Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations

Abstract
1. Introduction
2. Computational Method
3. Results and Discussion
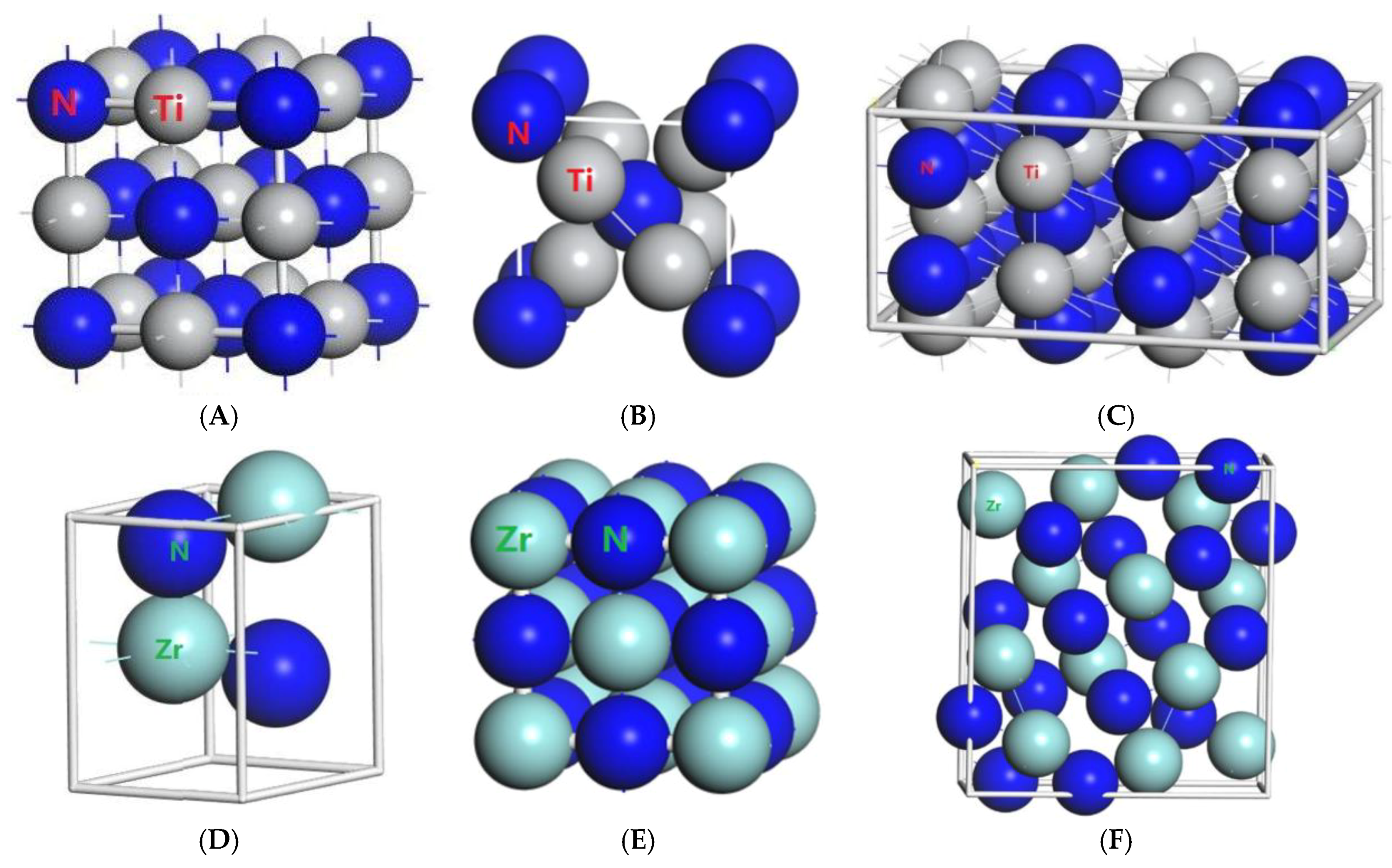
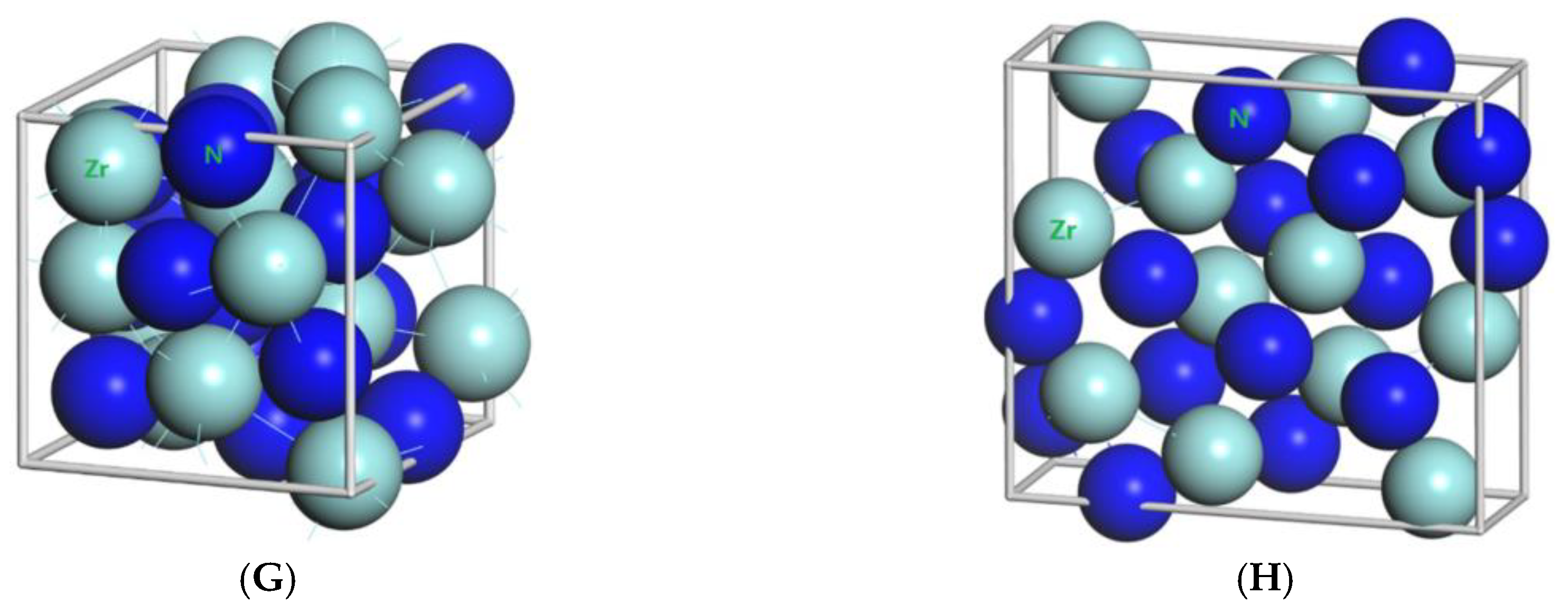
3.1. Result of the Elastic Properties
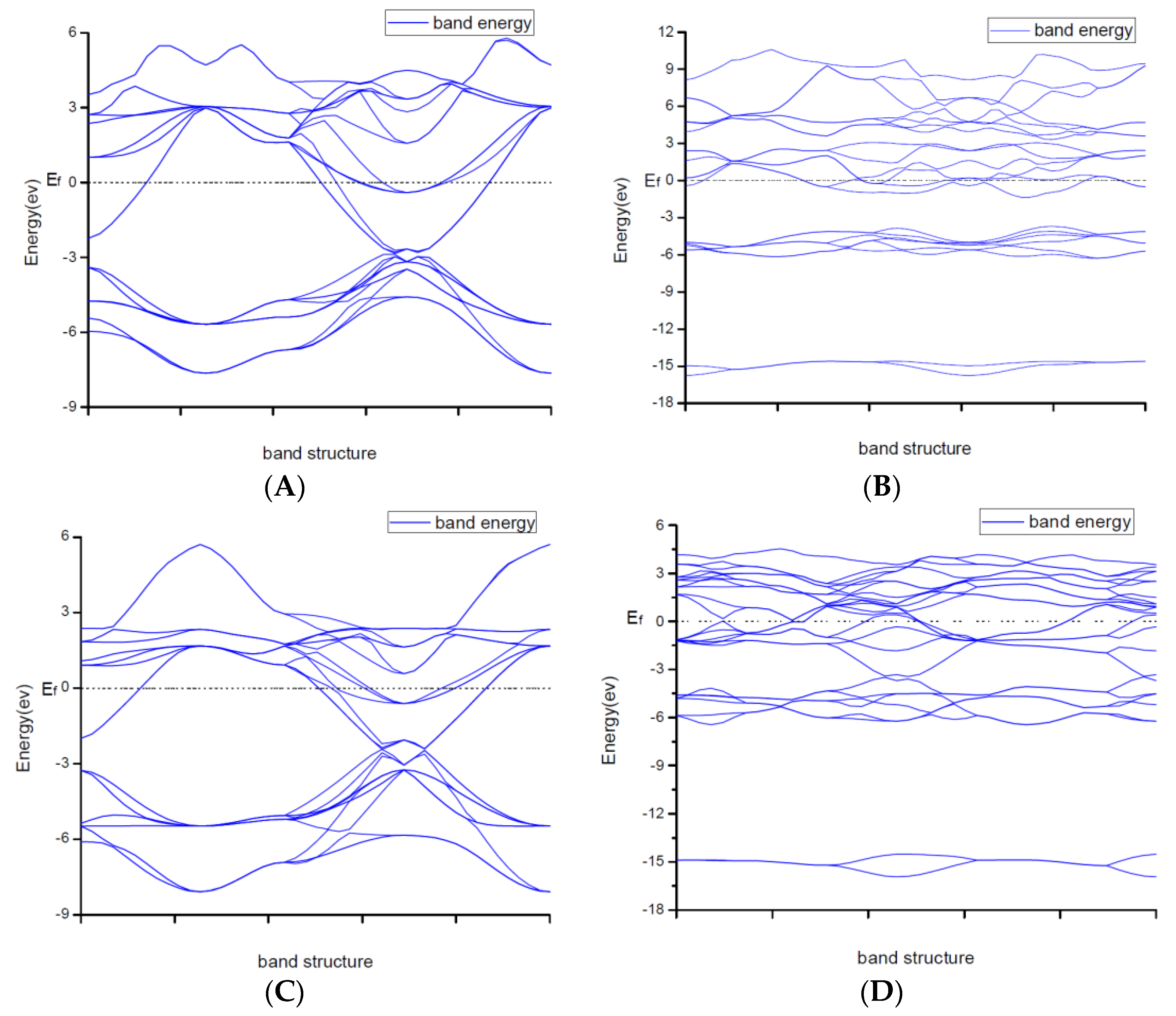
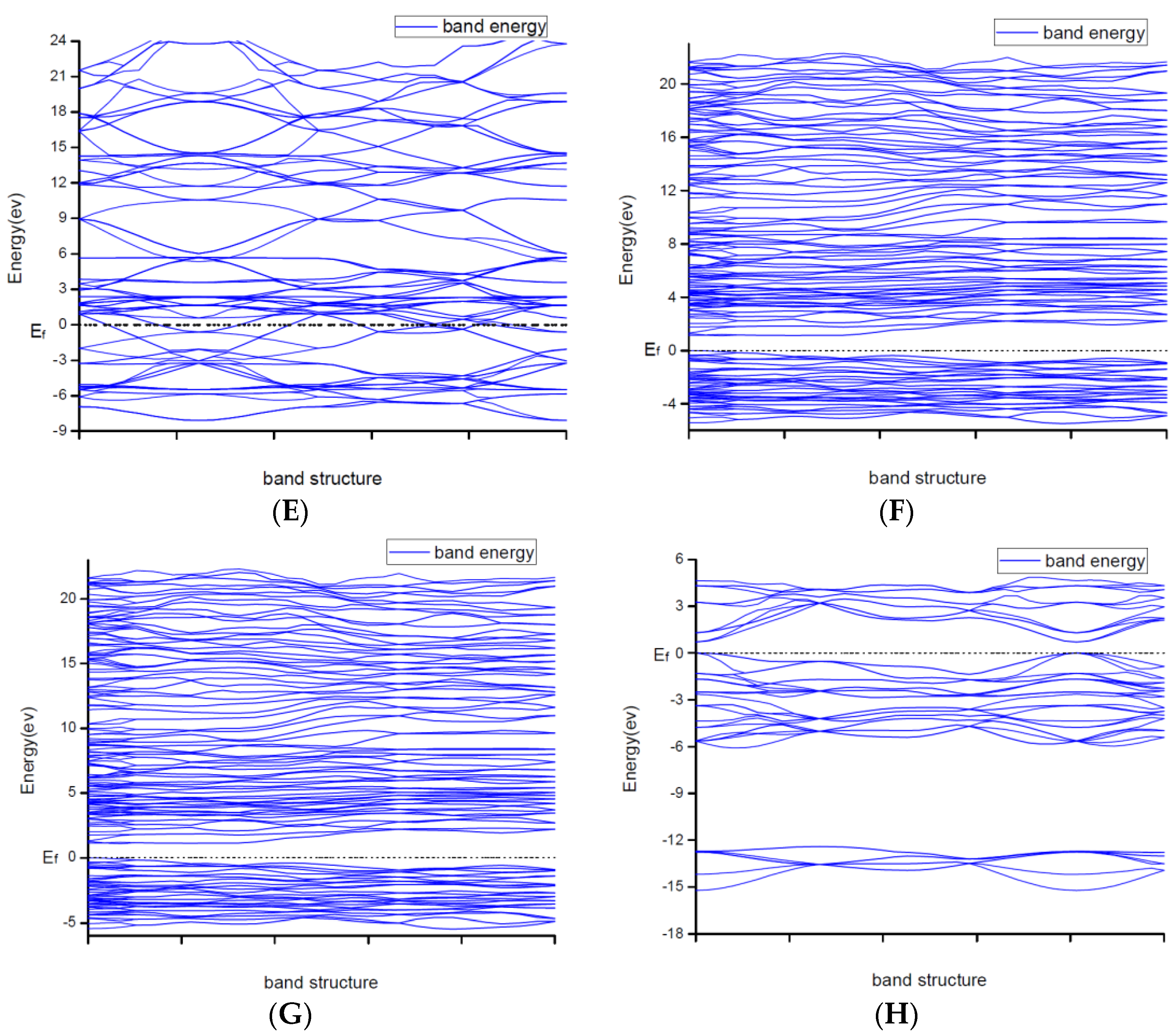
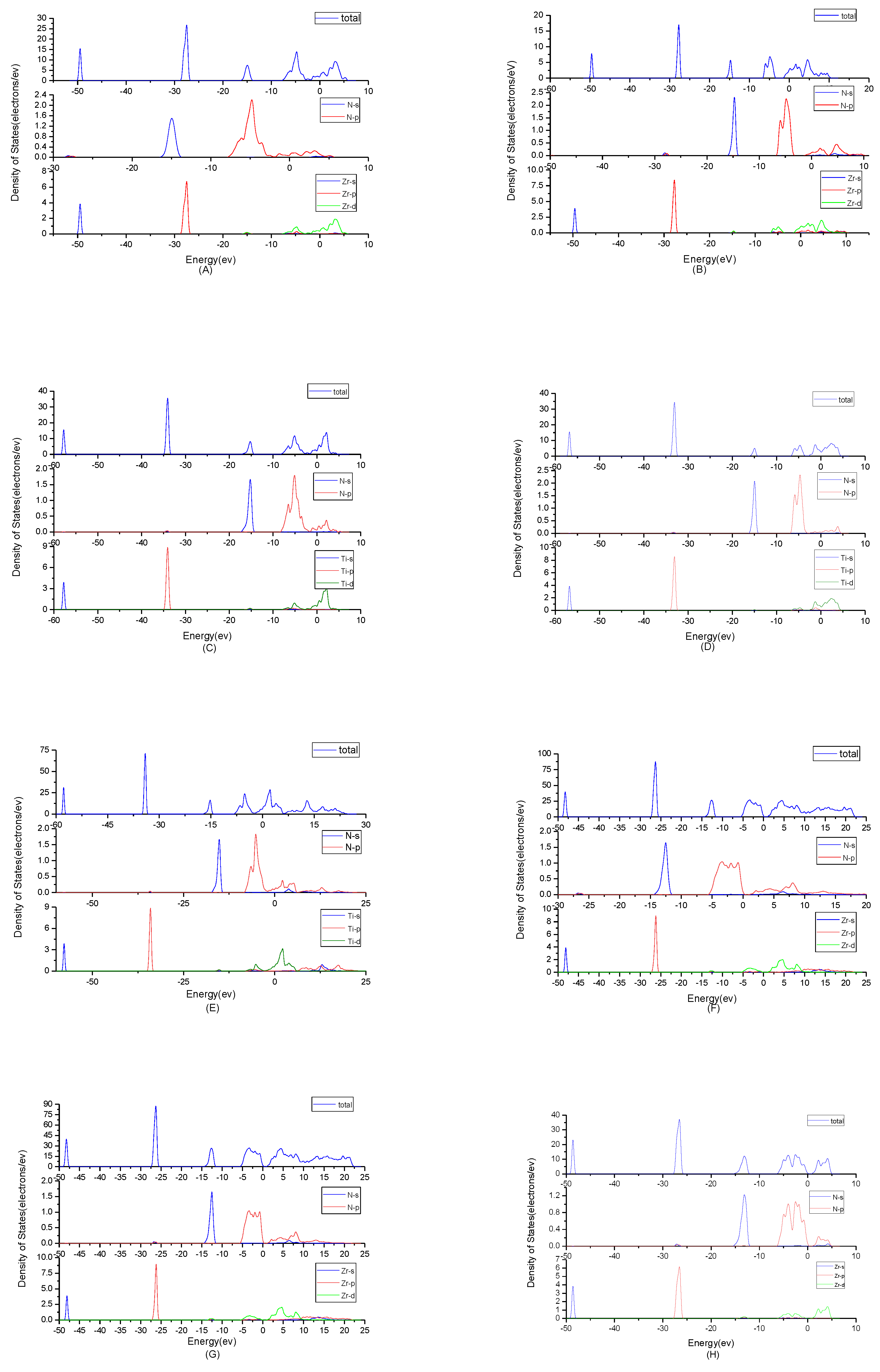
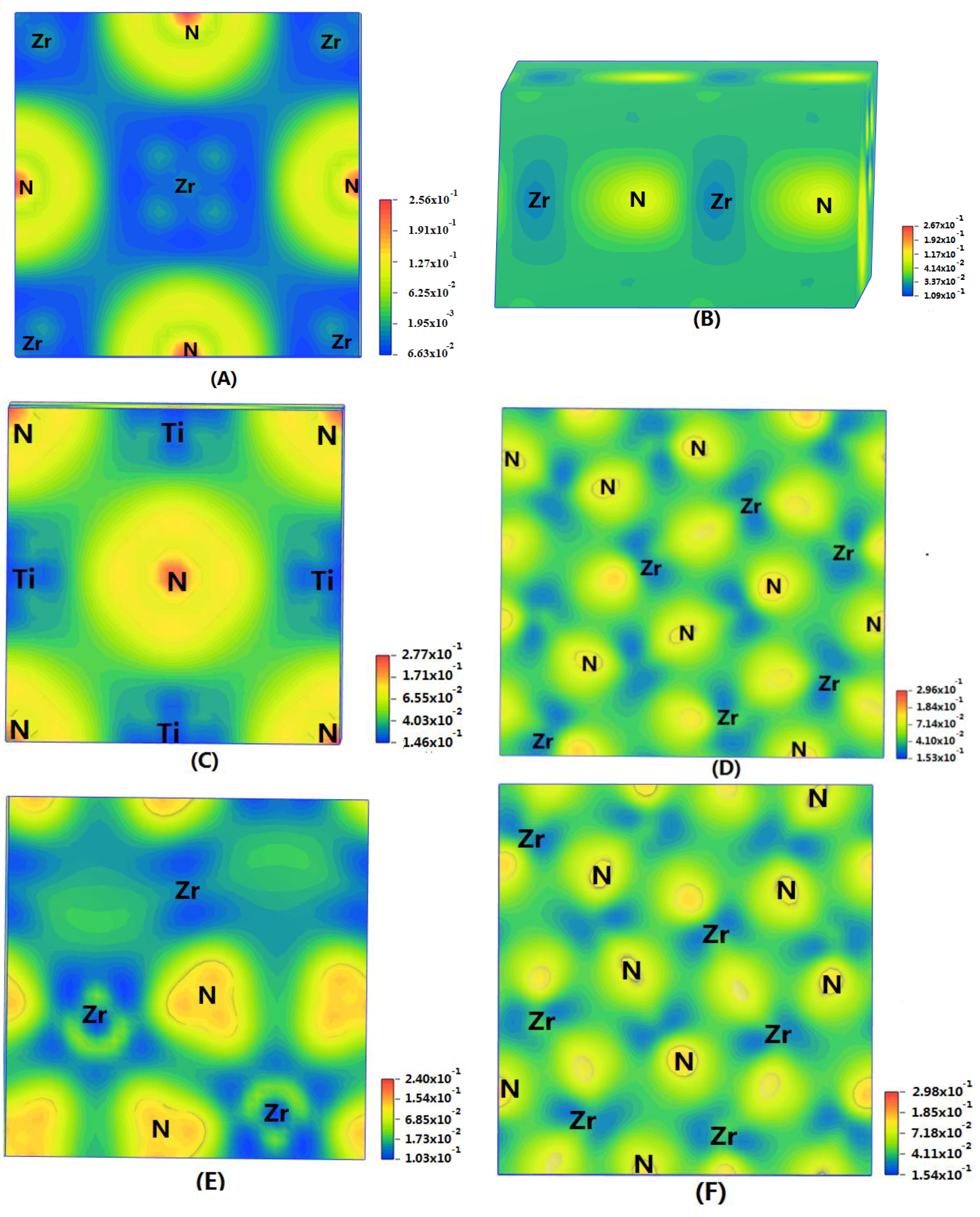
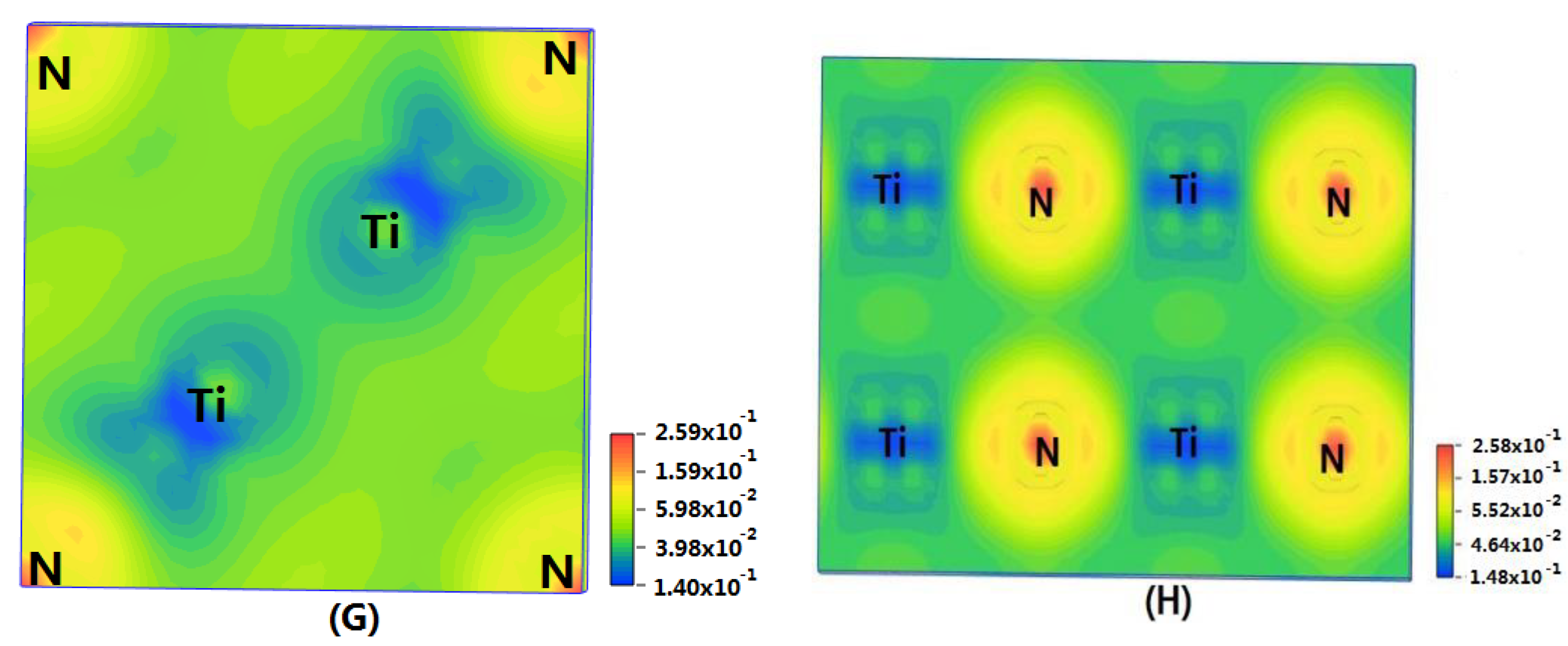
3.2. Result of the Electronic Properties
3.3. Superconducting Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Veprek, S.J. The search for novel, superhard materials. Vac. Sci. Technol. A 1999, 17, 2401. [Google Scholar] [CrossRef]
- Léger, J.M.; Haines, J. The search for superhard materials. Endeavour 1997, 21, 121–124. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Gregoryanz, E. Synthesis of superhard materials. Mater. Today 2005, 8, 44–51. [Google Scholar] [CrossRef]
- Kaner, R.B.; Gilman, J.J.; Tolbert, S.H. Designing superhard materials. Mater. Sci. 2005, 308, 1268. [Google Scholar]
