Ab Initio Screening of Doped Mg(AlH4)2 Systems for Conversion-Type Lithium Storage

 ,
,  ,
, 
Abstract
1. Introduction
2. Methods
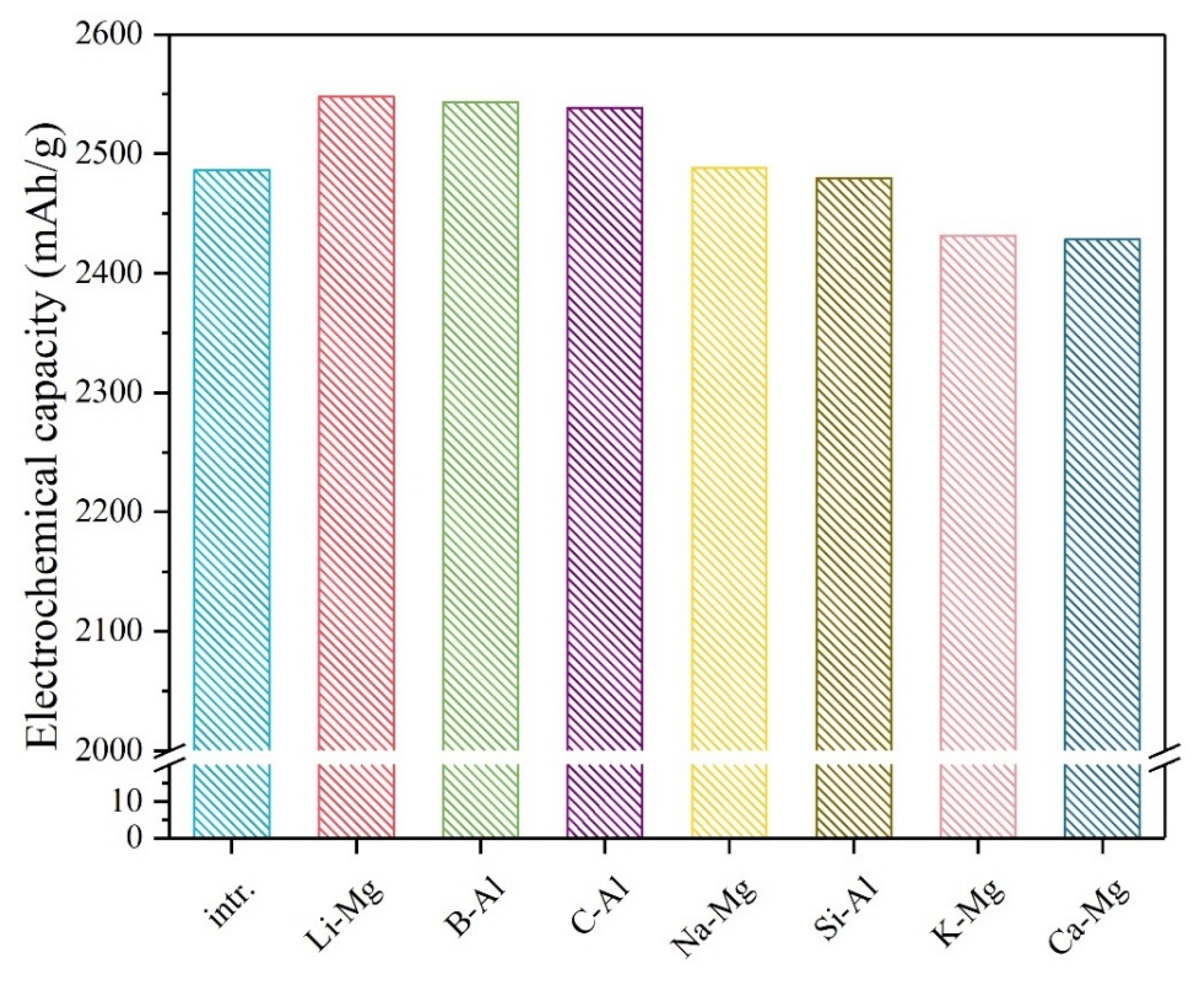
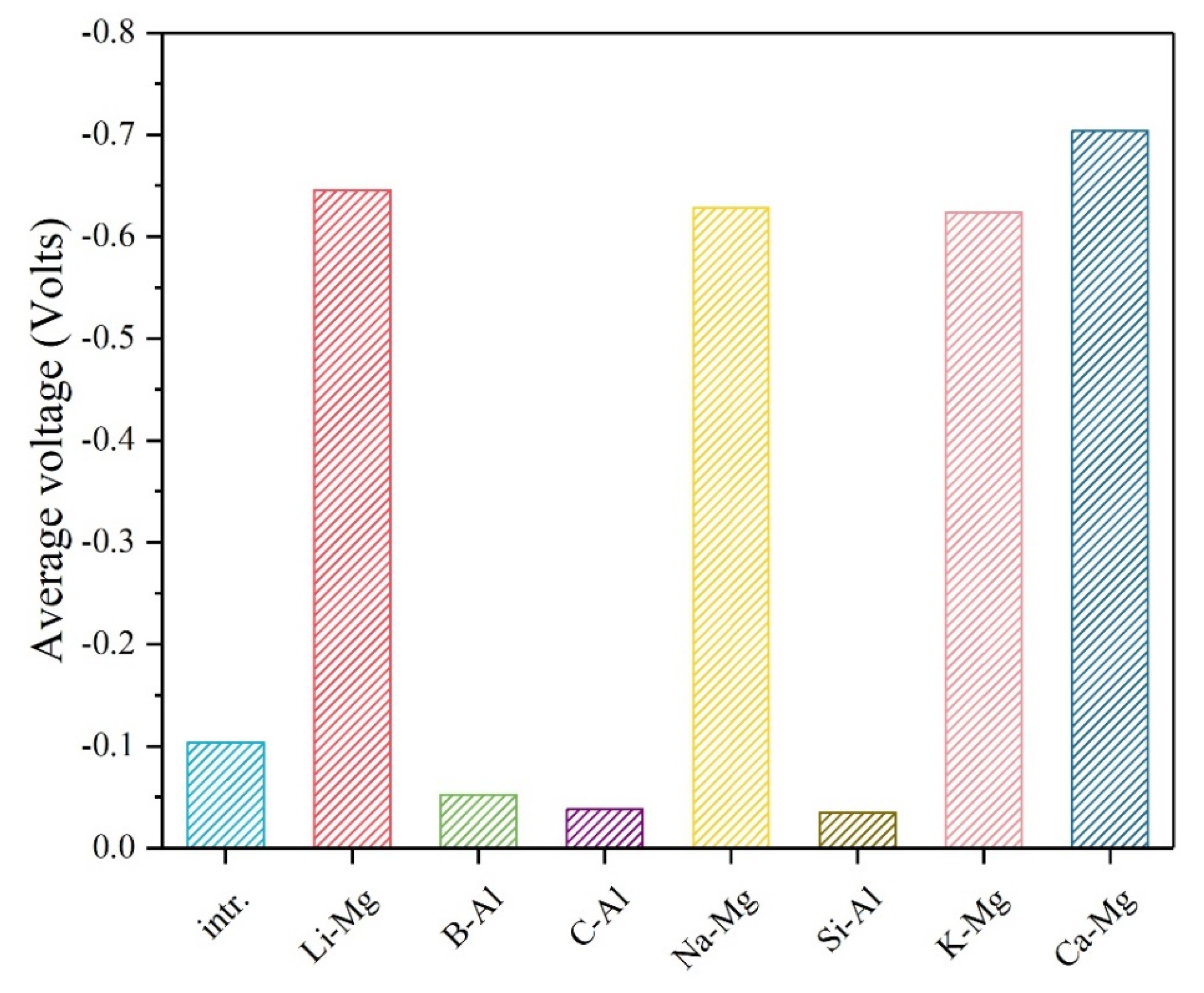
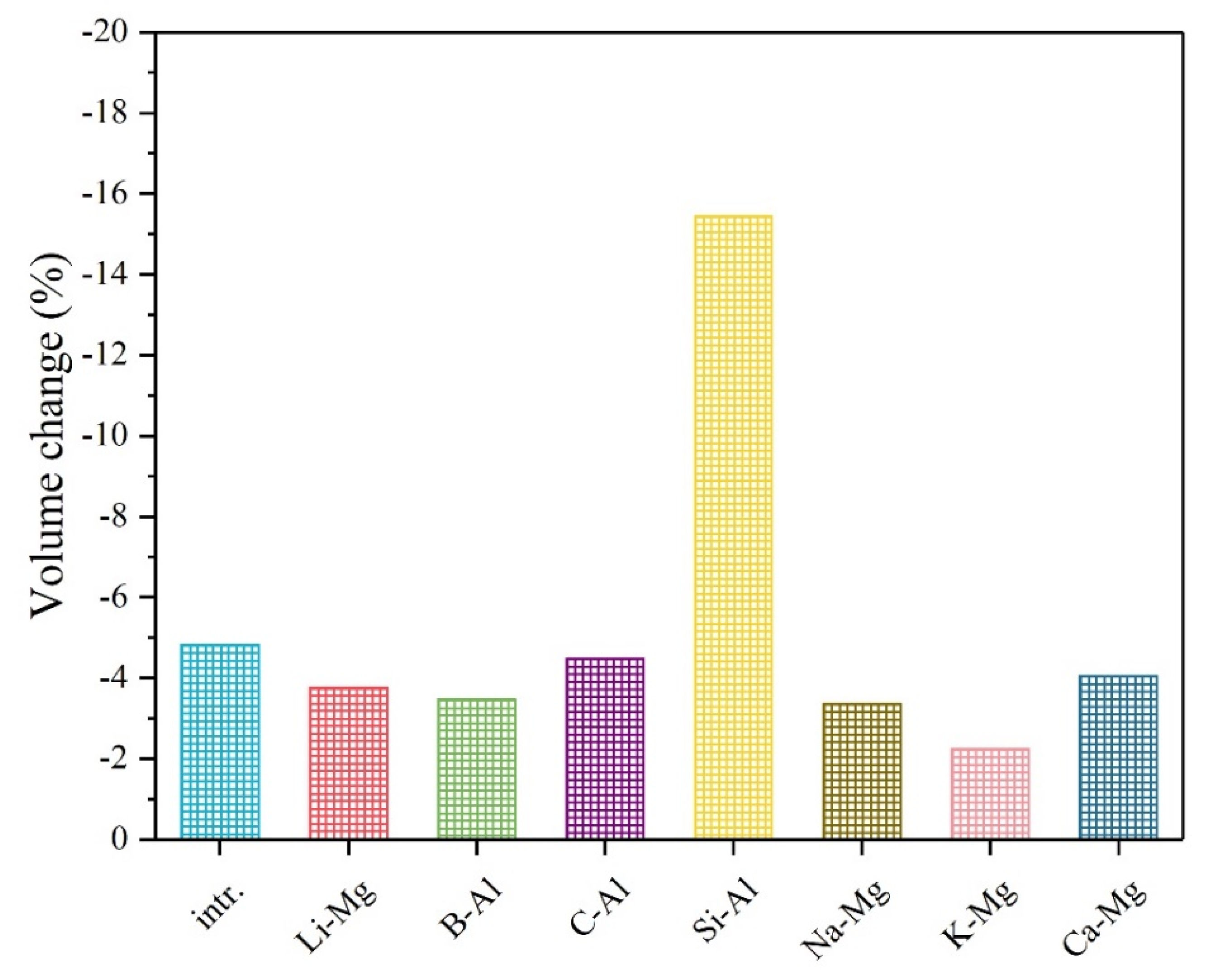
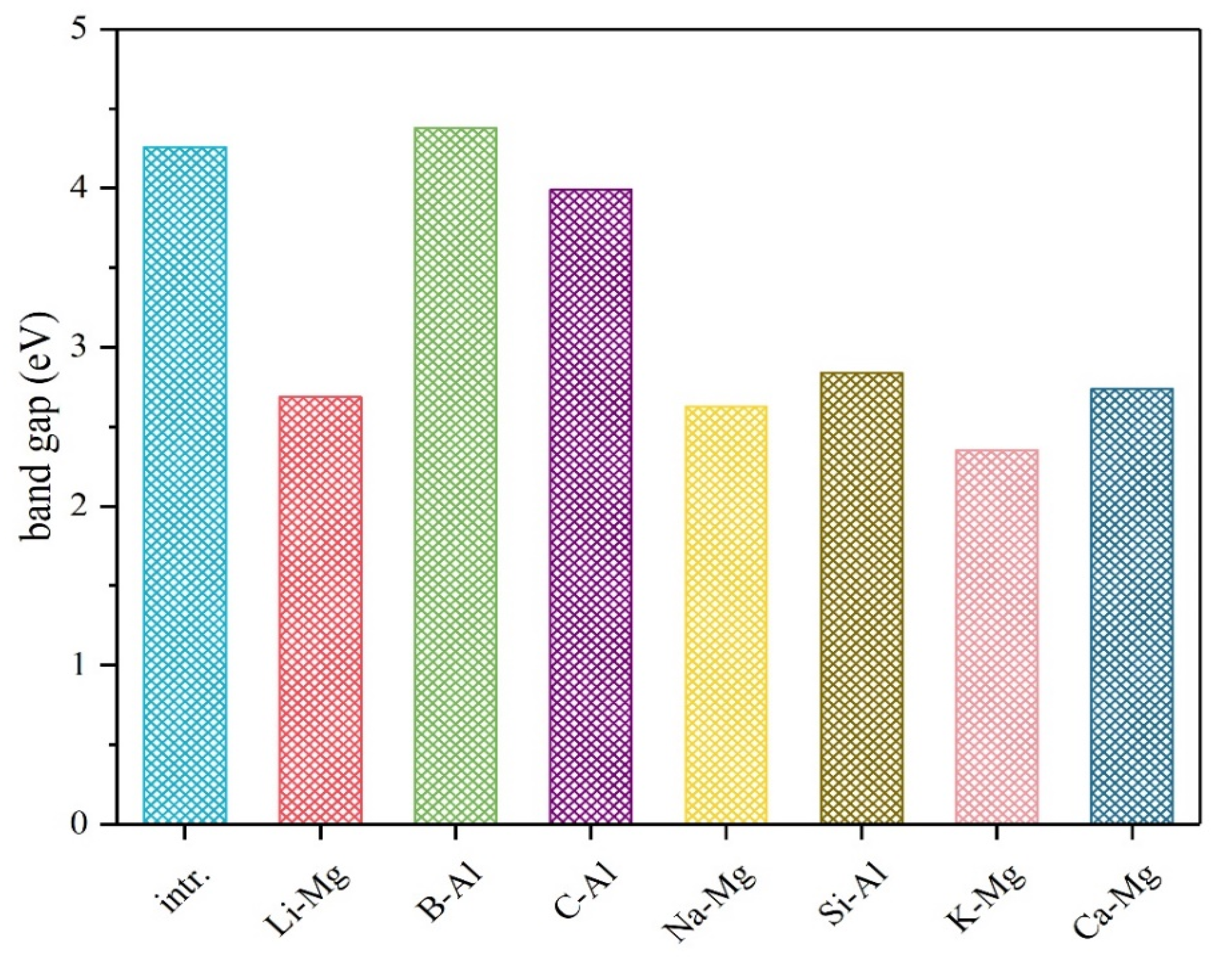
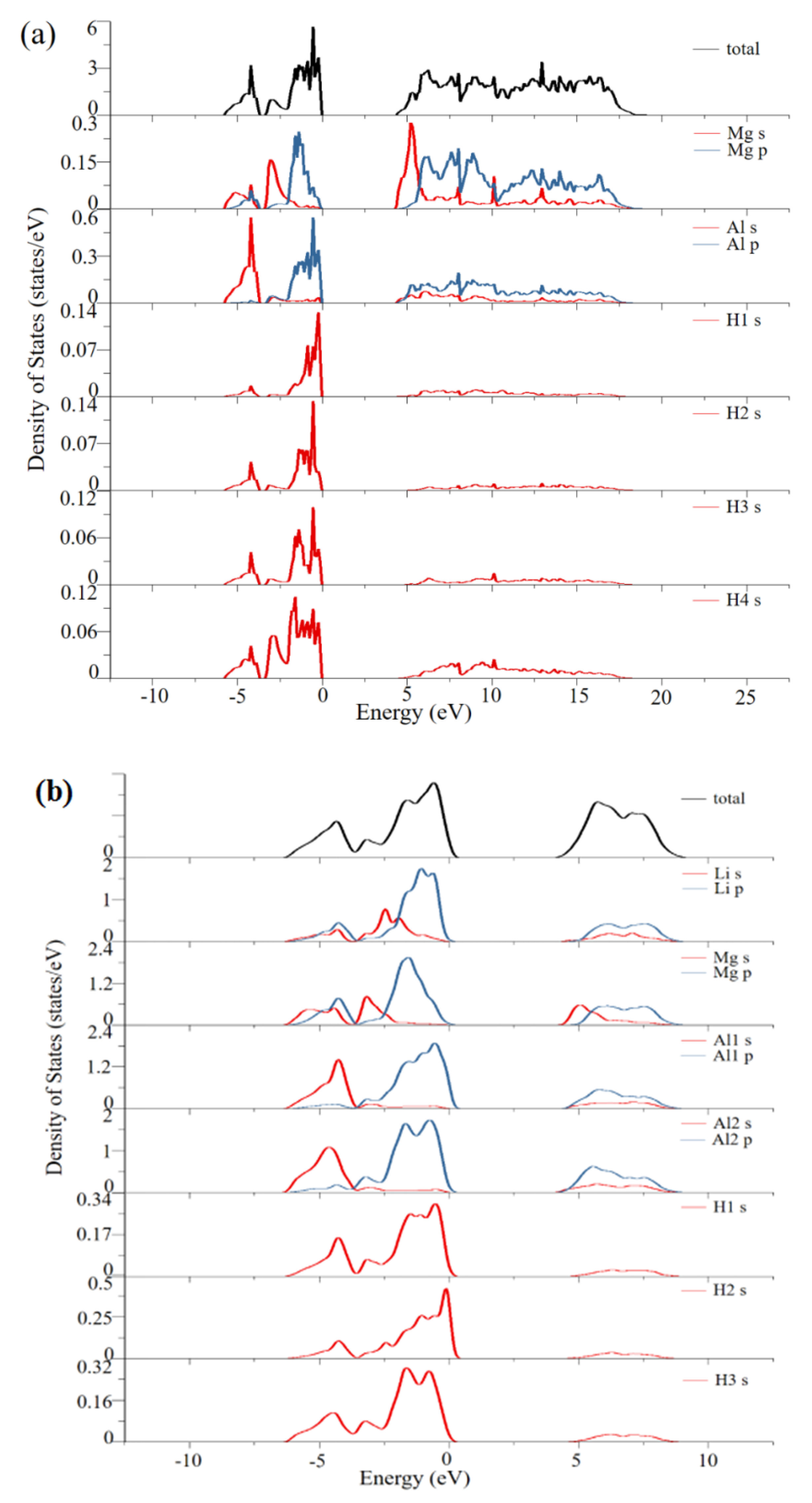
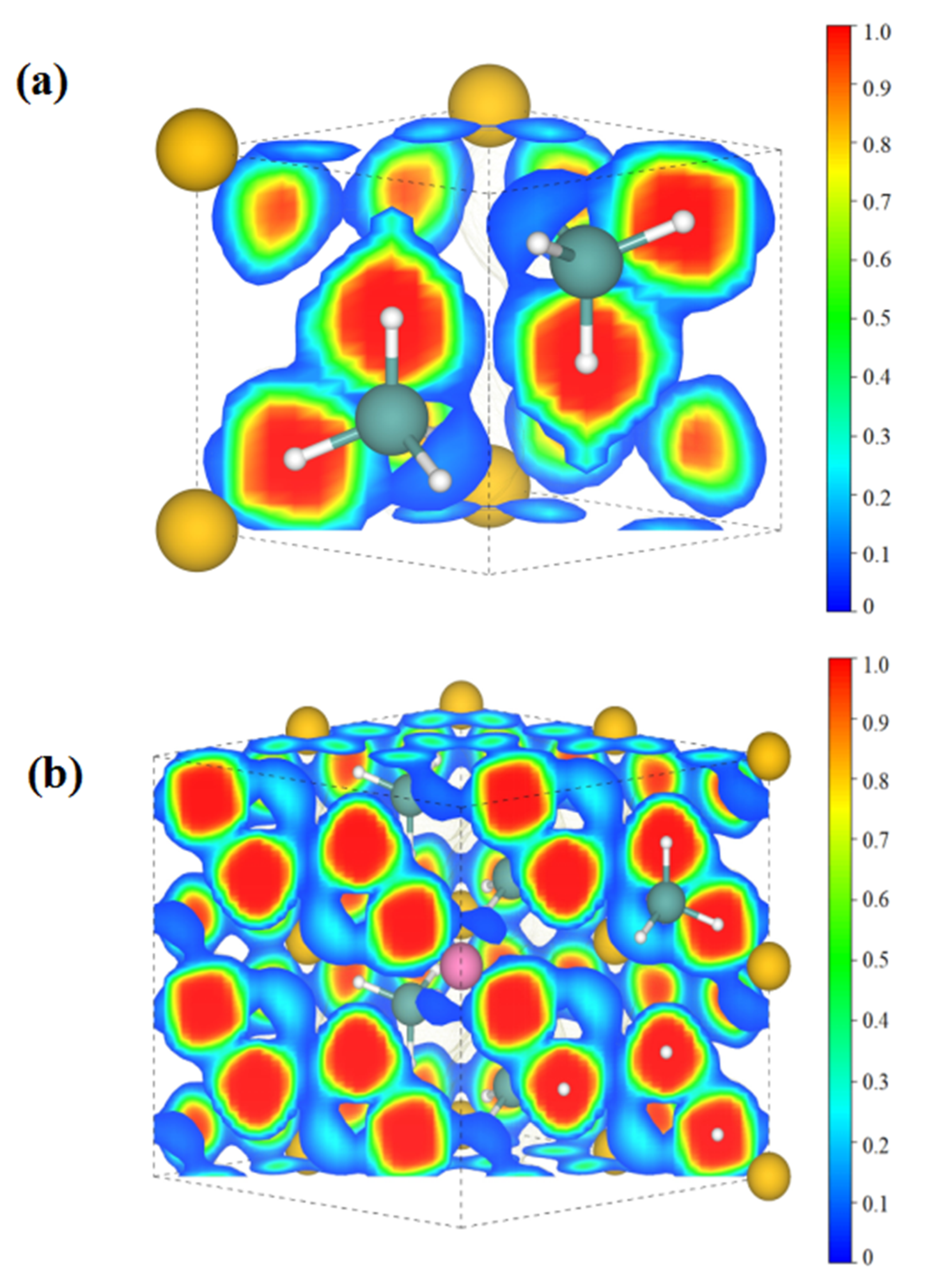
3. Results and Discussion
4. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armand, M.; Tarascon, J.-M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Wu, F.X.; Lee, J.; Yushin, T.G. Li-ion battery materials: present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Yoon, G.; Kim, D.; Prak, I.; Chang, D.; Kim, B.; Lee, B.; Oh, K.; Kang, K. Using First-principles calculations for the advancement of materials for rechargeable batteries. Adv. Funct. Mater. 2017, 27, 1702887. [Google Scholar] [CrossRef]
