Towards Developing Bioresponsive, Self-Assembled Peptide Materials: Dynamic Morphology and Fractal Nature of Nanostructured Matrices

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Statement of Significance
2. Introduction
3. Results
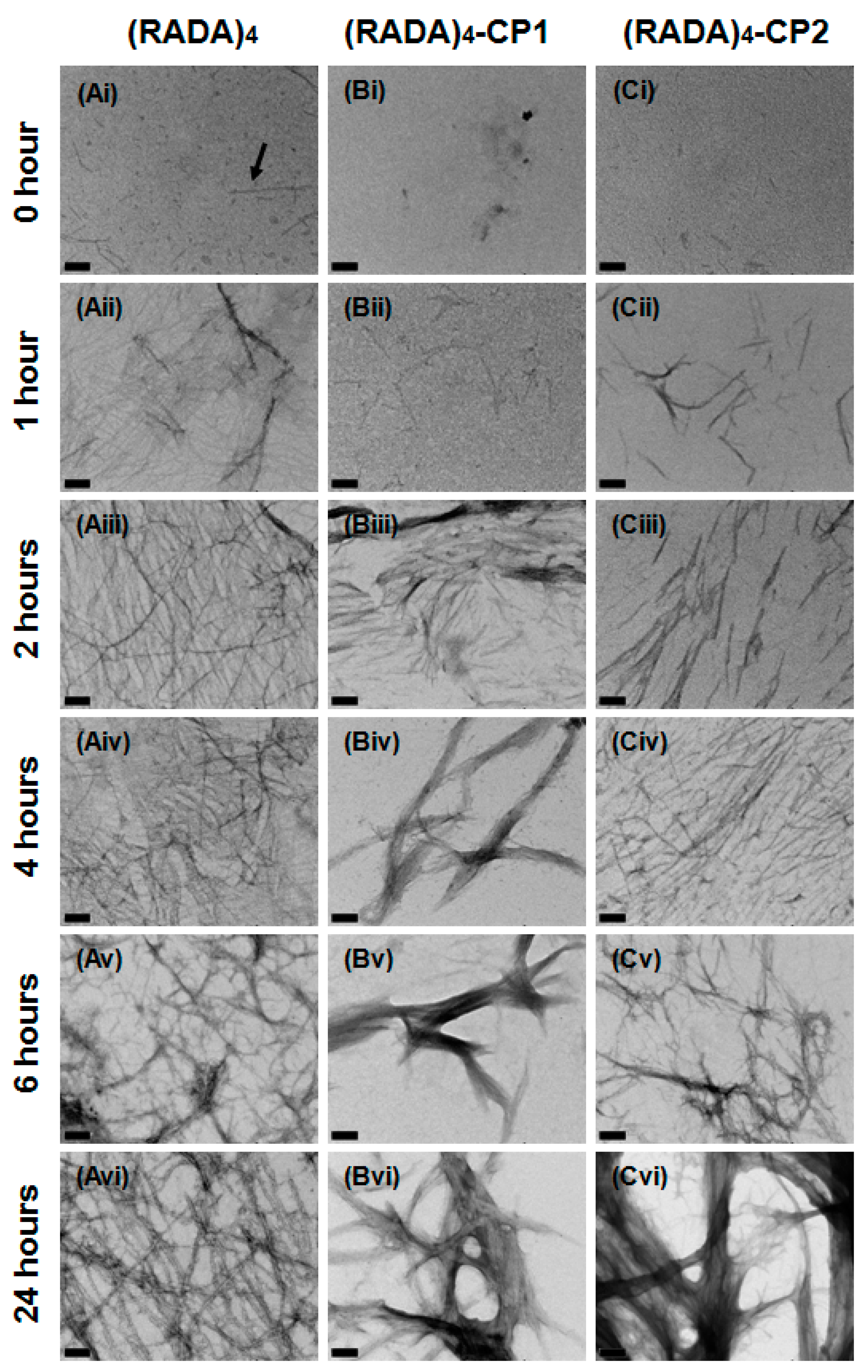
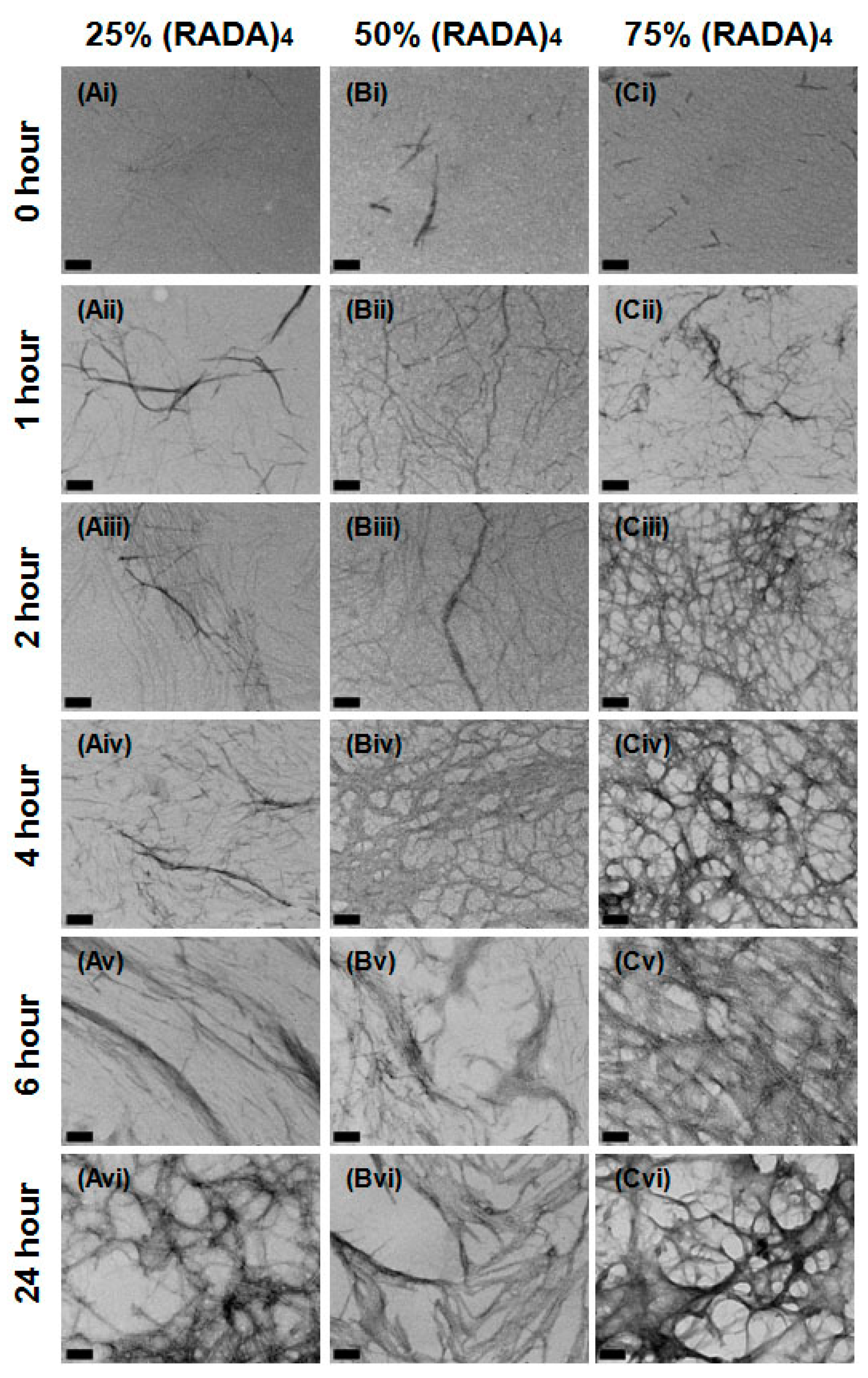
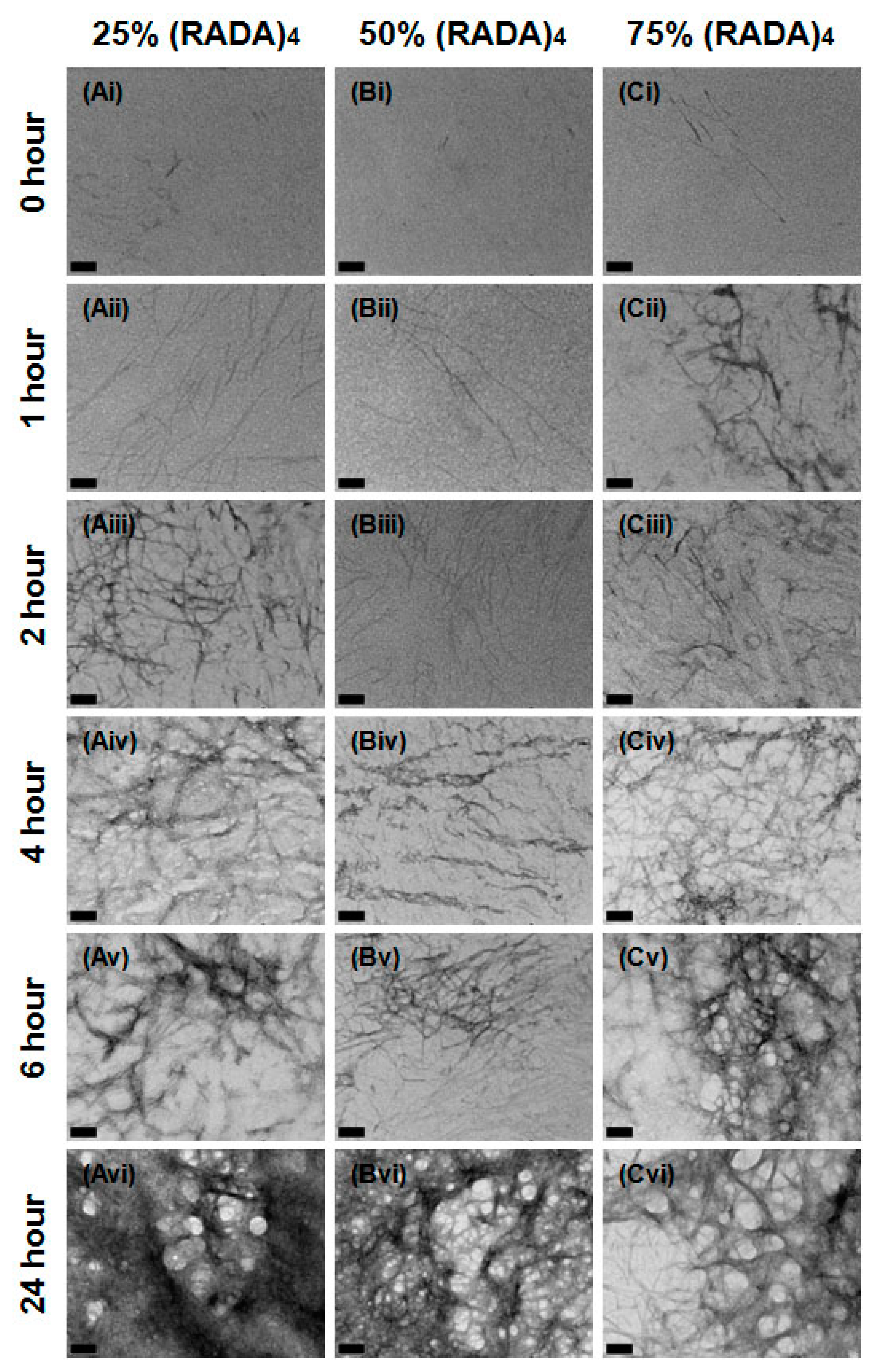
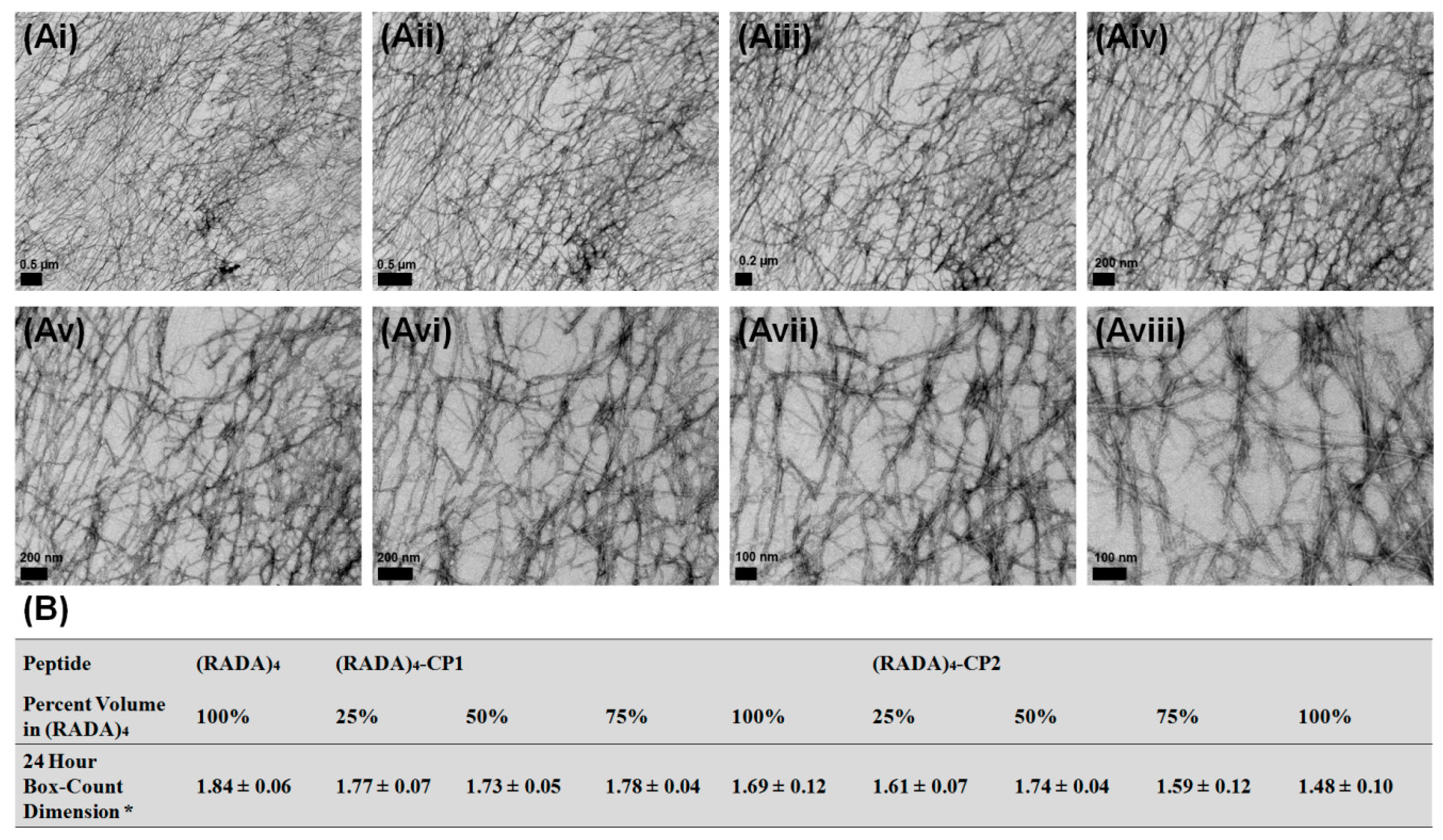
3.1. Nanoscale Morphology
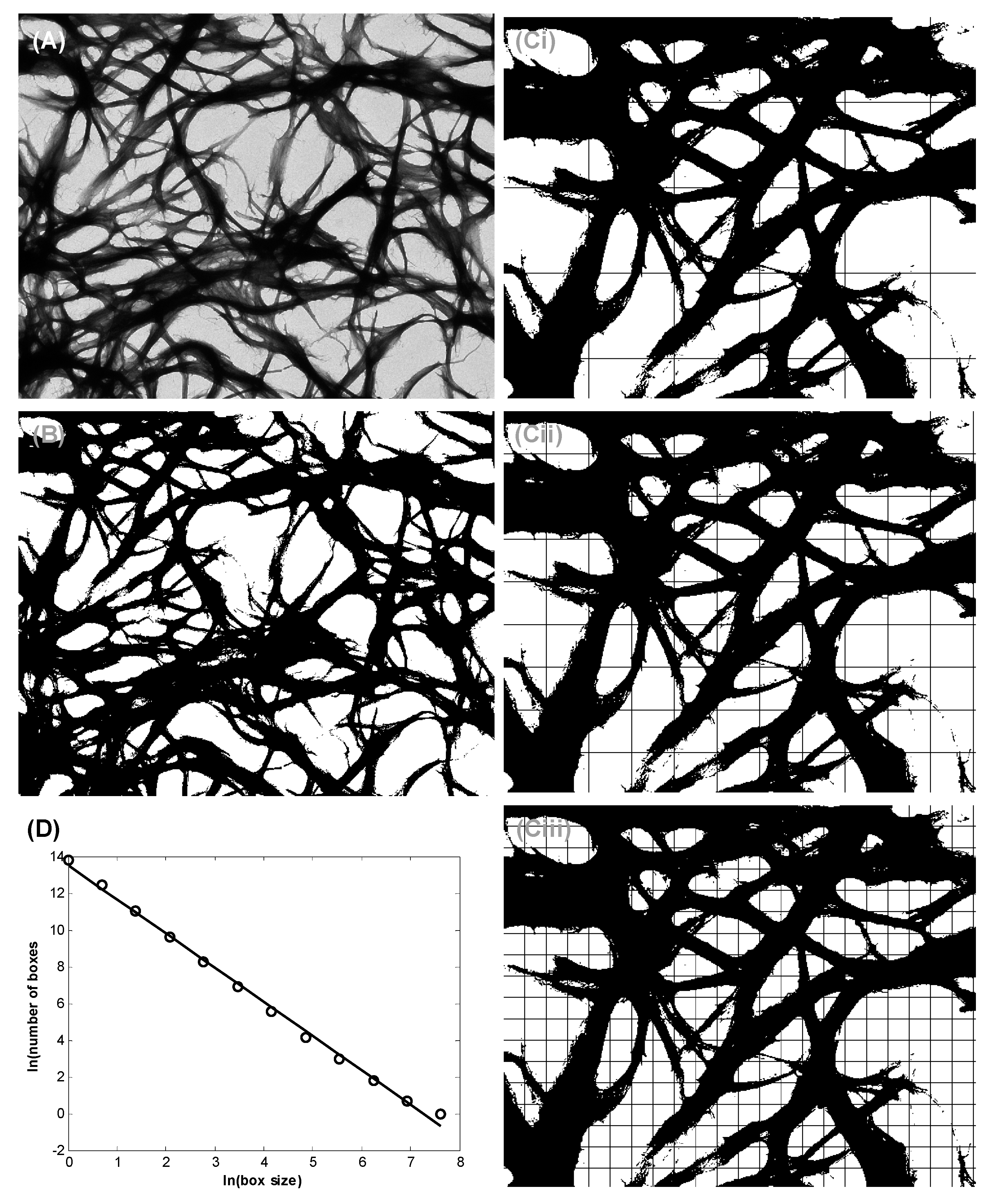
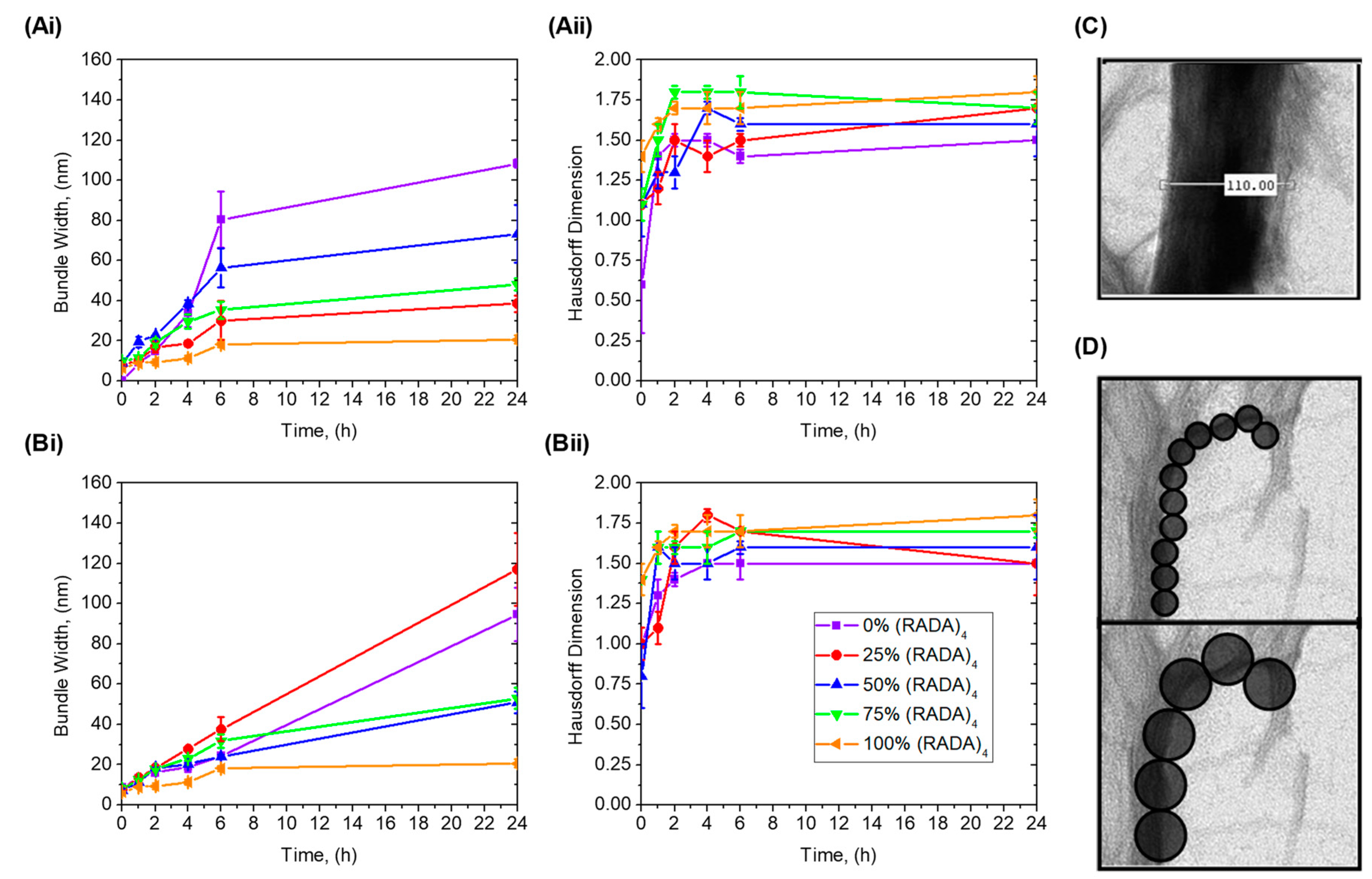
3.2. Bundle Thickness and Fractal Dimension Analysis
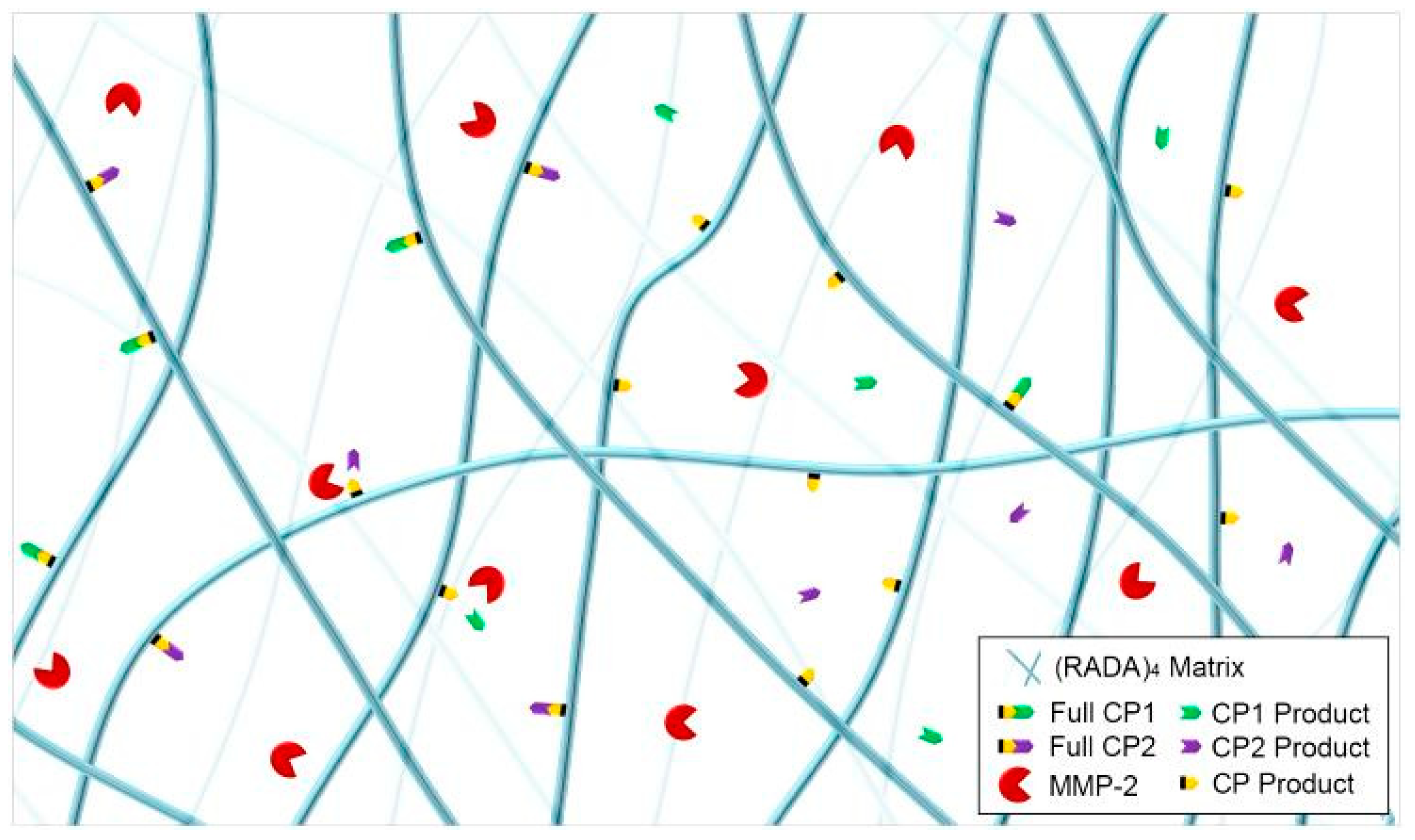
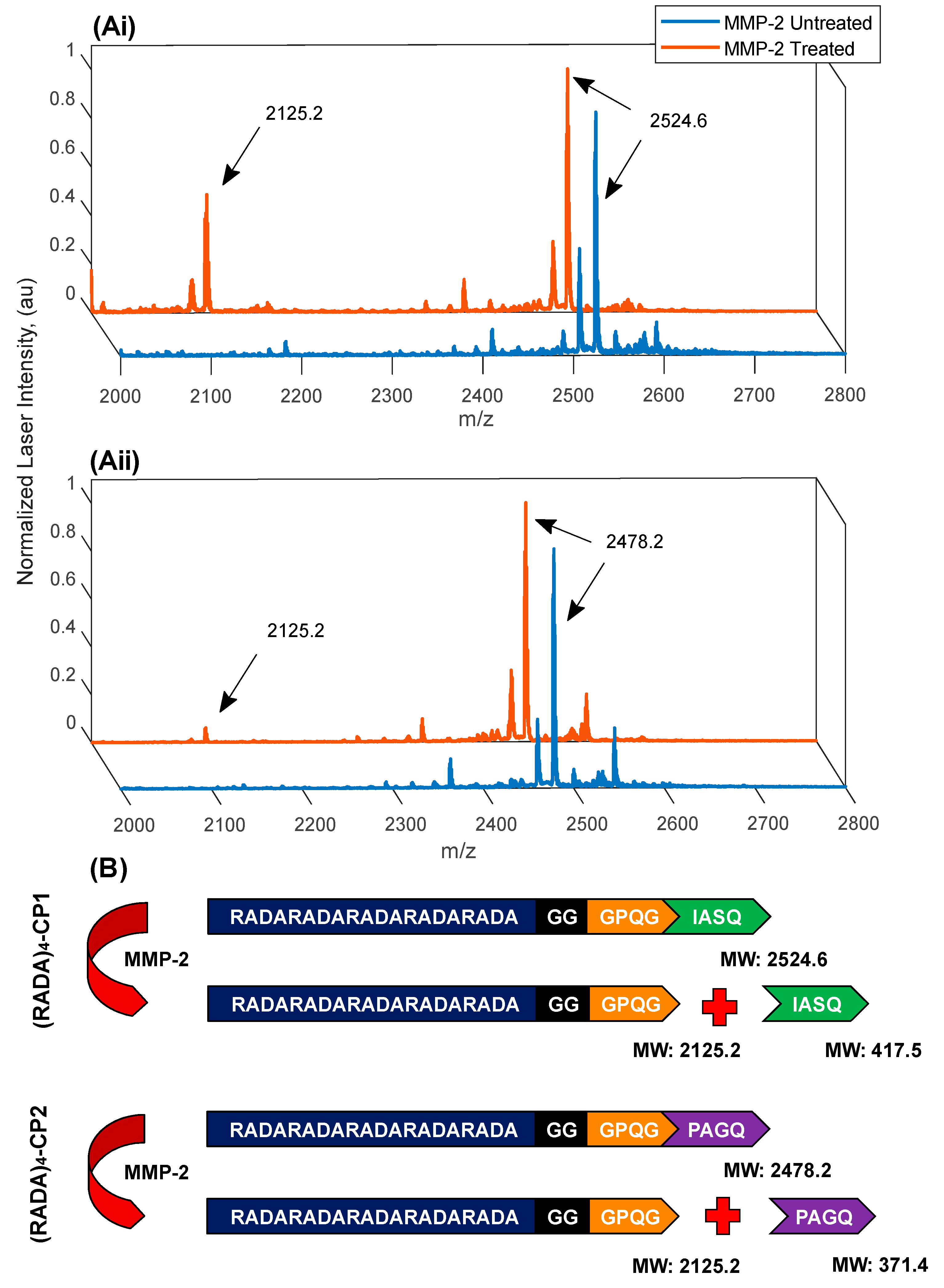
3.3. MMP-2 Induced Peptide Cleavage
4. Discussion
5. Materials and Methods
5.1. Materials
5.2. Peptide Synthesis
5.3. MMP-2 Enzymatic Treatment
5.4. Scanning Electron Microscopy
5.5. Transmission Electron Microscopy
5.6. Nanofiber Bundle and Hausdorff Dimension Analyses
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uhlig, T.; Kyprianou, T.; Martinelli, F.G.; Oppici, C.A.; Heiligers, D.; Hills, D.; Calvo, X.R.; Verhaert, P. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPA Open Proteom. 2014, 4, 58–69. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Vashist, A.; Ahmad, S. Hydrogels: Smart materials for drug delivery. Orient. J. Chem. 2013, 29, 861–870. [Google Scholar] [CrossRef]