- Richter, A.; Smith, R.; Dubrovinskaia, N.; Mcgee, E. Mechanical properties of superhard materials synthesised at various pressure-temperature conditions investigated by nanoindentation. High Press. Res. 2006, 26, 99–109. [Google Scholar] [CrossRef]
- Zalnezhad, E.; Sarhan, A.D.; Hamdi, M. Optimizing the PVD TiN thin film coating’s parameters on aerospace AL7075-T6 alloy for higher coating hardness and adhesion with better tribological properties of the coating surface. Int. J. Adv. Manuf. Technol. 2013, 64, 281–290. [Google Scholar] [CrossRef]
- Gardy, J.; Hassanpour, A.; Lai, X.; Ahmed, M.H. Synthesis of Ti(SO4)O solid acid nano-catalyst and its application for biodiesel production from used cooking oil. Appl. Catal. A Gen. 2016, 527, 81–95. [Google Scholar] [CrossRef]
- Siow, P.C.; Ghani, J.A.; Ghazali, M.J.; Jaafar, T.R.; Selamat, M.A.; Haron, C.H.C. Characterization of TiCN and TiCN/ZrN coatings for cutting tool application. Ceram. Inter. 2013, 39, 1293–1298. [Google Scholar] [CrossRef]
- Weht, R.; Filippetti, A.; Pickett, W.E. Electron doping in the honeycomb bilayer superconductors (Zr, Hf) NCl. Europhys. Lett. 2007, 48, 320–325. [Google Scholar] [CrossRef]