- Oumellal, Y.; Rougier, A.; Nazri, G.A.; Tarascon, J.-M.; Aymard, L. Metal hydrides for lithium-ion batteries. Nat. Mater. 2008, 7, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Aymard, L.; Oumellal, Y.; Bonnet, J.-P. Metal hydrides: An innovative and challenging conversion reaction anode for lithium-ion batteries. Beilstein J. Nanotechnol. 2015, 6, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Hu, Z.; Ma, J.; Ma, H.; Dai, L.; Zhang, J. A rechargeable iodine-carbon battery that exploits ion intercalation and iodine redox chemistry. Nat. Commun. 2017, 8, 527. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Sarkar, A.D.; Maark, T.A.; Jiang, X.; Deshpande, M.D.; Bououdina, M.; Ahuja, R. Pure and Li-doped NiTiH: potential anode materials for Li-ion rechargeable batteries. Appl. Phys. Lett. 2013, 103, 033902. [Google Scholar] [CrossRef]
- Qian, Z.; Jiang, X.; Sarkar, A.D.; Maark, T.A.; Deshpande, M.D.; Bououdina, M.; Johansson, B.; Ahuja, R. Screening study of light-metal and transition-metal-doped NiTiH hydrides as Li-ion battery anode materials. Solid State Ion. 2014, 258, 88–91. [Google Scholar] [CrossRef]