- Sharpe, L.A.; Daily, A.M.; Horava, S.D.; Peppas, N.A. Therapeutic applications of hydrogels in oral drug delivery. Expert Opin. Drug Deliv. 2014, 11, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Cascone, S.; Lamberti, G.; AngelaBarba, A.; Larsson, A. Swellable Hydrogel-based Systems for Controlled Drug Delivery. In Smart Drug Delivery System; InTech Open Limited: London, UK, 2016. [Google Scholar]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1879. [Google Scholar] [CrossRef] [PubMed]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Anderson, D.G.; Levenberg, S.; Langer, R. Nanoliter-scale synthesis of arrayed biomaterials and application to human embryonic stem cells. Nat. Biotechnol. 2004, 22, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Flaim, C.J.; Chien, S.; Bhatia, S.N. An extracellular matrix microarray for probing cellular differentiation. Nat. Methods 2005, 2, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- MacPhee, C.E.; Woolfson, D.N. Engineered and designed peptide-based fibrous biomaterials. Curr. Opin. Solid State Mater. Sci. 2004, 8, 141–149. [Google Scholar] [CrossRef]
- Stupp, S.I. Biomaterials for Regenerative Medicine. MRS Bull. 2005, 30, 546–553. [Google Scholar] [CrossRef]
- Holmes, T.C. Novel peptide-based biomaterial scaffolds for tissue engineering. Trends Biotechnol. 2002, 20, 16–21. [Google Scholar] [CrossRef]
