Mechanical and Surface-Chemical Properties of Polymer Derived Ceramic Replica Foams



Abstract
1. Introduction
2. Materials and Methods
2.1. Foam Preparation
2.2. Characterization
3. Results and Discussion
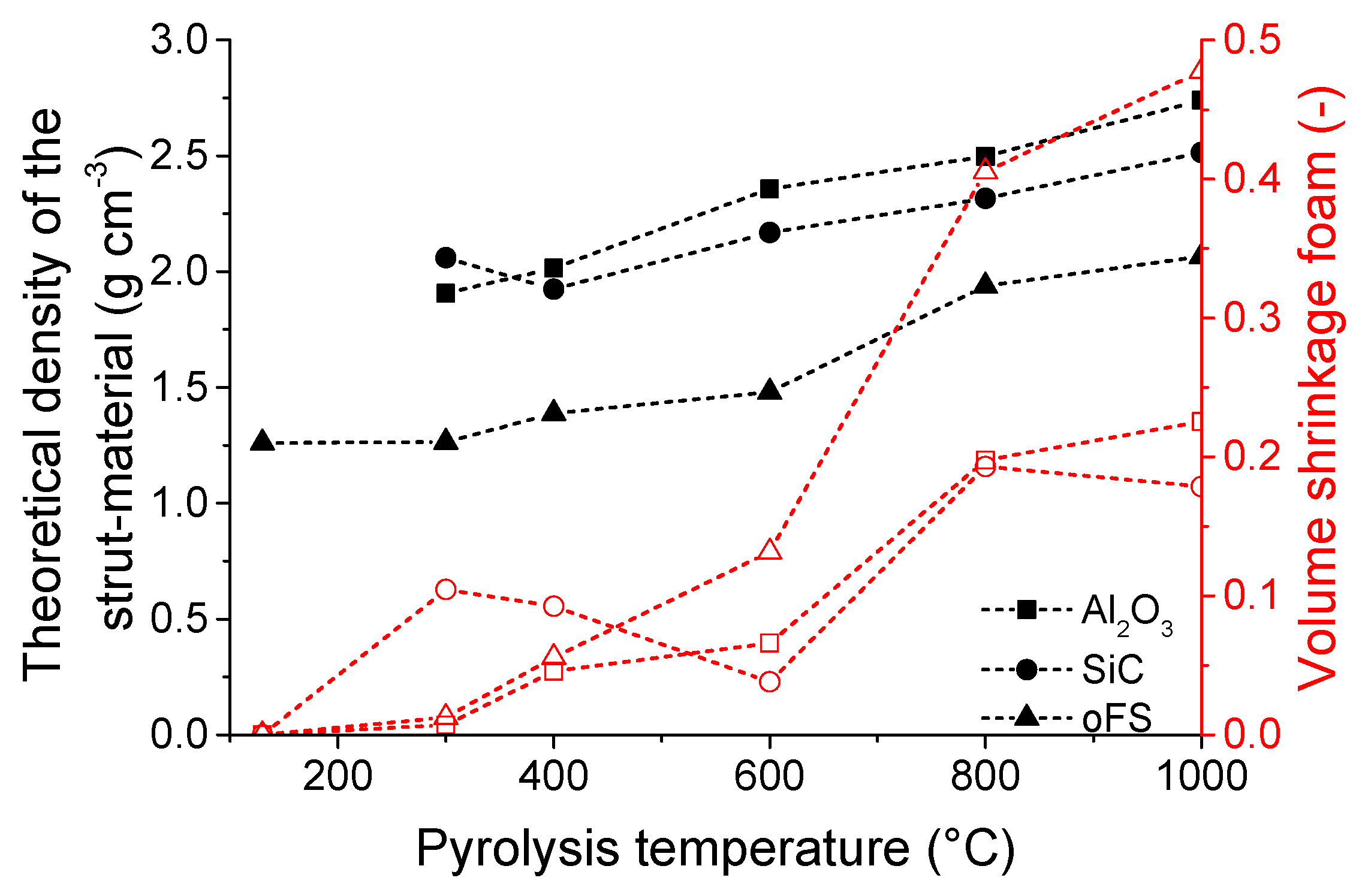
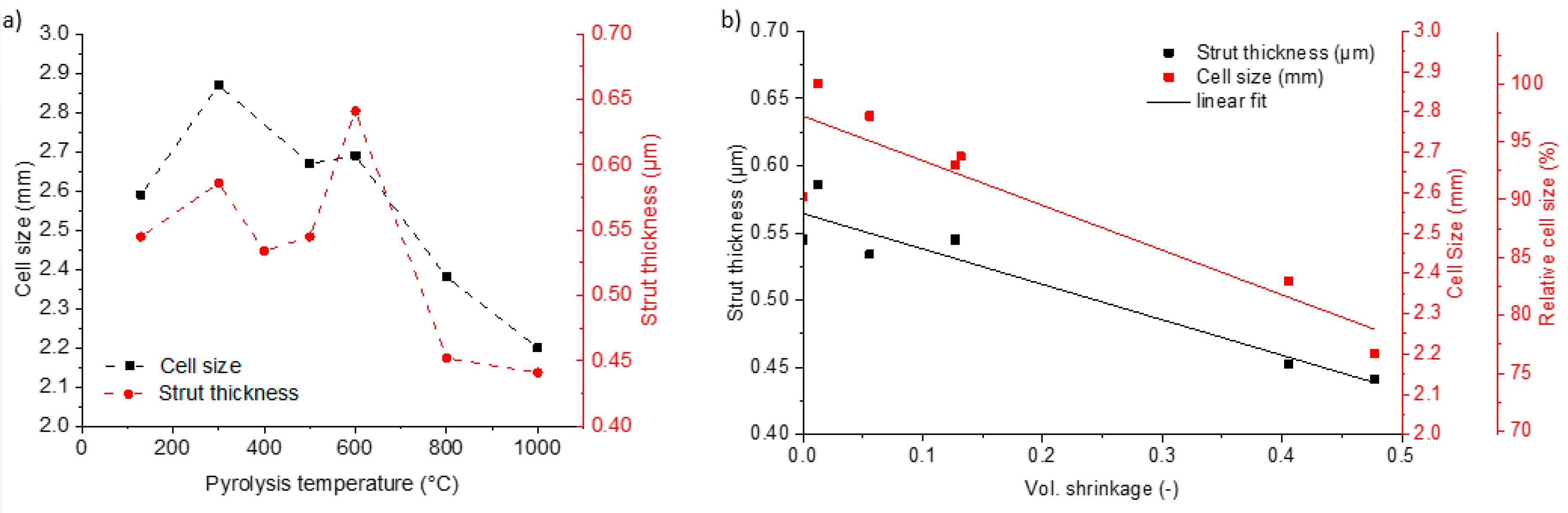
3.1. Microstructure and Shrinkage
3.2. Compositional and Structural Change
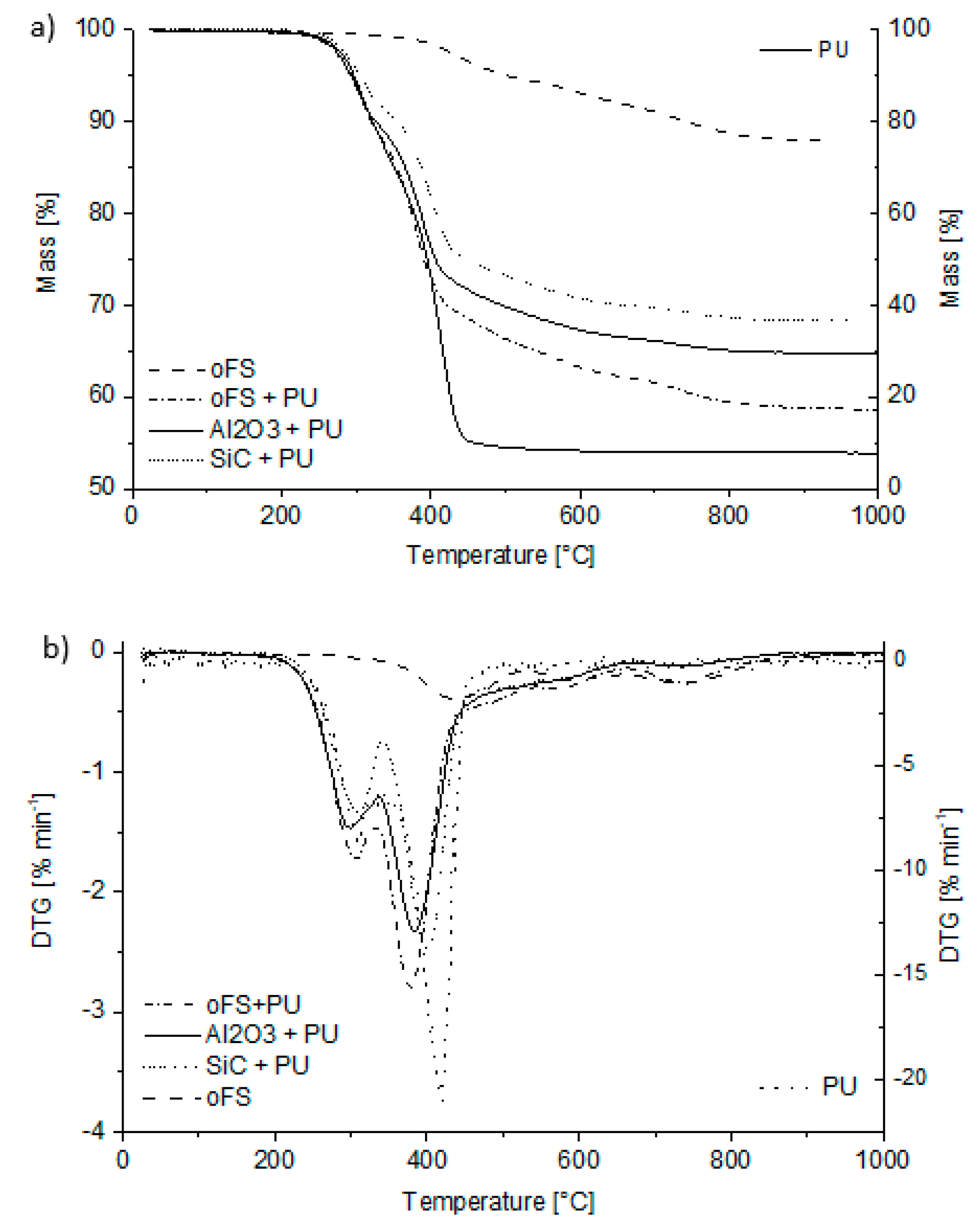
3.2.1. Thermal Analysis
- < 400 °C: Formation and release of water and ethanol due to remained crosslinking active groups like Si–OH and/or Si–OC2H5
- ~550–1000 °C: Redistribution reactions involving the exchange of Si-O, Si-H and Si-C bonds with the release of volatile silicon compounds (e.g., Me3SiOSiMe3);~600–700 °C: Possible release of small amounts of higher linear or cyclic polysiloxanes and/or tetramethylsilanes
- Radical reactions and release of gaseous products with formation of an open porous network.In dependence of the bonded groups the following temperature steps are ascertainable:~300–700 °C: Cleavage of Si–(C6H5) bonds and release of benzene (for PhSiO1.5)~600–900 °C: Cleavage of Si–(CH3) bonds and release of methane (for MeSiO1.5)~600–1100 °C: Dehydrogenation
- > 800 °C: Formation of aromatic carbon/ free carbon
- > 1000 °C–1600 °C: Enrichment of SiO4- and SiC4-units and precipitation of nanocrystals of SiO2 and SiC due to further chemical bond redistribution
- > 1200 °C–1500 °C: Carbothermal reduction of SiO2 and C according to the following reaction equations [16]:SiO2 + 3 C → SiC + 3 COIf y > 1 + x: SiOxCy → SiC + x CO + (y − x − 1) CIf y < 1 + x: SiOxCy → [(x + y − 1)/2] SiC + [(x + y −1)/2 CO + (x – y + 1) SiC
- > 1550 °C: Graphitization/ crystallization of the carbon and crystallization of SiC
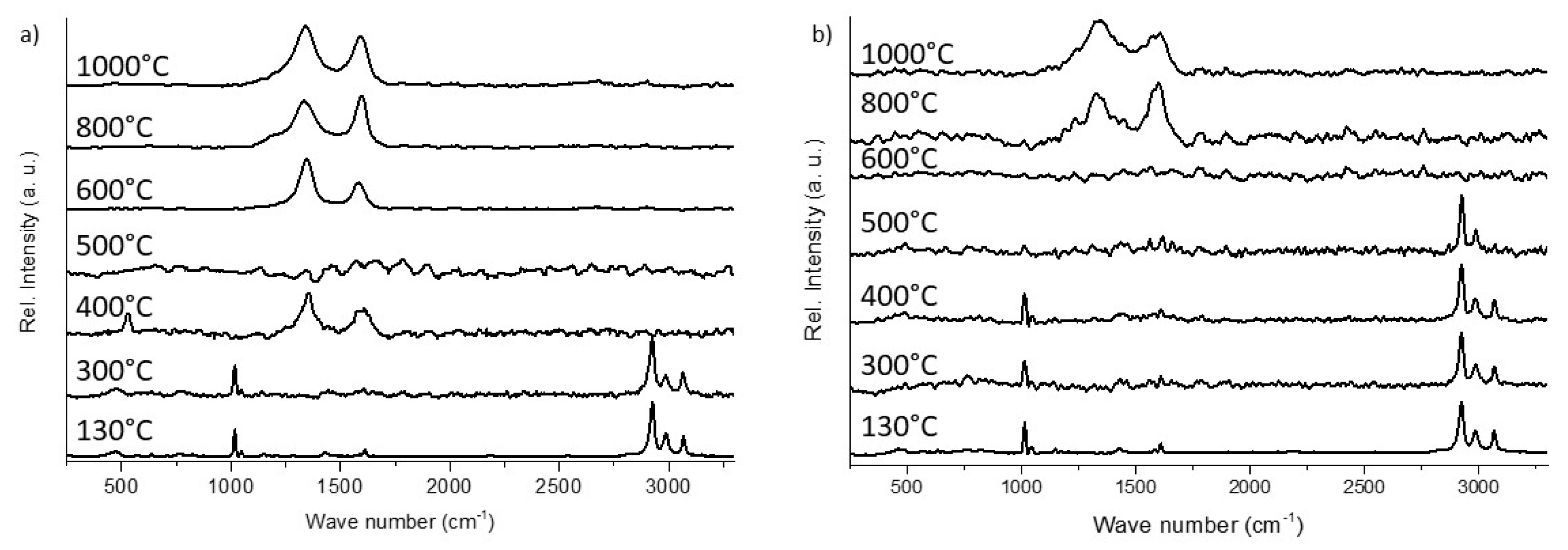
3.2.2. Raman Microscopy