- Sartori, S.; Cuevas, F.; Latroche, M. Metal hydrides used as negative electrode materials for Li-ion batteries. Appl. Phys. A 2016, 122, 135. [Google Scholar] [CrossRef]
- Hou, Z.F. First-principles investigation of Mg(AlH4)2 complex hydride. J. Power Sources 2006, 159, 111–115. [Google Scholar] [CrossRef]
- Krehl, M.; Schulze, K.; Petzow, G. The influence of gas atmospheres on the first-stage sintering of high-purity niobium powders. Phys. Chem. Chem. Phys. 1984, 15, 1111–1116. [Google Scholar] [CrossRef]
- Deligoz, E.; Colakoglu, K.; Ozisik, H.; Cifti, Y.O. The first principles investigation of lattice dynamical and thermodynamical properties of Al2Ca and Al2Mg compounds in the cubic Laves structure. Comput. Mater. Sci. 2013, 68, 27–31. [Google Scholar] [CrossRef]
- Qian, Z.; Raghubanshi, H.; Hudson, M.S.L.; Srivastava, O.N.; Liu, X.; Ahuja, R. Ab initio insight into graphene nanofibers to destabilize hydrazineborane for hydrogen release. Chem. Phys. Lett. 2017, 669, 110–114. [Google Scholar] [CrossRef]
