Assessment of Migration of Human MSCs through Fibrin Hydrogels as a Tool for Formulation Optimisation


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fibrin Gel Preparation and Synthesis
- (a)
- A total of 200 μL of formed fibrin gel material was deposited on borosilicate glass coverslips for the physical-chemical characterisations.
- (b)
- For the different cellular studies, cells were added within thrombin solution and fibrin gels containing 25,000 cells/gel were formed on 13 mm diameter circular borosilicate glass coverslips (631-0159, VWR, Lutterworth, UK).
- (c)
- For cell migration experiments, cellular spheroids (50,000 cell) were encapsulated within fibrin gels in multi-wells.
2.2. ScanningElectron Microscopy
2.3. Rheological Characterisation
2.4. Cell Culture
2.5. Live/Dead Cell Staining
2.6. PrestoBlue® Cell Viability Reagent
2.7. Spheroid Outgrowth Studies
2.8. Statistical Analysis
3. Results
3.1. SEM Analysis
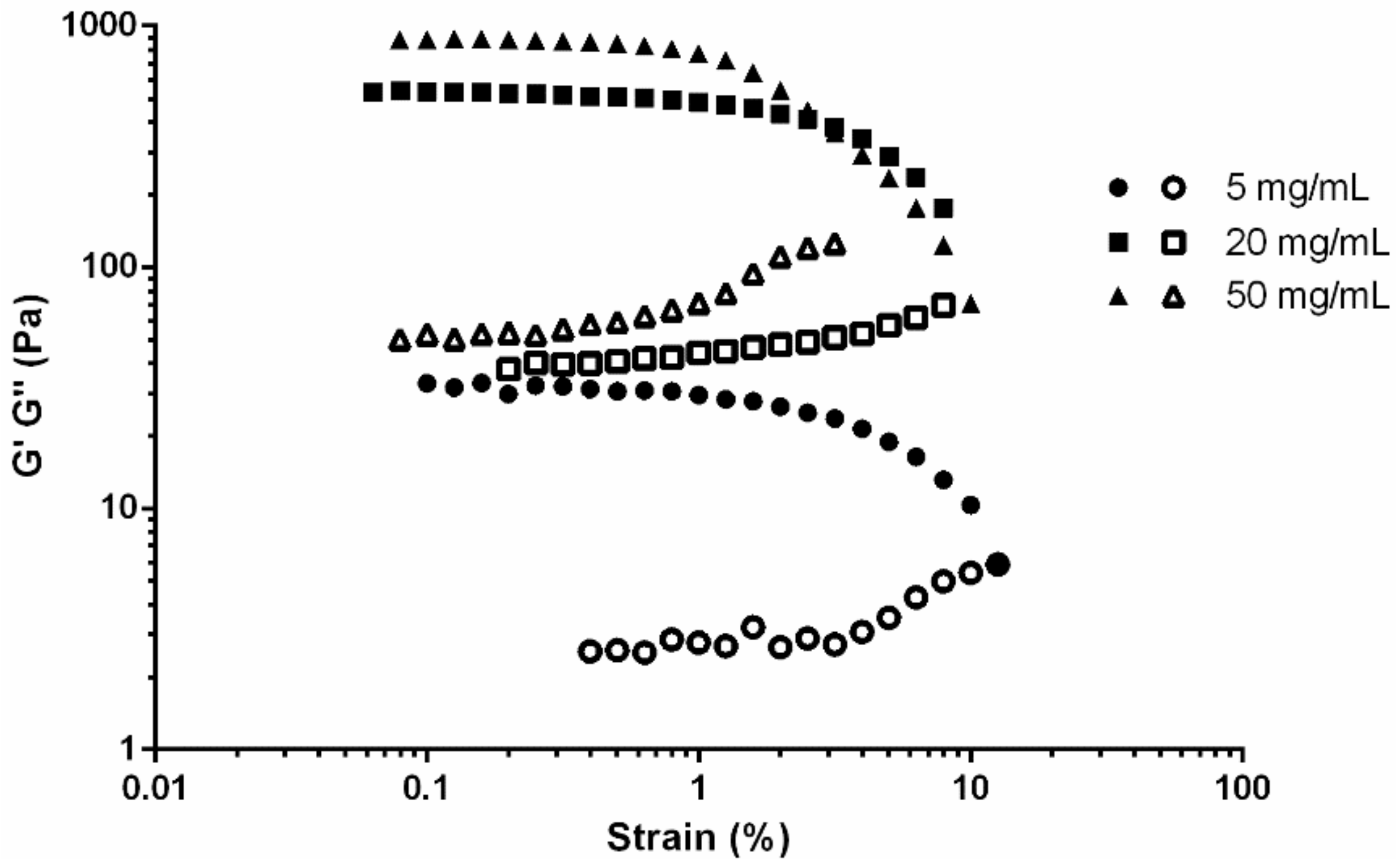
3.2. Viscoelasticity of Fibrin Gels
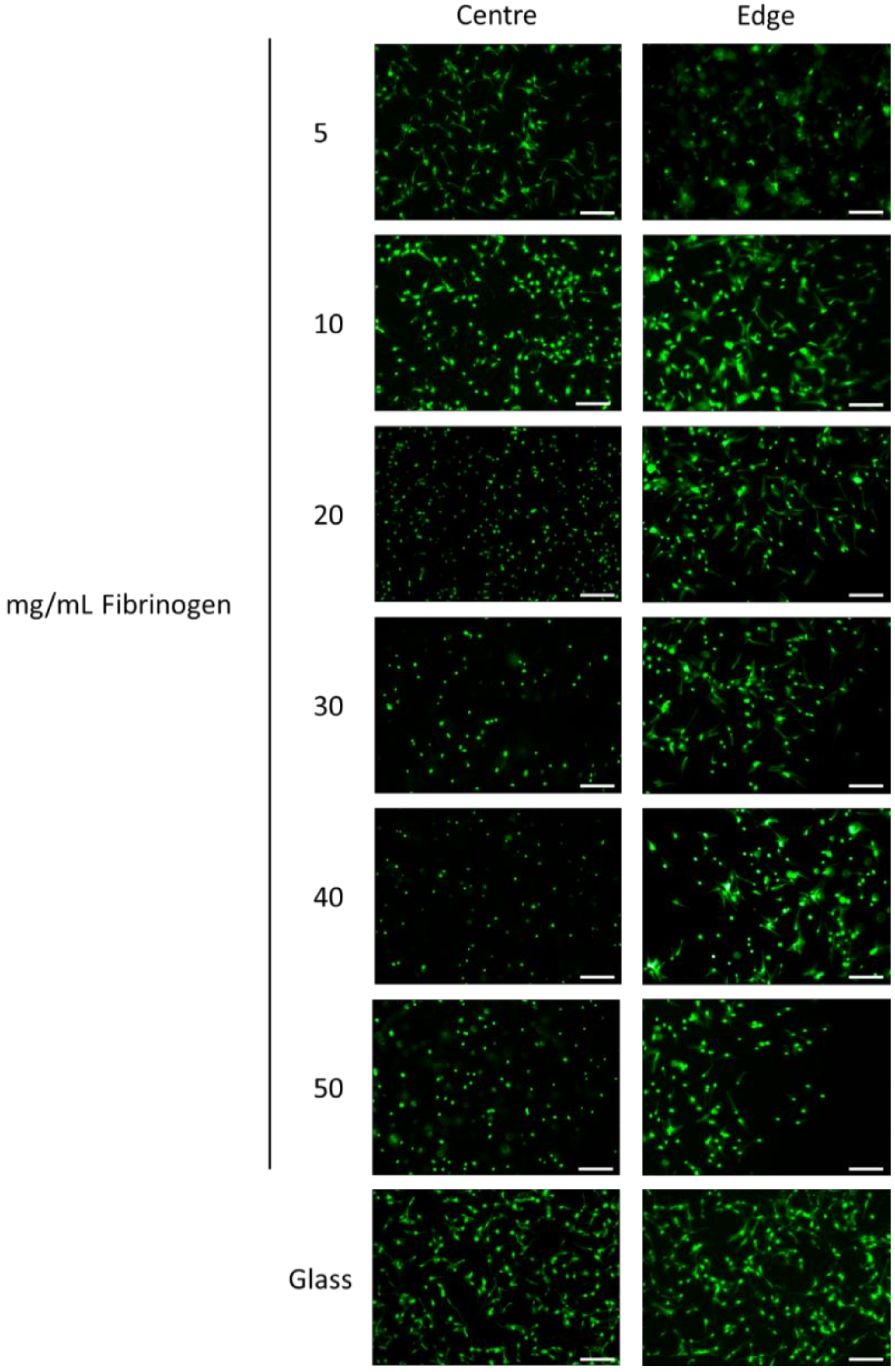
3.3. Viability and Morphology of hMSCs within Fibrin Gels
3.4 Encapsulated hMSC Proliferation
3.5 Encapsulated hMSC Migration
4. Discussion
4.1 Fibrinogen Concentration Influenced Gel Structure
4.2 Rheological Characterisation of Fibrin Gels
4.3 Cytocompatibility of Fibrin Gels
- (a)
- Cells at the edge could be able to obtain more nutrients and oxygen via diffusion and mass transport as a result [27], thus making them more mobile.