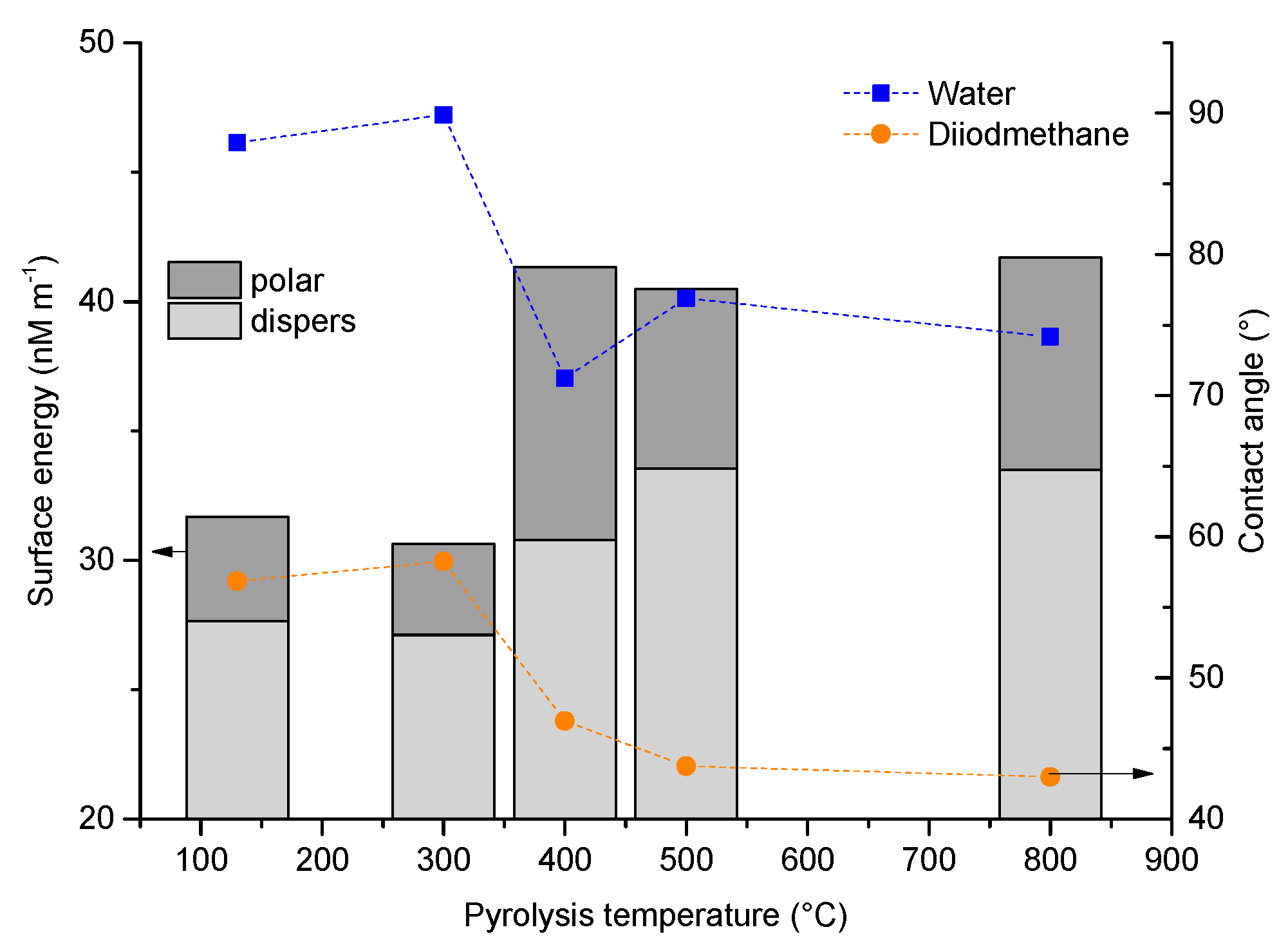
3.2.3. Wetting Behavior as a Function of Pyrolysis Temperature
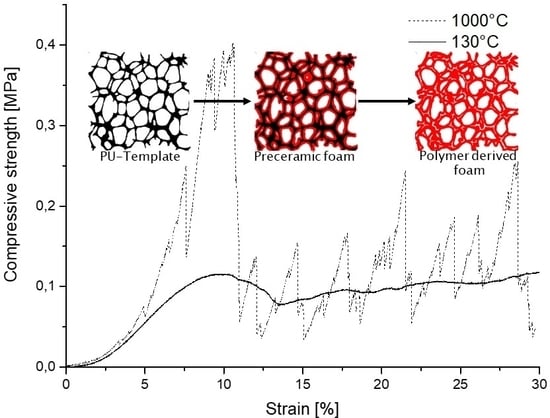
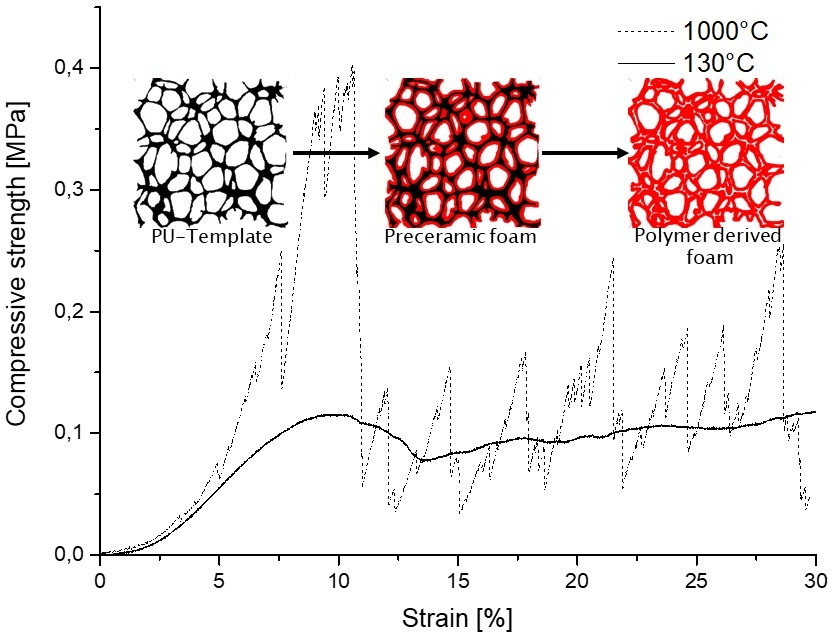
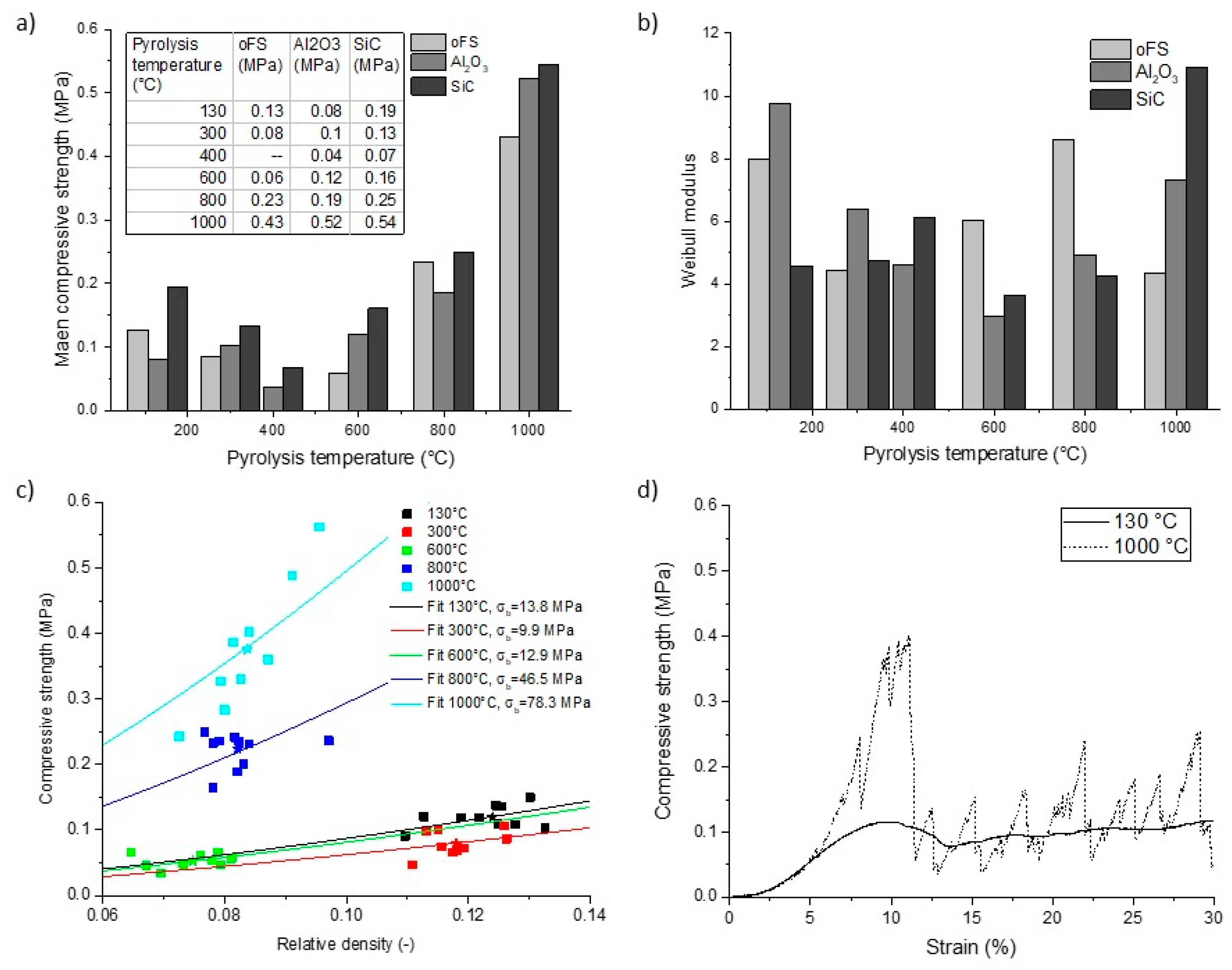
3.2.4. Compressive Strength as a Function of Pyrolysis Temperature
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yajima, S.; Hayashi, J.; Omori, M.; Okamura, K. Development of a silicon carbide fibre with high tensile strength. Nature 1976, 261, 683–685. [Google Scholar] [CrossRef]
- Yajima, S.; Hasegawa, Y.; Okamura, K.; Matsuzawa, T. Development of high tensile strength silicon carbide fibre using an organosilicon polymer precursor. Nature 1978, 273, 525–527. [Google Scholar] [CrossRef]
- Verbeek, W. Production of Shaped Articles of Homogeneous Mixtures of Silicon Carbide and Nitride. U.S. Patent No. 3,853,567, 2 April 1973. [Google Scholar]
- Colombo, P.; Mera, G.; Riedel, R.; Sorarù, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Noll, W. Zur Chemie und Technologie der Silicone. Angew. Chem. 1954, 66, 41–55. [Google Scholar] [CrossRef]
- Wan, J.; Gasch, M.J.; Mukherjee, A.K. InSitu Densification Behavior in the Pyrolysis Consolidation of Amorphous Si-N-C Bulk Ceramics from Polymer Precursors. J. Am. Ceram. Soc. 2001, 84, 2165–2169. [Google Scholar] [CrossRef]
- Cordelair, J.; Greil, P. Electrical conductivity measurements as a microprobe for structure transitions in polysiloxane derived Si–O–C ceramics. J. Eur. Ceram. Soc. 2000, 20, 1947–1957. [Google Scholar] [CrossRef]
- Bois, L.; Maquet, J.; Babonneau, F.; Bahloul, D. Structural Characterization of Sol-Gel Derived Oxycarbide Glasses. 2.: Study of the Thermal Stability of the Silicon Oxycarbide Phase. Chem. Mater. 1995, 7, 975–981. [Google Scholar] [CrossRef]
