Surface-Attached Poly(oxanorbornene) Hydrogels with Antimicrobial and Protein-Repellent Moieties: The Quest for Simultaneous Dual Activity


Abstract
1. Introduction
2. Experiment
2.1. Materials
2.2. Instrumentation.
2.3. Synthesis
3. Results and Discussion
3.1. Material Design
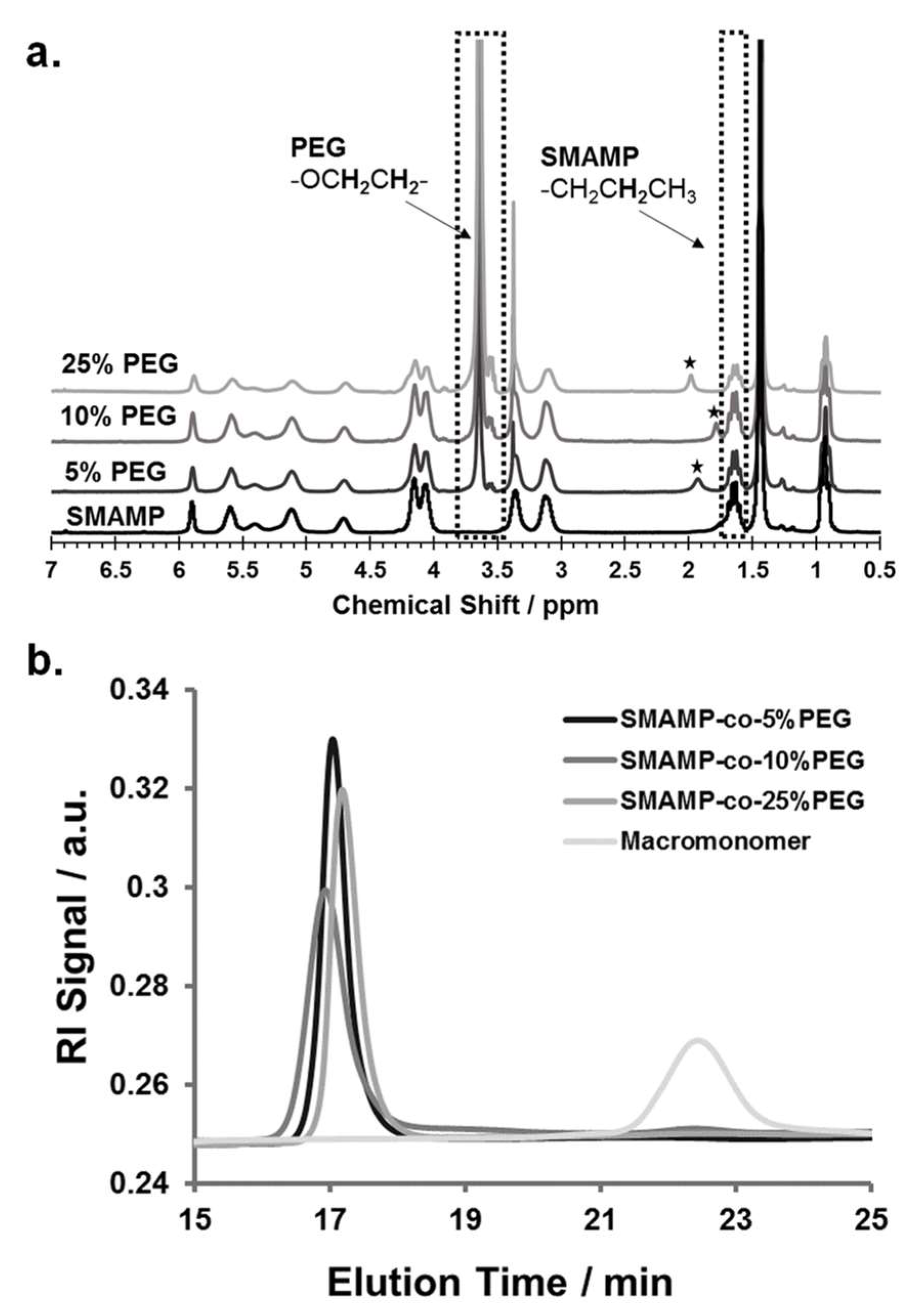
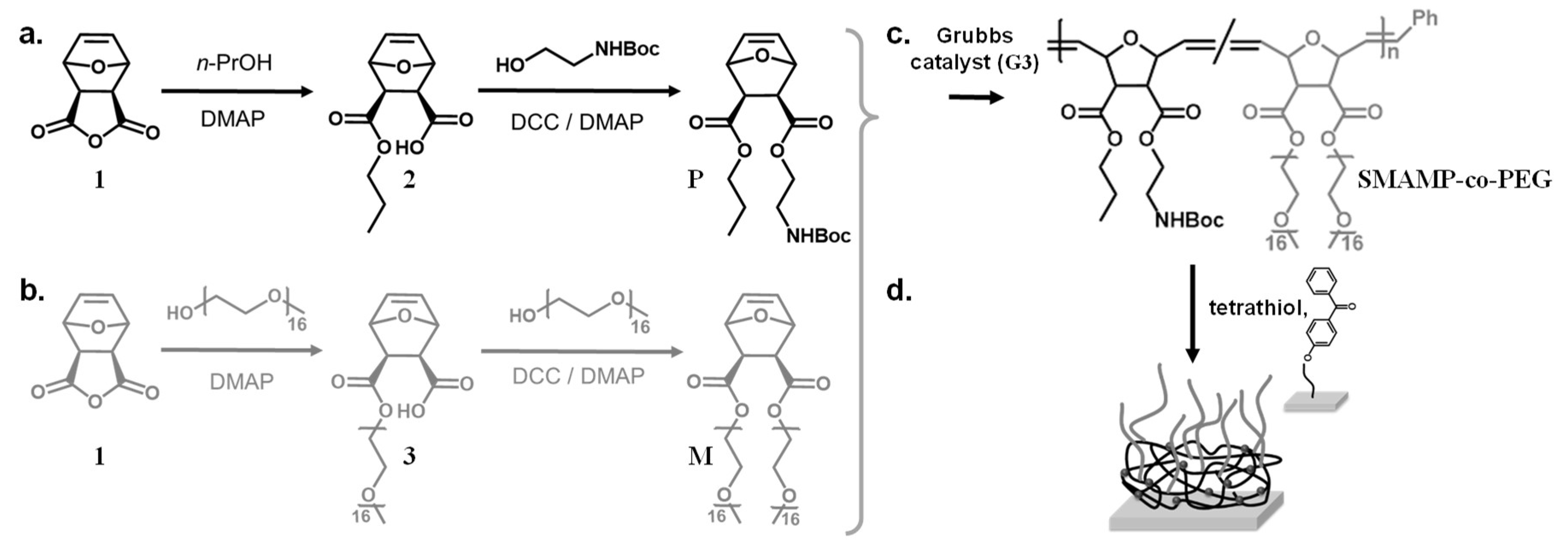
3.2. Monomer Synthesis
3.3. Polymer Synthesis
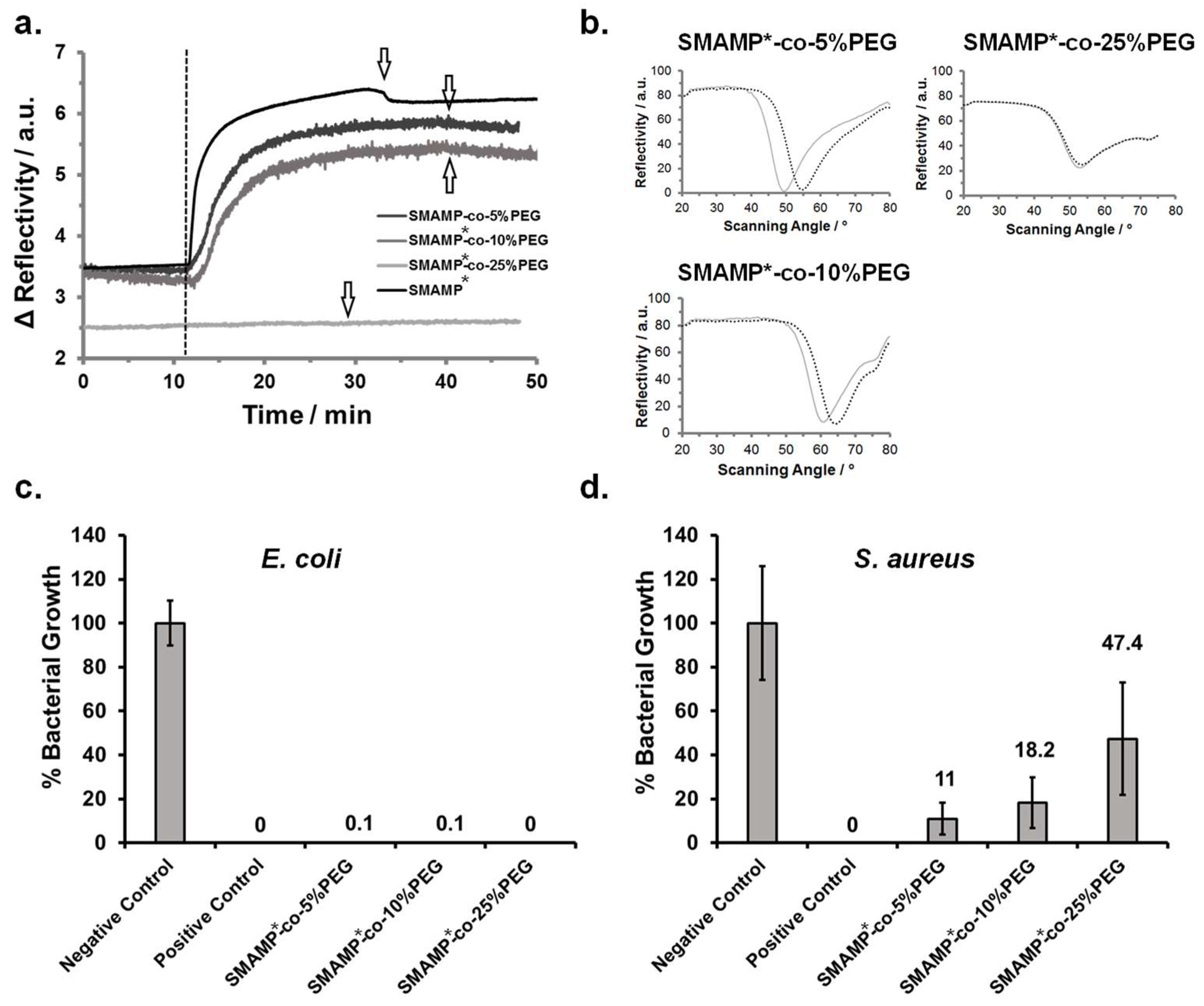
3.4. Synthesis and Physical Characterization of Surface-Attached Polymer Networks.
3.5. Protein Adhesion and Antimicrobial Activity of Surface-Attached Polymer Networks.
3.6. Discussion
4. Conclusion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Tackling drug-resistant infections globally: Final report and recommendations. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 19 May 2016).
- Tiller, J.C. Antimicrobial surfaces. Adv. Polym. Sci. 2011, 240, 193–217. [Google Scholar]
- Timofeeva, L.; Kleshcheva, N. Antimicrobial polymers: Mechanism of action, factors of activity, and applications. Appl. Microbiol. Biotechnol. 2011, 89, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Afacan, N.J.; Yeung, A.T.Y.; Pena, O.M.; Hancock, R.E.W. