Acid Mesoporous Carbon Monoliths from Lignocellulosic Biomass Waste for Methanol Dehydration


 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Method
2.1. ACMs Preparation
2.2. ACM Characterization
2.3. Methanol Dehydration
3. Results and Discussion
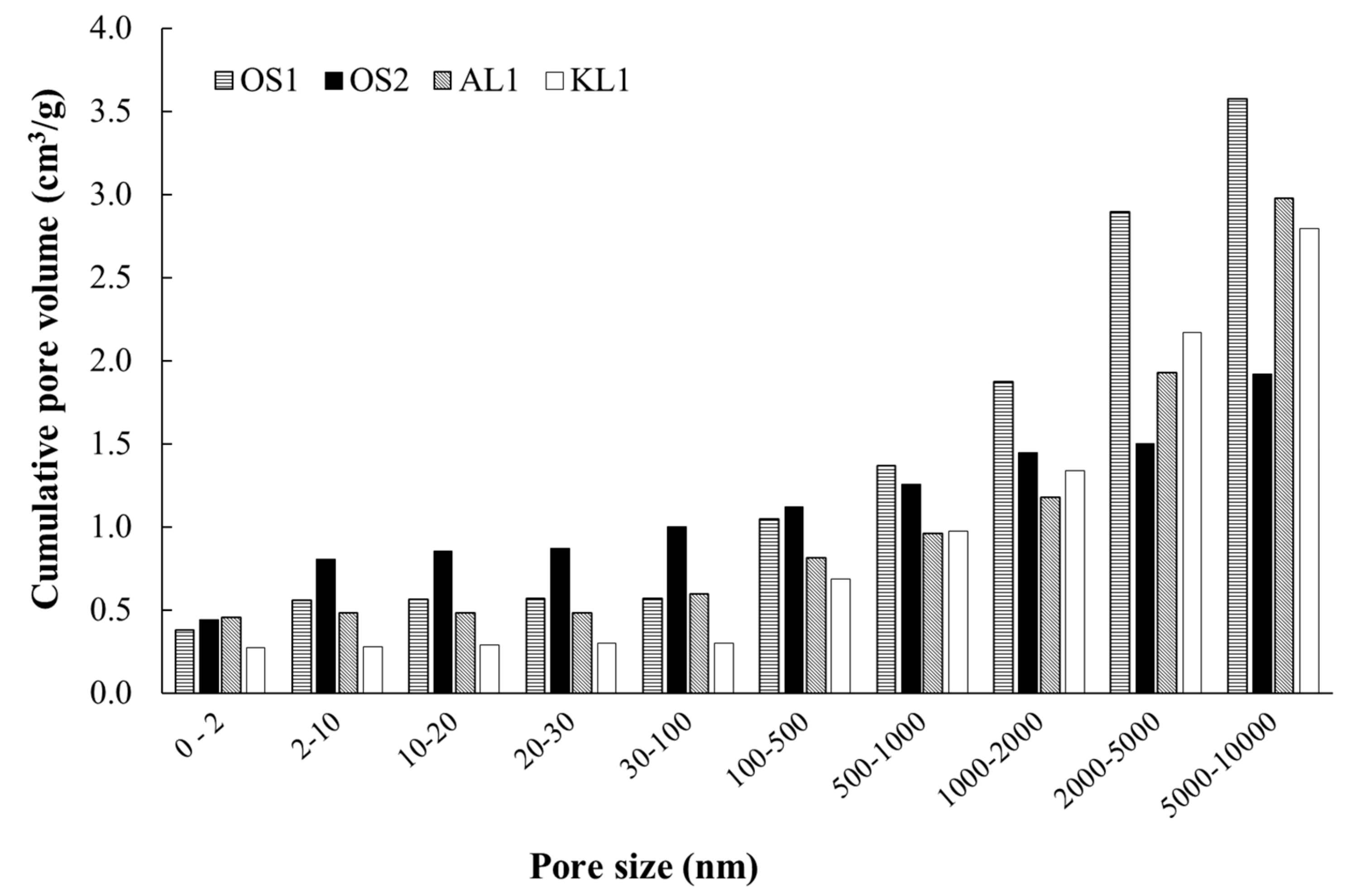
3.1. ACMs Characterization
3.2. Methanol Decomposition
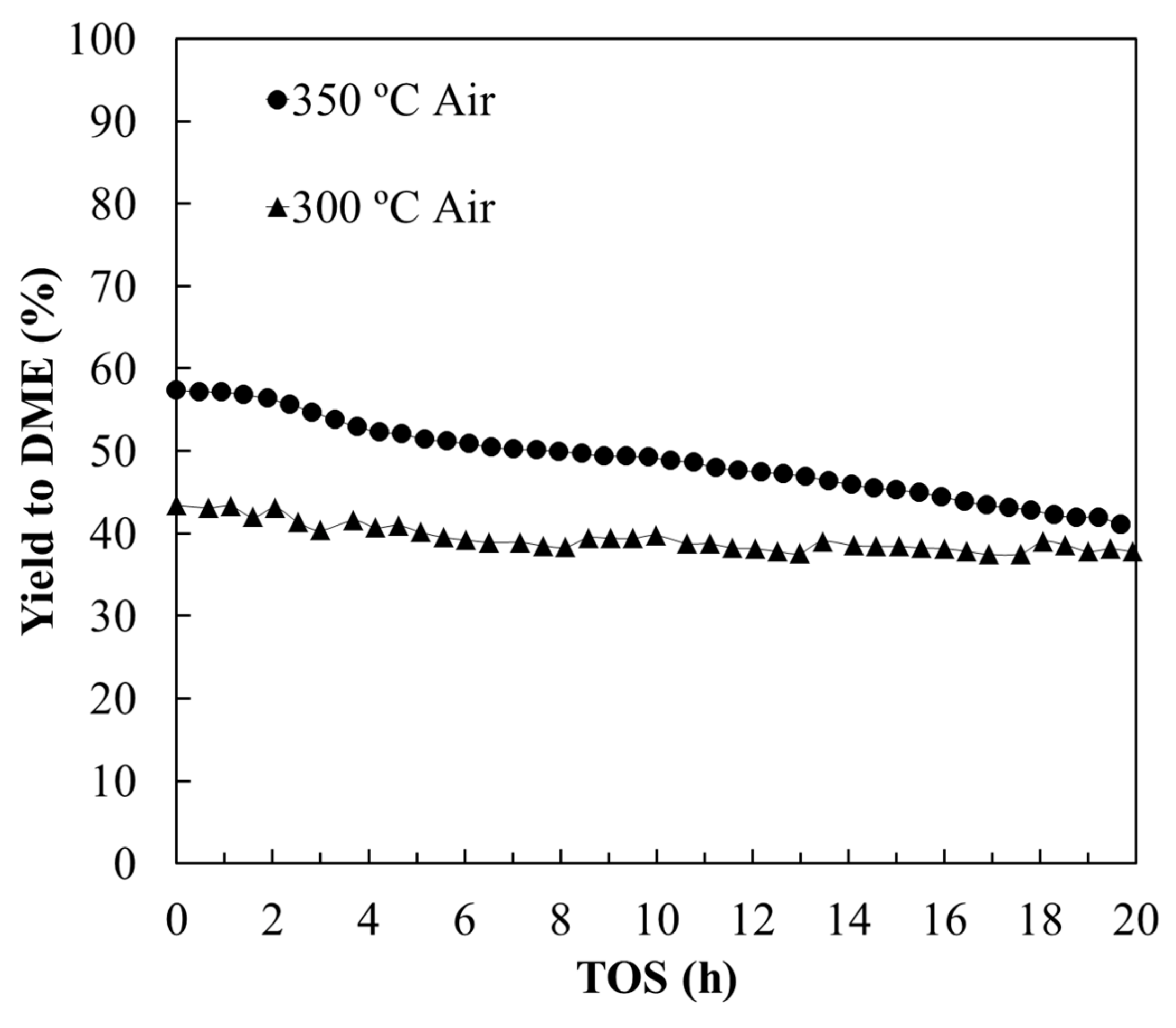
3.2.1. Stability Study
3.2.2. Influence of Water Vapor in the Reaction Mixture
3.3. Kinetic Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pérez-Uriarte, P.; Ateka, A.; Gayubo, A.G.; Cordero-Lanzac, T.; Aguayo, A.T.; Bilbao, J. Deactivation kinetics for the conversion of dimethyl ether to olefins over a HZSM-5 zeolite catalyst. Chem. Eng. J. 2017, 311, 367–377. [Google Scholar] [CrossRef]
- Aguayo, T.; Gayubo, A.G.; Vivanco, R.; Olazar, M.; Bilbao, J. Role of acidity and microporous structure in alternative catalysts for the transformation of methanol into olefins. Appl. Catal. A Gen. 2005, 283, 197–207. [Google Scholar] [CrossRef]
- Rodríguez-reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Zawadzki, J.; Azambre, B.; Heintz, O.; Krztoń, A.; Weber, J. IR study of the adsorption and decomposition of methanol on carbon surfaces and carbon-supported catalysts. Carbon 2000, 38, 509–515. [Google Scholar] [CrossRef]
- Jasiñska, J.; Krzyžyñska, B.; Kozłowski, M. Influence of activated carbon modifications on their catalytic activity in methanol and ethanol conversion reactions. Cent. Eur. J. Chem. 2011, 9, 925–931. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Parejo-Pérez, C.; López Ramón, M. Dehydration of methanol to dimethyl ether catalyzed by oxidized activated carbons with varying surface acidic character. Carbon 2001, 39, 869–875. [Google Scholar] [CrossRef]
- Rosas, J.M.; Bedia, J.; Rodríguez-Mirasol, J.; Cordero, T. Preparation of hemp-derived activated carbon monoliths. Adsorption of water vapor. Ind. Eng. Chem. Res. 2008, 47, 1288–1296. [Google Scholar] [CrossRef]
- Bedia, J.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Pd supported on mesoporous activated carbons with high oxidation resistance as catalysts for toluene oxidation. Appl. Catal. B Environ. 2010, 94, 8–18. [Google Scholar] [CrossRef]