- (b)
- Greater diffusion of media into the gels at the edge may make the gels locally less stiff, and allow greater spreading, possibly accentuated through cells accessing plasmin, allowing for some matrix remodelling.
4.4 Spheroid Outgrowth Experiments
4.5 Limitations of the Current Study
5. Conclusions
Author Contributions
Funding
Acknowledgments
Open Data
Conflicts of Interest
References
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Ahmed, T.A.E.; Dare, E.V.; Hincke, M. Fibrin: A versatile scaffold for tissue engineering applications. Tissue Eng. Part B Rev. 2008, 14, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Humar, R.; Zimmerli, L.; Battegay, E. Angiogenesis and hypertension: An update. J. Hum. Hypertens. 2009, 23, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Yuk, S.H. Polymeric protein delivery systems. Prog. Polym. Sci. 2007, 32, 669–697. [Google Scholar] [CrossRef]
- Bensaïd, W.; Triffitt, J.T.; Blanchat, C.; Oudina, K.; Sedel, L.; Petite, H. A biodegradable fibrin scaffold for mesenchymal stem cell transplantation. Biomaterials 2003, 24, 2497–2502. [Google Scholar]
- Mizushima, T.; Takahashi, H.; Takeyama, H.; Naito, A.; Haraguchi, N.; Uemura, M.; Nishimura, J.; Hata, T.; Takemasa, I.; Yamamoto, H.; et al. A clinical trial of autologous adipose-derived regenerative cell transplantation for a postoperative enterocutaneous fistula. Surg. Today 2016, 46, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Park, K.J.; Cho, Y.B.; Yoon, S.N.; Song, K.H.; Kim, D.S.; Jung, S.H.; Kim, M.; Yoo, H.-W.; Kim, I.; et al. Autologous adipose tissue-derived stem cells treatment demonstrated favorable and sustainable therapeutic effect for Crohn’s fistula. Stem Cells 2013, 31, 2575–2581. [Google Scholar] [CrossRef] [PubMed]
- Ravari, H.; Hamidi-Almadari, D.; Salimifar, M.; Bonakdaran, S.; Parizadeh, M.R.; Koliakos, G. Treatment of non-healing wounds with autologous bone marrow cells, platelets, fibrin glue and collagen matrix. Cytotherapy 2011, 13, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.-G.; Kwon, O.-R.; Kim, Y.-S.; Choi, Y.-J.; Tak, D.-H. Adipose-derived mesenchymal stem cells with microfracture versus microfracture alone: 2-year follow-up of a prospective randomized trial. Arthrosc. J. Arthrosc. Relat. Surg. 2016, 32, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Khademhosseini, A.; Langer, R. A decade of progress in tissue engineering. Nat. Protoc. 2016, 11, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Eyrich, D.; Brandl, F.; Appel, B.; Wiese, H.; Maier, G.; Wenzel, M.; Staudenmaier, R.; Goepferich, A.; Blunk, T. Long-term stable fibrin gels for cartilage engineering. Biomaterials 2007, 28, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ossendorf, C.; Kaps, C.; Kreuz, P.C.; Burmester, G.R.; Sittinger, M.; Erggelet, C. Treatment of posttraumatic and focal osteoarthritic cartilage defects of the knee with autologous polymer-based three-dimensional chondrocyte grafts: 2-year clinical results. Arthritis Res. Ther. 2007, 9, R41. [Google Scholar] [CrossRef] [PubMed]
- Uehara, K.; Zhao, C.; Gingery, A.; Thoreson, A.R.; An, K.-N.; Amadio, P.C. Effect of fibrin formulation on initial strength of tendon repair and migration of bone marrow stromal cells in vitro. J. Bone Jt. Surg. Am. 2015, 97, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Ludeña, D.; Fernández, A.; Aranda, J.L.; Varela, G.; Iglesias, J. Autologous fibrin scaffolds cultured dermal fibroblasts and enriched with encapsulated bFGF for tissue engineering. J. Biomed. Mater. Res. A 2011, 99, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Jockenhoevel, S.; Zund, G.; Hoerstrup, S.P.; Chalabi, K.; Sachweh, J.S.; Demircan, L.; Messmer, B.J.; Turina, M. Fibrin gel-advantages of a new scaffold in cardiovascular tissue engineering. Eur. J. Cardiothorac. Surg. 2001, 19, 424–430. [Google Scholar] [CrossRef]
- Camci-Unal, G.; Annabi, N.; Dokmeci, M.R.; Liao, R.; Khademhosseini, A. Hydrogels for cardiac tissue engineering. NPG Asia Mater. 2014, 6, e99. [Google Scholar] [CrossRef]