- Gelain, F.; Unsworth, L.D.; Zhang, S. Slow and sustained release of active cytokines from self-assembling peptide scaffolds. J. Controll. Release 2010, 145, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kisiday, J.; Jin, M.; Kurz, B.; Hung, H.; Semino, C.; Zhang, S.; Grodzinsky, A.J. Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: Implications for cartilage tissue repair. Proc. Natl. Acad. Sci. USA 2002, 99, 9996–10001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Holmes, T.C.; DiPersio, C.M.; Hynes, R.O.; Su, X.; Rich, A. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials 1995, 16, 1385–1393. [Google Scholar] [CrossRef]
- Holmes, T.C.; de Lacalle, S.; Su, X.; Liu, G.; Rich, A.; Zhang, S. Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc. Natl. Acad. Sci. USA 2000, 97, 6728–6733. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S.; Zhang, S. Long-term three-dimensional neural tissue cultures in functionalized self-assembling peptide hydrogels, matrigel and collagen I. Acta Biomater. 2013, 9, 5162–5169. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.; Luo, Y.; Cheung, A.C.Y.; Nagai, Y.; Zhang, S.; Kobler, J.B.; Zeitels, S.M.; Langer, R. Incorporation of a matrix metalloproteinase-sensitive substrate into self-assembling peptides—A model for biofunctional scaffolds. Biomaterials 2008, 29, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Hauser, C.A.E.; Zhang, S. Designer self-assembling peptide nanofiber biological materials. Chem. Soc. Rev. 2010, 39, 2780–2790. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Englander, O.; Paravastu, A.; Oates, W.S. An effective continuum approach for modeling non-equilibrium structural evolution of protein nanofiber networks. J. Chem. Phys. 2011, 135, 055102. [Google Scholar] [CrossRef] [PubMed]
- Law, B.; Tung, C.-H. Proteolysis: A biological process adapted in drug delivery, therapy, and imaging. Bioconjug. Chem. 2009, 20, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.E.; Huang, L.L.; Piro, E.T.; Cantley, L.C. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 2001, 19, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Fields, G.B. Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Peptide Sci. 1996, 40, 399–416. [Google Scholar] [CrossRef]
- Netzel-Arnett, S.; Sang, Q.X.; Moore, W.G.I.; Navre, M.; Birkedal-Hansen, H.; Van Wart, H.E. Comparative sequence specificities of human 72- and 92-kDa gelatinases (type IV collagenases) and PUMP (matrilysin). Biochemistry 1993, 32, 6427–6432. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, S.G.; Shoichet, M.S. Synthesis of enzyme-degradable, peptide-cross-linked dextran hydrogels. Bioconjug. Chem. 2007, 18, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Fatin-Rouge, N.; Starchev, K.; Buffle, J. Size effects on diffusion processes within agarose gels. Biophys. J. 2004, 86, 2710–2719. [Google Scholar] [CrossRef]