- Van Setten, M.J.; De Wijs, G.A.; Popa, V.A.; Brocks, G. Ab initio study of Mg(AlH4)2. Phys. Rev. B 2005, 72, 073107. [Google Scholar] [CrossRef]
- Silvestri, L.; Farina, L.; Meggiolaro, D.; Panero, S.; Padella, F. Reactivity of sodium alanates in lithium batteries. J. Phys. Chem. C 2015, 119, 28766–28775. [Google Scholar] [CrossRef]
- Shi, B.; Song, Y.; Dai, J.H.; Yu, H.Z. Influence of Ti and Al dopants on the dehydrogenation characteristics of Mg(BH4)2: electronic structure mechanisms. J. Phys. Chem. C 2012, 116, 12001–12007. [Google Scholar] [CrossRef]
- Zhao, T.S.; Wang, Q.; Jena, P. Cluster-inspired design of high-capacity anode for Li-ion batteries. ACS Energy Lett. 2016, 1, 202–208. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, L.Y.; Yang, F.S.; Jiang, Z.; Zhang, Z.X. Influences of interstitial nitrogen with high electronegativity on structure and hydrogen storage properties of Mg-based metal hydride: A theoretical study. Int. J. Hydrog. Energy 2016, 41, 18550–18561. [Google Scholar] [CrossRef]