- Černý, M.; Chlup, Z.; Strachota, A.; Halasová, M.; Rýglová, Š.; Schweigstillová, J.; Svítilová, J.; Havelcová, M. Changes in structure and in mechanical properties during the pyrolysis conversion of crosslinked polymethylsiloxane and polymethylphenylsiloxane resins to silicon oxycarbide glass. Ceram. Int. 2015, 41, 6237–6247. [Google Scholar] [CrossRef]
- Hurwitz, F.I.; Heimann, P.; Farmer, S.C.; Hembree, D.M. Characterization of the pyrolytic conversion of polysilsesquioxanes to silicon oxycarbides. J. Mater. Sci. 1993, 28, 6622–6630. [Google Scholar] [CrossRef]
- Ma, J.; Shi, L.; Shi, Y.; Luo, S.; Xu, J. Pyrolysis of polymethylsilsesquioxane. J. Appl. Polym. Sci. 2002, 85, 1077–1086. [Google Scholar] [CrossRef]
- Scheffler, M.; Gambaryan-Roisman, T.; Takahashi, T.; Kaschta, J.; Muenstedt, H.; Buhler, P.; Greil, P. Pyrolytic Decomposition of Preceramic Organo Polysiloxanes. In Innovative Processing and Synthesis of Ceramics, Glasses, and Composites IV: Proceedings of the Innovative Processing and Synthesis of Ceramics, Glasses, and Composites Symposium at the 102nd Annual Meeting of the American Ceramic Society, St. Louis, MI, USA, 29 April–3 May 2000; Bansal, N.P., Ed.; American Ceramic Society: Westerville, OH, USA, 2000; pp. 239–250. ISBN 1-57498-111-0. [Google Scholar]
- Bahloul-Hourlier, D.; Latournerie, J.; Dempsey, P. Reaction pathways during the thermal conversion of polysiloxane precursors into oxycarbide ceramics. J. Eur. Ceram. Soc. 2005, 25, 979–985. [Google Scholar] [CrossRef]
- Belot, V.; Corriu, R.J.P.; Leclercq, D.; Mutin, P.H.; Vioux, A. Thermal reactions occurring during pyrolysis of crosslinked polysiloxane gels, precursors to silicon oxycarbide glasses. J. Non-Cryst. Solids 1992, 147–148, 52–55. [Google Scholar] [CrossRef]