- Saha, B.; Acharya, J.; Sands, T.D.; Waghmare, U.V. Electronic structure, phonons, and thermal properties of ScN, ZrN, and HfN: A first-principles study. J Appl. Phys. 2010, 107, 960. [Google Scholar] [CrossRef]
- Guo, Q.X.; Kwan, W.K.; Cheng, X.L.; Zhang, H. First-principles study of the structural and electronic properties of the cubic Zr3N4, under high pressure. Phys. Status Solidi 2010, 247, 67–71. [Google Scholar] [CrossRef]
- Wang, A.J.; Shang, S.L.; Du, Y.; Kong, Y.; Zhang, L.J.; Chen, L.; Liu, Z.K. Structural and elastic properties of cubic and hexagonal TiN and AlN from first-principles calculations. Comput. Mater. Sci. 2010, 48, 705–709. [Google Scholar] [CrossRef]
- Ivashchenko, V.I.; Turchi, P.E.A.; Shevchenko, V.I.; Olifan, E.I. First-principles study of phase stability of Ti2N under pressure. Phys. Rev. B 2012, 86, 2602–2607. [Google Scholar] [CrossRef]
- Kim, J.; Jhi, S.H.; Ryeol, L.K. Color of TiN and ZrN from first-principles calculations. J. Appl. Phys. 2011, 110, 4889. [Google Scholar] [CrossRef]