- Yvon, K.; Schefer, J.; Stucki, F. Structural studies of the hydrogen storage material Mg2NiH4. Inorg. Chem. 1981, 20, 2776–2778. [Google Scholar] [CrossRef]
- Mitov, M.; Chorbadzhiyska, E.; Nalbandian, L.; Hubenova, Y. Nickel-based electrodeposits as potential cathode catalysts for hydrogen production by microbial electrolysis. J. Power Sources 2017, 356, 467–472. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Vidya, R.; Fjellvåg, H.; Kjekshus, A. Huge-pressure-induced volume collapse in LiAlH4 and its implications to hydrogen storage. Phys. Rev. B 2005, 68, 212101. [Google Scholar] [CrossRef]
- Løvvik, O.M.; Opalka, S.M.; Brinks, H.W.; Hauback, B.C. Crystal structure and thermodynamic stability of the lithium alanates LiAlH4 and Li3AlH6. Phys. Rev. B 2004, 69, 134117. [Google Scholar] [CrossRef]
- Sai-Cheong, C.; Hiroyuki, M. Thermochemistry and crystal structures of lithium, sodium and potassium alanates as determined by ab initio simulations. J. Alloy. Compd. 2004, 372, 92–96. [Google Scholar] [CrossRef]
- Keyzer, E.N.; Lee, J.; Liu, Z.G.; Bond, A.D. A general synthetic methodology to access magnesium aluminate electrolyte systems for Mg batteries. J. Mater. Chem. A 2019, 7, 6. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Neuhauser, D.; Baer, R.; Rabani, E. Communication: Embedded fragment stochastic density functional theory. J. Chem. Phys. 2014, 141, 041102. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.; Ernzerhof, M.; Perdew, J.P. The adiabatic connection method: a non-empirical hybrid. Chem. Phys. Lett. 1997, 265, 115–120. [Google Scholar]
- Fossdala, A.; Brinksa, H.W.; Fichtnerb, M.; Hauback, B.C. Determination of the crystal structure of Mg(AlH4)2 by combined X-ray and neutron diffraction. J. Alloy. Compd. 2005, 387, 47–51. [Google Scholar] [CrossRef]
- Nisar, J.; Pathak, B.; Ahuja, R. Screened hybrid density functional study on Sr2Nb2O7 for visible light photocatalysis. Appl. Phys. Lett. 2012, 100, 181903. [Google Scholar] [CrossRef]