- Belot, V.; Corriu, R.J.P.; Leclercq, D.; Mutin, P.H.; Vioux, A. Thermal redistribution reactions in crosslinked polysiloxanes. J. Polym. Sci. Part A Polym. Chem. 1992, 30, 613–623. [Google Scholar] [CrossRef]
- Mutin, P.H. Control of the Composition and Structure of Silicon Oxycarbide and Oxynitride Glasses Derived from Polysiloxane Precursors. J. Sol-Gel Sci. Technol. 1999, 14, 27–38. [Google Scholar] [CrossRef]
- Bahloul, D.; Pereira, M.; Goursat, P.; Yive, N.S.C.K.; Corriu, R.J.P. Preparation of Silicon Carbonitrides from an Organosilicon Polymer I: Thermal Decomposition of the Cross-linked Polysilazane. J. Am. Ceram. Soc. 1993, 76, 1156–1162. [Google Scholar] [CrossRef]
- Schlüter, F.; Meyer, J.; Wilhelm, M.; Rezwan, K. Hierarchical emulsion based hybrid ceramics synthesized with different siloxane precursor and with embedded nickel nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2016, 492, 160–169. [Google Scholar] [CrossRef]
- Prenzel, T.; Wilhelm, M.; Rezwan, K. Pyrolyzed polysiloxane membranes with tailorable hydrophobicity, porosity and high specific surface area. Microporous Mesoporous Mater. 2013, 169, 160–167. [Google Scholar] [CrossRef]
- Zhang, Z. Catalytic Effect of Aluminum Acetylacetonate on Hydrolysis and Polymerization of Methyltrimethoxysilane. Langmuir 1997, 13, 473–476. [Google Scholar] [CrossRef]
- Budunoglu, H.; Yildirim, A.; Guler, M.O.; Bayindir, M. Highly transparent, flexible, and thermally stable superhydrophobic ORMOSIL aerogel thin films. ACS Appl. Mater. Interfaces 2011, 3, 539–545. [Google Scholar] [CrossRef]
- Rao, A.V.; Bhagat, S.D.; Hirashima, H.; Pajonk, G.M. Synthesis of flexible silica aerogels using methyltrimethoxysilane (MTMS) precursor. J. Colloid Interface Sci. 2006, 300, 279–285. [Google Scholar] [CrossRef]