- Brik, M.G.; Ma, C.G. First-principles studies of the electronic and elastic properties of metal nitrides XN (X = Sc, Ti, V, Cr, Zr, Nb). Comput. Mater. Sci. 2012, 51, 380–388. [Google Scholar] [CrossRef]
- Mattesini, M.; Ahuja, R.; Johansson, B. Cubic Hf3N4, and Zr3N4: A class of hard materials. Phys. Rev. B 2003, 68, 184108. [Google Scholar] [CrossRef]
- Kroll, P. Hafnium nitride with thorium phosphide structure: Physical properties and an assessment of the Hf-N, Zr-N, and Ti-N phase diagrams at high pressures and temperatures. Phys. Rev. Lett. 2003, 90, 125501. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Yin, G.; Li, J.; Zheng, Y.; Chen, L.; Jia, Y. Optical properties of cubic Ti3N4, Zr3N4, and Hf3N4. Appl. Phys. Lett. 2006, 89, 151908. [Google Scholar] [CrossRef]
- Xu, Y.X.; Chen, L.; Pei, F.; Chang, K.K.; Du, Y. Effect of the modulation ratio on the interface structure of TiAlN/TiN and TiAlN/ZrN multi-layers: First-principles and experimental investigations. Acta Mater. 2017, 130, 281–288. [Google Scholar] [CrossRef]
- Kobayashi, K. First-principles study of the electronic properties of transition metal nitride surfaces. Surf. Sci. 2001, 493, 665–670. [Google Scholar] [CrossRef]