- Bancaud, A.; Huet, S.; Daigle, N.; Mozziconacci, J.; Beaudouin, J.; Ellenberg, J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009, 28, 3785–3798. [Google Scholar] [CrossRef] [PubMed]
- Montague, P.R.; Friedlander, M.J. Expression of an intrinsic growth strategy by mammalian retinal neurons. Proc. Natl. Acad. Sci. USA 1989, 86, 7223–7227. [Google Scholar] [CrossRef] [PubMed]
- Santoso, S.; Hwang, W.; Hartman, H.; Zhang, S. Self-assembly of Surfactant-like Peptides with Variable Glycine Tails to Form Nanotubes and Nanovesicles. Nano Lett. 2002, 2, 687–691. [Google Scholar] [CrossRef]
- Von Maltzahn, G.; Vauthey, S.; Santoso, S.; Zhang, S. Positively Charged Surfactant-like Peptides Self-assemble into Nanostructures. Langmuir 2003, 19, 4332–4337. [Google Scholar] [CrossRef]
- Lomander, A.; Hwang, W.; Zhang, S. Hierarchical Self-Assembly of a Coiled-Coil Peptide into Fractal Structure. Nano Lett. 2005, 5, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Hochman-Mendez, C.; Cantini, M.; Moratal, D.; Salmeron-Sanchez, M.; Coelho-Sampaio, T. A Fractal Nature for Polymerized Laminin. PLoS ONE 2014, 9, e109388. [Google Scholar] [CrossRef] [PubMed]
- Taraballi, F.; Natalello, A.; Campione, M.; Villa, O.; Doglia, S.M.; Paleari, A.; Gelain, F. Glycine-spacers influence functional motifs exposure and self-assembling propensity of functionalized substrates tailored for neural stem cell cultures. Front. Neuroeng. 