- Rosas, J.M.; Bedia, J.; Rodríguez-Mirasol, J.; Cordero, T. HEMP-derived activated carbon fibers by chemical activation with phosphoric acid. Fuel 2009, 88, 19–26. [Google Scholar] [CrossRef]
- Rosas, J.M.; Ruiz-Rosas, R.; Rodríguez-Mirasol, J.; Cordero, T. Kinetic study of the oxidation resistance of phosphorus-containing activated carbons. Carbon 2012, 50, 1523–1537. [Google Scholar] [CrossRef]
- Bedia, J.; Rosas, J.M.; Márquez, J.; Rodríguez-Mirasol, J.; Cordero, T. Preparation and characterization of carbon based acid catalysts for the dehydration of 2-propanol. Carbon 2009, 47, 286–294. [Google Scholar] [CrossRef]
- Valero-Romero, M.J.; Calvo-Muñoz, E.M.; Ruiz-Rosas, R.; Rodríguez-Mirasol, J.; Cordero, T. Phosphorus-Containing Mesoporous Carbon Acid Catalyst for Methanol Dehydration to Dimethyl Ether. Ind. Eng. Chem. Res. 2019, 58, 4042–4053. [Google Scholar] [CrossRef]
- Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. NO reduction on carbon-supported chromium catalysts. Energy Fuel. 2010, 24, 3321–3328. [Google Scholar] [CrossRef]
- Guerrero-Pérez, M.O.; Rosas, J.M.; López-Medina, R.; Bañares, M.A.; Rodríguez-Mirasol, J.; Cordero, T. Lignocellulosic-derived catalysts for the selective oxidation of propane. Catal. Commun. 2011, 12, 989–992. [Google Scholar] [CrossRef]
- Bedia, J.; Ruiz-Rosas, R.; Rodríguez-Mirasol, J.; Cordero, T. A kinetic study of 2-propanol dehydration on carbon acid catalysts. J. Catal. 2010, 271, 33–42. [Google Scholar] [CrossRef]
- Bedia, J.; Barrionuevo, R.; Rodríguez-Mirasol, J.; Cordero, T. Ethanol dehydration to ethylene on acid carbon catalysts. Appl. Catal. B Environ. 2011, 103, 302–310. [Google Scholar] [CrossRef]
- Bedia, J.; Ruiz-Rosas, R.; Rodríguez-Mirasol, J.; Cordero, T. Kinetic Study of the Decomposition of 2-Butanol on Carbon-Based Acid Catalyst. AICHE J. 2010, 56, 1557–1568. [Google Scholar] [CrossRef]
- Govender, S.; Friedrich, H. Monoliths: A Review of the Basics, Preparation Methods and Their Relevance to Oxidation. Catalysts 2017, 7, 62. [Google Scholar] [CrossRef]