- Gonze, X.; Beuken, J.M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.M.; Torrent, M. First-principles computation of material properties: The ABINIT software project. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Shang, S.L.; Wang, Y.; Kim, D.; Liu, Z.K. First-principles thermodynamics from phonon and Debye model: Application to Ni and Ni3Al. Comput. Mater. Sci. 2010, 47, 1040–1048. [Google Scholar] [CrossRef]
- Wang, B.T.; Shi, H.; Li, W.D.; Zhang, P. First-principles study of ground-state properties and high pressure behavior of ThO2. J. Nucl. Mater. 2010, 399, 181–188. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, A.J. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.; Léger, J.M.; Bocquillon, G. Synthesis and design of superhard materials. Annu. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Ghebouli, B.; Fatmi, M.; Ghebouli, M.A.; Choutri, H.; Louail, L.; Chihi, T.; Bin-Omran, S. Theoretical study of the structural, elastic, electronic and optical properties of XCaF3, (X = K and Rb). Solid State Sci. 2015, 43, 9–14. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Greaves, G.N.; Greer, A.L.; Lakes, R.S.; Rouxel, T. Poisson’s ratio and modern materials. Nat. Mater. 2011, 10, 823–837. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Space Group | a (Å) | b (Å) | c (Å) | V (Å3) | Cutoff Energy (eV) | K Point |
|---|---|---|---|---|---|---|---|
| TiN | Fmm | 4.25 | - | - | 76.77 | 750 | 20 × 20 × 20 |
| Ti2N | I41/amz | 4.149 | 4.149 | 8.786 | 74.20 | 600 | 8 × 8 × 13 |
| P42/mm | 4.945 | 4.945 | 3.034 | 151.26 | 680 | 9 × 9 × 4 | |
| ZrN | Fmm | 4.573 | - | - | 95.76 | 800 | 20 × 20 × 20 |
| P63mc | 3.128 | 3.128 | 5.013 | 42.49 | 720 | 22 × 22 × 6 | |
| Zr3N4 | I3d | 6.74 | - | - | 306.18 | 500 | 15 × 15 × 15 |
| Pna21 | 9.729 | 10.818 | 3.281 | 345.32 | 480 | 15 × 15 × 15 | |
| Pnam | 9.788 | 10.854 | 3.300 | 350.59 | 490 | 6 × 17 × 5 |
| Compound | Data Type | S.G | a (Å) | b (Å) | c (Å) |
|---|---|---|---|---|---|
| TiN | TW | Fmm | 4.246 | - | - |
| Exp. | Fmm | 4.250 | - | - | |
| Cal. | Fmm | 4.256 [12] | - | - | |
| Ti2N | TW | I41/amdz | 6.003 | 6.003 | 8.502 |
| TW | P42/mnm | 4.952 | 4.952 | 3.034 | |