- Zhang, H.; D’Angelo Nunes, P.; Wilhelm, M.; Rezwan, K. Hierarchically ordered micro/meso/macroporous polymer-derived ceramic monoliths fabricated by freeze-casting. J. Eur. Ceram. Soc. 2016, 36, 51–58. [Google Scholar] [CrossRef]
- Liu, H.; Xu, J.; Li, Y.; Li, B.; Ma, J.; Zhang, X. Fabrication and Characterization of an Organic-Inorganic Gradient Surface made by Polymethylsilsesquioxane (PMSQ). Macromol. Rapid Commun. 2006, 27, 1603–1607. [Google Scholar] [CrossRef]
- Schelm, K.; Schwidder, M.; Samuel, J.; Scheffler, F.; Scheffler, M. Tailored Surface Properties of Ceramic Foams for Liquid Multiphase Reactions. Adv. Eng. Mater. 2017, 19, 1700418. [Google Scholar] [CrossRef]
- Zhang, H.; Fidelis, C.L.; Serva, A.L.T.; Wilhelm, M.; Rezwan, K. Water-based freeze casting: Adjusting hydrophobic polymethylsiloxane for obtaining hierarchically ordered porous SiOC. J. Am. Ceram. Soc. 2017, 100, 1907–1918. [Google Scholar] [CrossRef]
- Studart, A.R.; Gonzenbach, U.T.; Tervoort, E.; Gauckler, L.J. Processing Routes to Macroporous Ceramics: A Review. J. Am. Ceram. Soc. 2006, 89, 1771–1789. [Google Scholar] [CrossRef]
- Ceron-Nicolat, B.; Fey, T.; Greil, P. Processing of Ceramic Foams with Hierarchical Cell Structure. Adv. Eng. Mater. 2010, 12, 884–892. [Google Scholar] [CrossRef]
- Vogt, U.F.; Györfy, L.; Herzog, A.; Graule, T.; Plesch, G. Macroporous silicon carbide foams for porous burner applications and catalyst supports. J. Phys. Chem. Solids 2007, 68, 1234–1238. [Google Scholar] [CrossRef]
- Nangrejo, M.R.; Edirisinghe, M.J. Porosity and Strength of Silicon Carbide Foams Prepared Using Preceramic Polymers. J. Porous Mater. 2002, 9, 131–140. [Google Scholar] [CrossRef]
- Bao, X.; Nangrejo, M.R.; Edirisinghe, M.J. Preparation of silicon carbide foams using polymeric precursor solutions. J. Mater. Sci. 2000, 35, 4365–4372. [Google Scholar] [CrossRef]