- Valero-Romero, M.J.; Márquez-Franco, E.M.; Bedia, J.; Rodríguez-Mirasol, J.; Cordero, T. Hierarchical porous carbons by liquid phase impregnation of zeolite templates with lignin solution. Microporous Microporous Mater. 2014, 196, 68–78. [Google Scholar] [CrossRef]
- Vargas, D.P.; Giraldo, L.; Moreno-piraján, J.C. CO2 Adsorption on Activated Carbon Honeycomb-Monoliths: A Comparison of Langmuir and Tóth Models. Int. J. Mol. Sci. 2012, 13, 8388–8397. [Google Scholar] [CrossRef]
- Giraldo, L.; Bastidas-Barranco, M.; Moreno-Pirajan, J.C. Preparation of carbon monoliths from orange peel for NOx retention. Orient. J. Chem. 2014, 30, 1517–1528. [Google Scholar] [CrossRef][Green Version]
- Ibeh, P.O.; García-Mateos, F.J.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Activated carbon monoliths from lignocellulosic biomass waste for electrochemical applications. J. Taiwan Inst. Chem. Eng. 2019, 97, 480–488. [Google Scholar] [CrossRef]
- Molina-Sabio, M.; RodRíguez-Reinoso, F.; Caturla, F.; Sellés, M.J. Porosity in granular carbons activated with phosphoric acid. Carbon 1995, 33, 1105–1113. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Molina-Sabio, M.; Rodríguez-Reinoso, F. Modification of the porous structure along the preparation of activated carbon monoliths with H3PO4and ZnCl2. Microporous Microporous Mater. 2007, 103, 29–34. [Google Scholar] [CrossRef]
- Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A.; Quinn, D.F. Activated carbon monoliths for methane storage: Influence of binder. Carbon 2002, 40, 2817–2825. [Google Scholar] [CrossRef]
- Lim, J.W.; Choi, Y.; Yoon, H.S.; Park, Y.K.; Yim, J.H.; Jeon, J.K. Extrusion of honeycomb monoliths employed with activated carbon-LDPE hybrid materials. J. Ind. Eng. Chem. 2010, 16, 51–56. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Z.; Yang, J.; Huang, Z.; Liu, Z. Effect of preparation conditions on the properties of a coal-derived activated carbon honeycomb monolith. Carbon 2007, 45, 2836–2842. [Google Scholar] [CrossRef]
- Machnikowski, J.; Kierzek, K.; Lis, K.; Machnikowska, H.; Czepirski, L. Tailoring Porosity Development in Monolithic Adsorbents Made of KOH-Activated Pitch Coke and Furfuryl Alcohol Binder for Methane Storage. Energy Fuels 2010, 24, 3410–3414. [Google Scholar] [CrossRef]