- Dawson, J.A.; Naylor, A.J.; Eames, C.; Roberts, M.; Zhang, W.; Snaith, H.J.; Bruce, P.G.; Islam, M.S. Mechanisms of Lithium intercalation and conversion processes in organic−inorganic halide perovskites. ACS Energy Lett. 2017, 2, 1818–1824. [Google Scholar] [CrossRef]
- Jiang, G.Z.; Qian, Z.; Bououdina, M.; Ahuja, R.; Liu, X.F. Exploring pristine and Li-doped Mg2NiH4 compounds with potential lithium-storage properties: Ab initio insight. J. Alloy. Compd. 2018, 746, 140–146. [Google Scholar] [CrossRef]
- Qian, Z.; Jiang, G.Z.; Ren, Y.Y.; Nie, X.; Ahuja, R. Atomistic modeling of various doped Mg2NiH4 as conversion electrode materials for lithium storage. Crystals 2019, 9, 254. [Google Scholar] [CrossRef]
- Løvvik, O.M.; Molin, P.N. Density-functional band-structure calculations of magnesium alanate Mg(AlH4)2. Phys. Rev. B 2005, 72, 073201. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Doping Element | Mg Site | Al Site |
|---|---|---|
| Li | 0.002 | 2.439 |
| B | −0.410 | −0.757 |
| C | −0.430 | −0.870 |
| Na | 0.140 | 3.984 |
| Si | −0.301 | −0.895 |