2010, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, M.; Bushnak, I.; McDermot, M.T.; Unsworth, L.D. Toward a Mechanistic Understanding of Ionic Self-Complementary Peptide Self-Assembly: Role of Water Molecules and Ions. Biomacromolecules 2013, 14, 3943–3950. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.E.; Sarkar, A.; Wampler, J.E. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 1990, 214, 253–260. [Google Scholar] [CrossRef]
- Pal, D.; Chakrabarti, P. Cis peptide bonds in proteins: Residues involved, their conformations, interactions and locations. J. Mol. Biol. 1999, 294, 271–288. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, M.W.; Thornton, J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991, 218, 397–412. [Google Scholar] [CrossRef]
- Kogan, M.J.; Dalcol, I.; Gorostiza, P.; Lopez-Iglesias, C.; Pons, R.; Pons, M.; Sanz, F.; Giralt, E. Supramolecular Properties of the Proline-Rich γ-Zein N-Terminal Domain. Biophys. J. 2002, 83, 1194–1204. [Google Scholar] [CrossRef]
- Creasey, R.G.; Voelcker, N.H.; Schultz, C.J. Investigation of self-assembling proline- and glycine-rich recombinant proteins and peptides inspired by proteins from a symbiotic fungus using atomic force microscopy and circular dichroism spectroscopy. Biochim. Biophys. Acta 2012, 1824, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carneado, J.; Kogan, M.J.; Castel, S.; Giralt, E. Potential Peptide Carriers: Amphipathic Proline-Rich Peptides Derived from the N-Terminal Domain of