- Jordá-Beneyto, M.; Lozano-Castelló, D.; Suárez-García, F.; Cazorla-Amorós, D.; Linares-Solano, Á. Advanced activated carbon monoliths and activated carbons for hydrogen storage. Microporous Microporous Mater. 2008, 112, 235–242. [Google Scholar] [CrossRef]
- Paola Vargas, D.; Giraldo, L.; Silvestre-Albero, J.; Moreno-Piraján, J.C. CO2 adsorption on binderless activated carbon monoliths. Adsorption 2011, 17, 497–504. [Google Scholar] [CrossRef]
- Vargas, D.D.P.; Giraldo, G.L.; Moreno, P.J.C. Enthalpic characterisation of activated carbon monoliths obtained from lignocellulosic materials. J. Therm. Anal. Calorim. 2013, 111, 1067–1072. [Google Scholar] [CrossRef]
- García-Blanco, A.A.; De Oliveira, J.C.A.; López, R.; Moreno-piraján, J.C.; Giraldo, L.; Zgrablich, G.; Sapag, K. A study of the pore size distribution for activated carbon monoliths and their relationship with the storage of methane and hydrogen. Colloids Surf. A Physicochem. Eng. Asp. 2010, 357, 74–83. [Google Scholar] [CrossRef]
- Djeridi, W.; Ouederni, A.; Wiersum, A.D.; Llewellyn, P.L.; Mir, L. El High pressure methane adsorption on microporous carbon monoliths prepared by olives stones. Mater. Lett. 2013, 99, 184–187. [Google Scholar] [CrossRef]
- Taer, E.; Deraman, M.; Taslim, R.; Iwantono. Preparation of Binderless Activated Carbon Monolith from Pre-Carbonization Rubber Wood Sawdust by Controlling of Carbonization and Activation Condition. AIP Conf. Proc. 2013, 1554, 33–37. [Google Scholar]
- Ubago-Pérez, R.; Carrasco-Marín, F.; Fairén-Jiménez, D.; Moreno-Castilla, C. Granular and monolithic activated carbons from KOH-activation of olive stones. Microporous Microporous Mater. 2006, 92, 64–70. [Google Scholar] [CrossRef]
- Bedia, J.; Rosas, J.M.; Vera, D.; Rodríguez-Mirasol, J.; Cordero, T. Isopropanol decomposition on carbon based acid and basic catalysts. Catal. Today 2010, 158, 89–96. [Google Scholar] [CrossRef]
