+ T-cell response. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Ding, Y.; Mujeeb, A.; Zhao, Y.; Nie, G. Tumor Microenvironment Targeting and Responsive Peptide-Based Nanoformulations for Improved Tumor Therapy. Mol. Pharmacol. 2017, 92, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Künnapuu, K.; Langel, Ü. Cell-penetrating peptides with intracellular organelle targeting. Expert Opin. Drug Deliv. 2017, 14, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Collado Camps, E.; Brock, R. An opportunistic route to success: Towards a change of paradigm to fully exploit the potential of cell-penetrating peptides. Bioorg. Med. Chem. 2018, 26, 2780–2787. [Google Scholar] [CrossRef] [PubMed]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Sequence | Characteristics |
|---|---|---|
| Tat | (Y)GKKKRRQRRR 1 | Cationic |
| Penetratin (Pen) 2 | RQIKIWFQNRRMKWKK | Cationic |
| Polyarginine (R5, R8 or R9) | RRRRR(RRR(R)) | Cationic |
| Pep-1 | KETWWETWWTEWSQPKKKRKV | Amphipathic |
| Proline-rich peptide (Pro) | VRLPPPVRLPPPVRLPPP | Proline-rich |
| Tat-HA2 | CRRRQRRKKRGGDIMGEWGNEIFGAIAGFLG | Cationic fusogenic |
| Hph-1 | YARVRRRGPRR | Cationic |
| HP4 | RRRRPRRRTTRRRR | Cationic |
| LAH4 | KKALLALALHHLAHLALHLALALKKA | Histidine-rich cationic amphipathic |
| LAH4-L1 | KKALLAHALHLLALLALHLAHALKKA | Histidine-rich cationic amphipathic |
| Vectofusin-1 | KKALLHAALAHLLALAHHLLALLKKA | Histidine-rich cationic amphipathic |
| Low molecular weight protamine (LMWP) | VSRRRRRRGGRRRR | Cationic |
| Virus | CPP | Cargo | Cell Lines Tested | Effect | Ref. |
|---|---|---|---|---|---|
| Adeno-associated virus type 2 and 8 | Pen, Tat-HA2, LAH4 | Viral genome with GFP gene | HEK-293T, HepG2, NIH-3T3, BMDC, MSC, Huh7 |
| [47] |
| Adenovirus | Tat-CAR, VP22-CAR, R9-CAR, Pen-CAR, Tat | LacZ or GFP gene or hTERT promoter in the genome | H4IIE, BNL, RT-101, T-36274, RKO, SAOS-2, SKLU-1, MCF-7, HT1080, HepG2, Huh7, HeLa, HT29, KLN205, P388D.1, RAW264.7, DC2.4, Jurkat, ISC |
| [50] |
| Adenovirus | HP4, Tat, Pen, Hph-1 | Viral genome with GFP or IL-12N220L gene | A375, CT26, B16F10, U-87MG, HeLa, A549, K562, C6Bu1, UCB-MSC, BM-MSC, AT-MSC, BMDC |
| [44] |
| Adenovirus | Branched oligomeric Tat, Hph-1, Pen, HP4 | Viral genome with eGFP, human bone morphogenetic protein 2, or brain-derived neurotrophic factor gene | BM-MSC, UCB-MSC |
| [48] |
| Adenovirus, retrovirus | Pen, Tat | Viral genome with GFP, β-galactosidase, eNOS, or VEGF gene | COS-7, HUVEC, BAEC |
| [46] |
| Adenovirus, pseudotyped lentivirus | Tat from HIV-1 and HIV-2, Pen | Viral genome with GFP gene | COS-7, SKOV3.ip1, HEY, PC-3, MG-63 |
| [52] |
| Pseudotyped lentiviruses and HIV-1-derived VLPs | LAH4-L1 | Plasmid with eGFP gene | HCT116, HSC |
| [45] |
| Pseudotyped lentiviruses | Vectofusin-1 | Plasmid with GFP gene | UCB-HSC, BM-HSC, activated human T cells |
| [43] |
| Pseudotyped retroviruses | Vectofusin-1 | Plasmid with eGFP gene | UCB-HSC, MPB-HSC |
| [51] |
| Lentiviral vectors targeted to CD4 and CD8 and pseudotyped lentiviruses | Vectofusin-1 | Plasmid for expression of chimeric antigen receptor and reporter molecule: truncated version of the low-affinity nerve growth factor receptor on VP surface | Human T lymphocytes |
| [49] |
| Virus | CPP | Place of CPP Incorporation | Cargo | Cell Lines Tested | Effect | Ref. |
|---|---|---|---|---|---|---|