γ-Zein. Angew. Chem. Int. Ed. 2004, 43, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo-Rodríguez, A.; Pujals, S.; Delgado, D.; Solinís, M.A.; Gascón, A.R.; Giralt, E.; Pedraz, J.L. A proline-rich peptide improves cell transfection of solid lipid nanoparticle-based non-viral vectors. J. Control. Release 2009, 133, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, H.; Elston, N.; Zietsch, B. Fractal analysis: Pitfalls and revelations in neuroscience. In Fractals in Biology and Medicine; Birkhäuser: Basel, Switzerland, 2005. [Google Scholar]
- ABI 433A Peptide Synthesizer; User Guide; Applied Biosystems: Waltham, MA, USA, 2004.
- Dawes, C.J. Biological Techniques for Transmission and Scanning Electron Microscopy, 2nd ed.; Ladd Research Industries: Williston, VT, USA, 1979. [Google Scholar]
- Barroso, M.M.S.; Freire, E.; Limaverde, G.S.; Rocha, G.M.; Batista, E.J.; Weissmüller, G.; Andrade, L.R.; Coelho-Sampaio, T. Artificial Laminin Polymers Assembled in Acidic pH Mimic Basement Membrane Organization. J. Biol. Chem. 2008, 283, 11714–11720. [Google Scholar] [CrossRef] [PubMed]
- French, P. Hausdorff Dimension by the Box Counting Method. Available online: http://www.mathworks.com/matlabcentral/fileexchange/15918-hausdorff-dimension-by-the-box-counting-method (accessed on 30 January 2018).
- Costa, A. Hausdorff (Box-Counting) Fractal Dimension. Available online: http://www.mathworks.com/matlabcentral/fileexchange/30329-hausdorff--box-counting--fractal-dimension (accessed on 30 January 2018).