- Nangrejo, M.R.; Bao, X.; Edirisinghe, M.J. Silicon carbide–titanium carbide composite foams produced using a polymeric precursor. Int. J. Inorg. Mater. 2001, 3, 37–45. [Google Scholar] [CrossRef]
- Nangrejo, M.R.; Bao, X.; Edirisinghe, M.J. The structure of ceramic foams using polymeric precursors. J. Mater. Sci. Lett. 2000, 19, 787–789. [Google Scholar] [CrossRef]
- Nangrejo, M.R.; Bao, X.; Edirisinghe, M.J. Preparation of silicon carbide–silicon nitride composite foams from pre-ceramic polymers. J. Eur. Ceram. Soc. 2000, 20, 1777–1785. [Google Scholar] [CrossRef]
- Sorarù, G.D.; Campostrini, R.; Ejigu, A.A.; Zera, E.; Jana, P. Processing and characterization of polymer derived SiOC foam with hierarchical porosity by HF etching. J. Ceram. Soc. Jpn. 2016, 124, 1023–1029. [Google Scholar] [CrossRef][Green Version]
- Takahashi, T.; Colombo, P. SiOC Ceramic Foams through Melt Foaming of a Methylsilicone Preceramic Polymer. J. Porous Mater. 2003, 10, 113–121. [Google Scholar] [CrossRef]
- Takahashi, T.; Münstedt, H.; Modesti, M.; Colombo, P. Oxidation resistant ceramic foam from a silicone preceramic polymer/polyurethane blend. J. Eur. Ceram. Soc. 2001, 21, 2821–2828. [Google Scholar] [CrossRef]
- Takahashi, T.; Münstedt, H.; Colombo, P.; Modesti, M. Thermal evolution of a silicone resin/polyurethane blend from preceramic to ceramic foam. J. Mater. Sci. 2001, 36, 1627–1639. [Google Scholar] [CrossRef]
- Betke, U.; Dalicho, S.; Rannabauer, S.; Lieb, A.; Scheffler, F.; Scheffler, M. Impact of Slurry Composition on Properties of Cellular Alumina: A Computed Tomographic Study. Adv. Eng. Mater. 2017, 19, 1700138. [Google Scholar] [CrossRef]
- Schelm, K.; Fey, T.; Dammler, K.; Betke, U.; Scheffler, M. Hierarchical-Porous Ceramic Foams by a Combination of Replica and Freeze Technique. Adv. Eng. Mater. 2019, 10, 155. [Google Scholar] [CrossRef]