- Fawcett, J.W. Spinal cord repair: from experimental models to human application. Spinal Cord. 1998, 36, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Talbot, M.; Carrier, P.; Giasson, C.J.; Deschambeault, A.; Guérin, S.L.; Auger, F.A.; Bazin, R.; Germain, L. Autologous transplantation of rabbit limbal epithelia cultured on fibrin gels for ocular surface reconstruction. Mol. Vis. 2006, 12, 65–75. [Google Scholar] [PubMed]
- Alaminos, M.; Sánchez-Quevedo, M.D.C.; Munoz-Ávila, J.I.; Serrano, D.; Medialdea, S.; Carreras, I.; Campos, A. Construction of a complete rabbit cornea substitute using a fibrin-agarose scaffold. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3311–3317. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. J. Vis. Exp. 2014, 752, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Even-Ram, S.; Yamada, K.M. Cell migration in 3D matrix. Curr. Opin. Cell Biol. 2005, 17, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension—How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Guvendiren, M.; Burdick, J.A. Engineering synthetic hydrogel microenvironments to instruct stem cells. Curr. Opin. Biotechnol. 2013, 24, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cuddihy, M.J.; Kotov, N.A. Three-dimensional cell culture matrices: state of the art. Tissue Eng. Part B Rev. 2008, 14, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.A.; Swartz, M.A. Mechanobiology in the third dimension. Ann. Biomed. Eng. 2005, 33, 1469–1490. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Bröcker, E.B. The biology of cell locomotion within three-dimensional extracellular matrix. Cell Mol. Life Sci. 2000, 57, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.L.; Lee, S.; Stegemann, J.P. Influence of thrombin concentration on the mechanical and morphological properties of cell-seeded fibrin hydrogels. Acta Biomater. 2007, 3, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, R.I.; Weisel, J.W. Fibrin mechanical properties and their structural origins. Matrix Biol. 2017, 60–61, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Blombäck, B.; Bark, N. Fibrinopeptides and fibrin gel structure. Biophys. Chem. 2004, 112, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.A.; Griffith, M.; Hincke, M. Characterization and inhibition of fibrin hydrogel-degrading enzymes during development of tissue engineering scaffolds. Tissue Eng. 2007, 13, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Cole, M.; Tawil, B. Behavior of human dermal fibroblasts in three-dimensional fibrin clots: dependence on fibrinogen and thrombin concentration. Tissue Eng. 2004, 10, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.E.; Gabriel, D.A.; McDonagh, J. Influence of Ca2+ on the structure of reptilase-derived and thrombin-derived fibrin gels. Biochem. J. 1986, 239, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Hethershaw, E.L.; Cilia La Corte, A.L.; Duval, C.; Ali, M.; Grant, P.J.; Ariëns, R.A.S.; Philippou, H. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. J. Thromb. Haemost. 2014, 12, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Nair, C.H.; Shah, G.A.; Dhall, D.P. Effect of temperature, pH and ionic strength and composition on fibrin network structure and its development. Thromb. Res. 1986, 42, 809–816. [Google Scholar] [CrossRef]
- Weisel, J.W. Structure of fibrin: Impact on clot stability. J. Thromb. Haemost. 2007, 5, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.; Tawil, B.; Dunn, J.C.Y.; Wu, B.M. The behavior of human mesenchymal stem cells in 3D fibrin clots: dependence on fibrinogen concentration and clot structure. Tissue Eng. 2006, 12, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Geer, D.J.; Andreadis, S.T. A novel role of fibrin in epidermal healing: Plasminogen-mediated migration and selective detachment of differentiated keratinocytes. J. Investig. Dermatol. 2003, 121, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Schleef, R.R.; Birdwell, C.R. The effect of fibrin on endothelial cell migration in vitro. Tissue Cell 1982, 14, 629–636. [Google Scholar] [CrossRef]