- Carrasco-Marín, F.; Mueden, A.; Moreno-Castilla, C. Surface-Treated Activated Carbons as Catalysts for the Dehydration and Dehydrogenation Reactions of Ethanol. J. Phys. Chem. B 1998, 102, 9239–9244. [Google Scholar] [CrossRef]
- Turek, W.; Haber, J.; Krowiak, A. Dehydration of isopropyl alcohol used as an indicator of the type and strength of catalyst acid centres. Appl. Surf. Sci. 2005, 252, 823–827. [Google Scholar] [CrossRef]
- Trens, P.; Stathopoulos, V.; Hudson, M.J.; Pomonis, P. Synthesis and characterization of packed mesoporous tungsteno-silicates: application to the catalytic dehydrogenation of 2-propanol. Appl. Catal. A Gen. 2004, 263, 103–108. [Google Scholar] [CrossRef]
- Valero-Romero, M.J.; García-Mateos, F.J.; Rodríguez-Mirasol, J.; Cordero, T. Role of surface phosphorus complexes on the oxidation of porous carbons. Fuel Process. Technol. 2017, 157, 116–126. [Google Scholar] [CrossRef]
- Vishwanathan, V.; Roh, H.; Kim, J.; Jun, K. Surface properties and catalytic activity of TiO2–ZrO2 mixed oxides in dehydration of methanol to dimethyl ether. Catal. Lett. 2004, 96, 23–28. [Google Scholar] [CrossRef]
- Mollavali, M.; Yaripour, F.; Atashi, H.; Sahebdelfar, S. Intrinsic Kinetics Study of Dimethyl Ether Synthesis from Methanol on γ-Al2O3 Catalysts. Ind. Eng. Chem. Res. 2008, 467, 3265–3273. [Google Scholar] [CrossRef]
- Sierra, I.; Ereña, J.; Aguayo, A.T.; Ateka, A.; Bilbao, J. Kinetic Modelling for the Dehydration of Methanol to Dimethyl Ether over γ-Al2O3. Chem. Eng. Trans. 2013, 32, 613–618. [Google Scholar]
- Kasaie, M.; Sohrabi, M. Kinetic Study on Methanol Dehydration to Dimethyl Ether Applying Clinoptilolite Zeolite as the Reaction Catalyst. J. Mex. Chem. Soc. 2009, 53, 233–238. [Google Scholar]
- Sun, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Catalysis Chemistry of Dimethyl Ether Synthesis. ACS Catal. 2014, 4, 3346–3356. [Google Scholar] [CrossRef]
- Chang, C.D. Mechanism of Hydrocarbon Formation from Methanol. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Nederland, 1988; Volume 36, pp. 127–143. [Google Scholar]
- Ruiz-Rosas, R.; Bedia, J.; Rosas, J.M.; Lallave, M.; Loscertales, I.G.; Rodríguez-Mirasol, J.; Cordero, T. Methanol decomposition on electrospun zirconia nanofibers. Catal. Today 2012, 187, 77–87. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACMs Catalysts | PXPS (wt.%) | COTPD (mmol/g) | CO2 TPD (mmol/g) |