| K | 0.177 | 4.032 |
| Ca | −0.461 | 0.720 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Z.; Zhang, H.; Jiang, G.; Bai, Y.; Ren, Y.; Du, W.; Ahuja, R. Ab Initio Screening of Doped Mg(AlH4)2 Systems for Conversion-Type Lithium Storage. Materials 2019, 12, 2599. https://doi.org/10.3390/ma12162599
Qian Z, Zhang H, Jiang G, Bai Y, Ren Y, Du W, Ahuja R. Ab Initio Screening of Doped Mg(AlH4)2 Systems for Conversion-Type Lithium Storage. Materials. 2019; 12(16):2599. https://doi.org/10.3390/ma12162599
Chicago/Turabian StyleQian, Zhao, Hongni Zhang, Guanzhong Jiang, Yanwen Bai, Yingying Ren, Wenzheng Du, and Rajeev Ahuja. 2019. "Ab Initio Screening of Doped Mg(AlH4)2 Systems for Conversion-Type Lithium Storage" Materials 12, no. 16: 2599. https://doi.org/10.3390/ma12162599
APA StyleQian, Z., Zhang, H., Jiang, G., Bai, Y., Ren, Y., Du, W., & Ahuja, R. (2019). Ab Initio Screening of Doped Mg(AlH4)2 Systems for Conversion-Type Lithium Storage. Materials, 12(16), 2599. https://doi.org/10.3390/ma12162599