| Baculovirus | Two longer versions of Tat | Envelope protein GP64 or capsid protein VP39 | Viral genome with Luc or eGFP gene | Vero E6, U2OS, CHO-RD |
| [59] |
| Adenovirus | Tat | Fiber knob protein | Viral genome with GFP gene | RD, D65MG, U118MG, HeLa, A549 |
| [55] |
| Adenovirus | Tat | Fiber knob protein—HI loop or C-terminus | Viral genome with Luc gene | U937, Jurkat, CSMC, ASMC, LN444, SF295, SK HEP-1 |
| [56] |
| Adenovirus | Tat | Fiber knob protein—HI loop | Viral genome with eGFP gene | A549, CHO, CHO-CAR, T24, NIH-3T3, C39, HUVEC |
| [57] |
| Adenovirus | Tat | Hexon protein—hypervariable region 5 | Viral genome with GFP gene or complete oncolytic virus for in vivo assay | BON, CNDT2.5, SKOV-3, A549, MB49, 911, 1064SK, mel526, SK-N-SH, HUVEC |
| [58] |
| MS2 bacteriophage-derived VLPs | Tat | Tat incorporated via a linker at the N-terminus of coat protein | Pre-microRNA-122 | Hep3B, Huh7, HeLa, HepG2, Huh7 |
| [62] |
| Phage lambda | Longer version of Tat | D protein—N-terminus | Viral genome with eGFP or Luc gene | COS-1, VA13/2RA, HEK-293, NIH-3T3, HeLa, A431 |
| [63] |
| Recombinant bacteriophage PP7-derived VLPs | Tat | Coat protein | Pre-microRNA-23b | SK-HEP-1, COS-7 |
| [60] |
| Recombinant bacteriophage PP7-derived VLPs | LMWP | Coat protein | mRNA encoding GFP protein | RM-1 |
| [61] |
| Virus | CPP | Cargo | Cell Lines Tested | Effect | Ref. |
|---|---|---|---|---|---|
| Adenovirus | Tat, R8 | Viral genome with GFP or Luc gene | A549, HeLa, U937, B16BL6, CT26, RAW264.7, EL4, LN444, LNZ308, SF295 |
| [65] |
| Adenovirus | Tat, R8, Pro | Viral genome with Luc gene | A549, CT26, B16BL6 |
| [66] |
| Adenovirus | Pen, Tat, R9, Pep1 | Viral genome with LacZ gene | NIH-3T3 |
| [67] |
| Cowpea mosaic virus-derived VPs | R5 | No | HeLa |
| [68] |
| Hepatitis B VPs | NRPDSAQFWLHH | No | A431 |
| [69] |
| MS2 bacteriophage-derived VPs | Tat | Pre-microRNA-146a | HeLa, HepG2, Huh-7, PBMC |
| [70] |
| MS2 bacteriophage-derived VPs | Tat | Antisense RNA against hepatitis C virus regulatory regions | Huh-7 |
| [71] |
| P22 bacteriophage-derived VPs | Tat | Ziconotide peptide | RBMVEC |
| [72] |
| Qβ bacteriophage-derived targeted VPs | KYGRRRQRRKKRG | Epirubicin, GFP | GBM U87-MG |
| [73] |
| Turnip yellow mosaic virus | Tat, R8, Pep-1, Pen | Fluorescein dye conjugated to the interior of the capsid | BHK |
| [74] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Váňová, J.; Hejtmánková, A.; Kalbáčová, M.H.; Španielová, H. The Utilization of Cell-Penetrating Peptides in the Intracellular Delivery of Viral Nanoparticles. Materials 2019, 12, 2671. https://doi.org/10.3390/ma12172671
Váňová J, Hejtmánková A, Kalbáčová MH, Španielová H. The Utilization of Cell-Penetrating Peptides in the Intracellular Delivery of Viral Nanoparticles. Materials. 2019; 12(17):2671. https://doi.org/10.3390/ma12172671
Chicago/Turabian StyleVáňová, Jana, Alžběta Hejtmánková, Marie Hubálek Kalbáčová, and Hana Španielová. 2019. "The Utilization of Cell-Penetrating Peptides in the Intracellular Delivery of Viral Nanoparticles" Materials 12, no. 17: 2671. https://doi.org/10.3390/ma12172671
APA StyleVáňová, J., Hejtmánková, A., Kalbáčová, M. H., & Španielová, H. (2019). The Utilization of Cell-Penetrating Peptides in the Intracellular Delivery of Viral Nanoparticles. Materials, 12(17), 2671. https://doi.org/10.3390/ma12172671