- Yang, Y.-I.; Kim, H.-I.; Shelby, J.; Choi, M.-Y.; Jang, S.-H.; Kim, J.-T.; Jang, W.-H.; Choi, C.S.; Cheong, S.-H. Fibrin matrix-supported three-dimensional organ culture of adipose tissue for selective outgrowth, expansion, and isolation of adipose-derived stem cells. Acta Biomater. 2011, 7, 4109–4119. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, K.M.; Harunaga, J.S.; Doyle, A.D.; Yamada, K.M. Direct comparisons of the morphology, migration, cell adhesions, and actin cytoskeleton of fibroblasts in four different three-dimensional extracellular matrices. Tissue Eng. Part A 2011, 17, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Brown, A.C. Characterizing Cell Migration Within Three-dimensional In Vitro Wound Environments. J. Vis. Exp. 2017, 126, e56099. [Google Scholar] [CrossRef] [PubMed]
- Man, A.J.; Davis, H.E.; Itoh, A.; Leach, J.K.; Bannerman, P. Neurite outgrowth in fibrin gels is regulated by substrate stiffness. Tissue Eng. Part A 2011, 17, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.W.; Goodrich, L.R.; Frisbie, D.D.; McIlwraith, C.W.; Kisiday, J.D. Effect of scaffold dilution on migration of mesenchymal stem cells from fibrin hydrogels. Am. J. Vet. Res. 2012, 73, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Catelas, I.; Sese, N.; Wu, B.M.; Dunn, J.C.Y.; Helgerson, S.; Tawil, B. Human mesenchymal stem cell proliferation and osteogenic differentiation in fibrin gels in vitro. Tissue Eng. 2006, 12, 2385–2396. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Fox, J.; Afsari, F.; Lee, J.; Clough, S.; Knight, C.; Ashmore, J.; Ashton, J.; Preham, O.; Hoogduijn, M.; et al. Multiparameter Analysis of Human Bone Marrow Stromal Cells Identifies Distinct Immunomodulatory and Differentiation-Competent Subtypes. Stem Cell Rep. 2015, 4, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, P.B.; Solursh, M.; Reiter, R.S. Stage-related capacity for limb chondrogenesis in cell culture. Dev. Biol. 1977, 60, 69–82. [Google Scholar] [CrossRef]
- Fennema, E.; Rivron, N.; Rouwkema, J.; van Blitterswijk, C.; de Boer, J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 2013, 31, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; Mockros, L.F.; Weisel, J.W.; Lorand, L. Structural origins of fibrin clot rheology. Biophys. J. 1999, 77, 2813–2826. [Google Scholar] [CrossRef]
- Weisel, J.W. The mechanical properties of fibrin for basic scientists and clinicians. Biophys. Chem. 2004, 112, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Profumo, A.; Turci, M.; Damonte, G.; Ferri, F.; Magatti, D.; Cardinali, B.; Cuniberti, C.; Rocco, M. Kinetics of fibrinopeptide release by thrombin as a function of CaCl2 concentration: Different susceptibility of FPA and FPB and evidence for a fibrinogen isoform-specific effect at physiological Ca2+ concentration. Biochemistry 2003, 42, 12335–12348. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Lee, S.K.; Yoon, J.I.; Kim, D.E.; Kim, M.; Ha, H. Fibrin glue improves the therapeutic effect of MSCs by sustaining survival and paracrine function. Tissue Eng. Part A 2013, 19, 2373–2781. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salam, N.; Toumpaniari, S.; Gentile, P.; Marina Ferreira, A.; Dalgarno, K.; Partridge, S. Assessment of Migration of Human MSCs through Fibrin Hydrogels as a Tool for Formulation Optimisation. Materials 2018, 11, 1781. https://doi.org/10.3390/ma11091781
Salam N, Toumpaniari S, Gentile P, Marina Ferreira A, Dalgarno K, Partridge S. Assessment of Migration of Human MSCs through Fibrin Hydrogels as a Tool for Formulation Optimisation. Materials. 2018; 11(9):1781. https://doi.org/10.3390/ma11091781
Chicago/Turabian StyleSalam, Nasseem, Sotiria Toumpaniari, Piergiorgio Gentile, Ana Marina Ferreira, Kenneth Dalgarno, and Simon Partridge. 2018. "Assessment of Migration of Human MSCs through Fibrin Hydrogels as a Tool for Formulation Optimisation" Materials 11, no. 9: 1781. https://doi.org/10.3390/ma11091781
APA StyleSalam, N., Toumpaniari, S., Gentile, P., Marina Ferreira, A., Dalgarno, K., & Partridge, S. (2018). Assessment of Migration of Human MSCs through Fibrin Hydrogels as a Tool for Formulation Optimisation. Materials, 11(9), 1781. https://doi.org/10.3390/ma11091781