- Hausdorff, F. Dimension und äuβeres Maβ. Math. Ann. 1919, 79, 157–159. [Google Scholar] [CrossRef]
- Lopes, R.; Betrouni, N. Fractal and multifractal analysis: A review. Med. Image Anal. 2009, 13, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Theiler, J. Estimating fractal dimension. JOSA A 1990, 7, 1055–1073. [Google Scholar] [CrossRef]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koss, K.M.; Unsworth, L.D. Towards Developing Bioresponsive, Self-Assembled Peptide Materials: Dynamic Morphology and Fractal Nature of Nanostructured Matrices. Materials 2018, 11, 1539. https://doi.org/10.3390/ma11091539
Koss KM, Unsworth LD. Towards Developing Bioresponsive, Self-Assembled Peptide Materials: Dynamic Morphology and Fractal Nature of Nanostructured Matrices. Materials. 2018; 11(9):1539. https://doi.org/10.3390/ma11091539
Chicago/Turabian StyleKoss, Kyle M., and Larry D. Unsworth. 2018. "Towards Developing Bioresponsive, Self-Assembled Peptide Materials: Dynamic Morphology and Fractal Nature of Nanostructured Matrices" Materials 11, no. 9: 1539. https://doi.org/10.3390/ma11091539
APA StyleKoss, K. M., & Unsworth, L. D. (2018). Towards Developing Bioresponsive, Self-Assembled Peptide Materials: Dynamic Morphology and Fractal Nature of Nanostructured Matrices. Materials, 11(9), 1539. https://doi.org/10.3390/ma11091539