| Cal. | P42/mnm | 4.928 [13] | 4.928 [13] | 3.021 [13] | |
| ZrN | TW | Fmm | 4.591 | - | - |
| Cal. | Fmm | 4.59 | - | - | |
| TW | P63mc | 3.564 [10] | 3.564 [10] | 5.538 [10] | |
| Zr3N4 | TW | I3d | 6.783 | - | - |
| Pna21 | 9.823 | 10.843 | 3.291 | ||
| Pnam | 9.814 | 10.840 | 3.294 | ||
| Exp. | I3d | 6.740 [11] | - | - |
| Compound | S.G | C11 | C12 | C44 |
|---|---|---|---|---|
| TiN | Fmm | 563.93 | 133.28 | 165.91 |
| ZrN | Fmm | 536.17 | 105.19 | 121.21 |
| Compound | S.G | C11 | C12 | C13 | C33 | C44 | C66 |
|---|---|---|---|---|---|---|---|
| Ti2N | I41/amdz | 512.26 | 188.64 | 120.05 | 568.49 | 166.22 | 231.47 |
| P42/mnm | 341.57 | 147.80 | 103.12 | 429.89 | 141.11 | 146.57 | |
| ZrN | P63mc | 219.22 | 162.29 | 165.69 | 120.14 | 51.24 | 28.46 |
| S.G | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|
| I3d | 332.4 | 119.7 | - | - | - | - | 64.91 | - | - |
| Pna21 | 211.4 | 126.1 | 125.0 | 383.9 | 116.2 | 425.5 | 56.44 | 95.7 | 132.8 |
| Pnam | 215.2 | 126.9 | 126.8 | 389.3 | 109.1 | 428.5 | 55.7 | 96.6 | 134.9 |
| Compound | S.G | B | G | E | ν | G/B |
|---|---|---|---|---|---|---|