- Betke, U.; Scheffler, M. Reticulated Open-Celled Zinc Oxide Ceramic Foams: Manufacturing, Microstructure, Mechanical, and Thermal Properties. Adv. Mater. Sci. Eng. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Gibson, L.J. Cellular Solids. Structure and Properties, 2nd ed.; Cambridge University Press: Cambridge, UK, 1997; ISBN 0521499119. [Google Scholar]
- Owens, D.K.; Wendt, R.C. Estimation of the surface free energy of polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- DIN. DIN EN 623-2:1993-11, Hochleistungskeramik; Monolithische Keramik; Allgemeine und strukturelle Eigenschaften; Teil_2: Bestimmung von Dichte und Porosität; Deutsche Fassung EN_623-2:1993; Beuth Verlag GmbH: Berlin, Germany, 1993. [Google Scholar]
- Schmidt, H.; Koch, D.; Grathwohl, G.; Colombo, P. Micro-/Macroporous Ceramics from Preceramic Precursors. J. Am. Ceram. Soc. 2001, 84, 2252–2255. [Google Scholar] [CrossRef]
- Schmidt, H.; Koch, D.; Grathwohl, G. Development of Ceramic Membranes and Adsorbents from Silicon-Organic Precursors. Chem. Eng. Technol. 2000, 23, 959–964. [Google Scholar] [CrossRef]
- Singh, A.K.; Pantano, C.G. Surface chemistry and structure of silicon oxycarbide gels and glasses. J. Sol-Gel Sci. Technol. 1997, 8, 371–376. [Google Scholar] [CrossRef]
- Förch, R. (Ed.) Surface Design. Applications in Bioscience and Nanotechnology; Wiley-VCH: Weinheim, Germany, 2009; ISBN 978-3-527-40789-7. [Google Scholar]
- He, X.; Su, B.; Tang, Z.; Zhao, B.; Wang, X.; Yang, G.; Qiu, H.; Zhang, H.; Yang, J. The comparison of macroporous ceramics fabricated through the protein direct foaming and sponge replica methods. J. Porous Mater. 2012, 19, 761–766. [Google Scholar] [CrossRef]