|---|---|---|---|
| OS1 | 2.8 | 5.5 | 0.6 |
| OS2 | 3.2 | 6.4 | 0.4 |
| AL1 | 3.7 | 6.1 | 0.5 |
| KL1 | 4.5 | 9.5 | 0.7 |
| Ea (kJ/mol) | ln ko | |
|---|---|---|
| OS1 | 104 | 33.14 |
| OS2 | 116 | 36.34 |
| AL1 | 134 | 39.70 |
| KL1 | 136 | 50.99 |
| Model | Rate Expression | OF Values |
|---|---|---|
| SO | 3.387 × 10−3 | |
| ER | 1.025 × 10−3 | |
| LH | 0.961 × 10−3 |
| Sample | 10−5·k0,SR mol s−1g−1 | EaSR kJ mol−1 | K0,SR | ΔHSR kJ mol−1 | K0MeOH atm−1 | ΔHMeOH kJ mol−1 | K0H2O atm−1 | ΔH H2O kJ mol−1 | 105·kSR,300 mol s−1 g−1 | KMeOH,300 atm−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| AL1 | 49 | 99 | 12 | 21 | 0.037 | −15 | 0.007 | −26 | 395 | 0.8 |
| KL1 | 354 | 111 | 43 | 24 | 0.025 | −18 | 0.003 | −17 | 269 | 1.1 |
| OS1 | 24 | 96 | 29 | 27 | 0.027 | −17 | 0.011 | −33 | 445 | 1.1 |
| OS2 | 6.2 | 92 | 19 | 25 | 0.155 | −12 | 0.009 | −35 | 276 | 1.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibeh, P.O.; García-Mateos, F.J.; Ruiz-Rosas, R.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Acid Mesoporous Carbon Monoliths from Lignocellulosic Biomass Waste for Methanol Dehydration. Materials 2019, 12, 2394. https://doi.org/10.3390/ma12152394
Ibeh PO, García-Mateos FJ, Ruiz-Rosas R, Rosas JM, Rodríguez-Mirasol J, Cordero T. Acid Mesoporous Carbon Monoliths from Lignocellulosic Biomass Waste for Methanol Dehydration. Materials. 2019; 12(15):2394. https://doi.org/10.3390/ma12152394
Chicago/Turabian StyleIbeh, Paul O., Francisco J. García-Mateos, Ramiro Ruiz-Rosas, Juana María Rosas, José Rodríguez-Mirasol, and Tomás Cordero. 2019. "Acid Mesoporous Carbon Monoliths from Lignocellulosic Biomass Waste for Methanol Dehydration" Materials 12, no. 15: 2394. https://doi.org/10.3390/ma12152394
APA StyleIbeh, P. O., García-Mateos, F. J., Ruiz-Rosas, R., Rosas, J. M., Rodríguez-Mirasol, J., & Cordero, T. (2019). Acid Mesoporous Carbon Monoliths from Lignocellulosic Biomass Waste for Methanol Dehydration. Materials, 12(15), 2394. https://doi.org/10.3390/ma12152394