| ZrN | Fmm | 248.85 | 152.92 | 380.77 | 0.245 | 0.615 |
| P63mc | 196.59 | −569.37 | - | - | - | |
| TiN | Fmm | 276.83 | 184.18 | 452.24 | 0.2277 | 0.665 |
| Ti2N | I41/amdz | 272.27 | 187.96 | 458.40 | 0.220 | 0.690 |
| P42/mnm | 202.12 | 134.43 | 330.11 | 0.228 | 0.665 | |
| Zr3N4 | I3d | 190.6 | 79.18 | 208.7 | 0.318 | 0.415 |
| Pna21 | 186.5 | 93.69 | 240.8 | 0.285 | 0.502 | |
| Pnam | 187.6 | 94.74 | 243.3 | 0.284 | 0.505 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Yuan, X. Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations. Materials 2018, 11, 1640. https://doi.org/10.3390/ma11091640
Ji Y, Yuan X. Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations. Materials. 2018; 11(9):1640. https://doi.org/10.3390/ma11091640
Chicago/Turabian StyleJi, Yangqi, and Xiaoli Yuan. 2018. "Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations" Materials 11, no. 9: 1640. https://doi.org/10.3390/ma11091640
APA StyleJi, Y., & Yuan, X. (2018). Elastic Properties and Electronic Properties of MxNy (M = Ti, Zr) from First Principles Calculations. Materials, 11(9), 1640. https://doi.org/10.3390/ma11091640