- Colombo, P.; Griffoni, M.; Modesti, M. Ceramic Foams from a Preceramic Polymer and Polyurethanes: Preparation and Morphological Investigations. J. Sol-Gel Sci. Technol. 1998, 13, 195–199. [Google Scholar] [CrossRef]
- Montanaro, L.; Jorand, Y.; Fantozzi, G.; Negro, A. Ceramic foams by powder processing. J. Eur. Ceram. Soc. 1998, 18, 1339–1350. [Google Scholar] [CrossRef]
- Ashby, M.F.; Mehl Medalist, R.F. The mechanical properties of cellular solids. Metall. Trans. A 1983, 14, 1755–1769. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Filler Free (oFS) | Filled with Al2O3 | Filled with SiC | |||
|---|---|---|---|---|---|---|
| wt% | vol% | wt% | vol% | wt% | vol% | |
| MTES | 4.23 | 5.20 | 7.95 | 14.34 | 8.08 | 14.03 |
| SILRES® H62 C | 38.06 | 37.08 | 22.94 | 32.77 | 22.63 | 31.12 |
| SILRES® MK | 57.09 | 57.14 | 22.94 | 33.66 | 22.63 | 31.97 |
| Filler | – | – | 45.88 | 18.80 | 46.34 | 22.44 |
| Aluminum acetylacetonate | 0.57 | 0.52 | 0.23 | 0.31 | 0.22 | 0.28 |
| Oleic acid | 0.04 | 0.05 | 0.07 | 0.13 | 0.09 | 0.16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schelm, K.; Abreu Morales, E.; Scheffler, M. Mechanical and Surface-Chemical Properties of Polymer Derived Ceramic Replica Foams. Materials 2019, 12, 1870. https://doi.org/10.3390/ma12111870
Schelm K, Abreu Morales E, Scheffler M. Mechanical and Surface-Chemical Properties of Polymer Derived Ceramic Replica Foams. Materials. 2019; 12(11):1870. https://doi.org/10.3390/ma12111870
Chicago/Turabian StyleSchelm, Katja, Elena Abreu Morales, and Michael Scheffler. 2019. "Mechanical and Surface-Chemical Properties of Polymer Derived Ceramic Replica Foams" Materials 12, no. 11: 1870. https://doi.org/10.3390/ma12111870
APA StyleSchelm, K., Abreu Morales, E., & Scheffler, M. (2019). Mechanical and Surface-Chemical Properties of Polymer Derived Ceramic Replica Foams. Materials, 12(11), 1870. https://doi.org/10.3390/ma12111870