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr. Pharm. Des. 2012, 18, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Bazaka, K.; Jacob, M.V.; Crawford, R.J.; Ivanova, E.P. Efficient surface modification of biomaterial to prevent biofilm formation and the attachment of microorganisms. Appl. Microbiol. Biotechnol. 2012, 95, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.C.; Wiradharma, N.; Ong, Z.Y.; Coady, D.J.; Hedrick, J.L.; Yang, Y.Y. Emerging trends in macromolecular antimicrobials to fight multi-drug-resistant infections. Nano Today 2012, 7, 201–222. [Google Scholar] [CrossRef]
- Munoz-Bonilla, A.; Fernandez-Garcia, M. Polymeric materials with antimicrobial activity. Prog. Polym. Sci. 2012, 37, 281–339. [Google Scholar] [CrossRef]
- Siedenbiedel, F.; Tiller, J.C. Antimicrobial polymers in solution and on surfaces: Overview and functional principles. Polymers 2012, 4, 46–71. [Google Scholar] [CrossRef]
- Wessels, S.; Ingmer, H. Modes of action of three disinfectant active substances: A review. Regul. Toxicol. Pharmacol. 2013, 67, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Armentano, I.; Fortunati, E.; Mattioli, S.; Arciola, C.R.; Ferrari, D.; Amoroso, C.F.; Rizzo, J.; Kenny, J.M.; Imbriani, M.; Visai, L. The interaction of bacteria with engineered nanostructured polymeric materials: A review. Sci. World. J. 2014, 2014, 410423. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Jiang, S. Integrated antimicrobial and nonfouling zwitterionic polymers. Angew. Chem. Int. Ed. 2014, 53, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Salwiczek, M.; Qu, Y.; Gardiner, J.; Strugnell, R.A.; Lithgow, T.; McLean, K.M.; Thissen, H. Emerging rules for effective antimicrobial coatings. Trends Biotechnol. 2014, 32, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Swartjes, J.J.T.M.; Sharma, P.K.; van Kooten, T.G.; van der Mei, H.C.; Mahmoudi, M.; Busscher, H.J.; Rochford, E.T.J. Current developments in antimicrobial surface coatings for biomedical applications. Curr. Med. Chem. 2015, 22, 2116–2129. [Google Scholar] [CrossRef] [PubMed]
- Ganewatta, M.S.; Tang, C. Controlling macromolecular structures towards effective antimicrobial polymers. Polymer 2015, 63, A1–A29. [Google Scholar] [CrossRef]
- Hasan, J.; Crawford, R.J.; Ivanova, E.P. Antibacterial surfaces: The quest for a new generation of biomaterials. Trends Biotechnol. 2013, 31, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.; O'Brien-Simpson, N.M.; Connal, L.A. Antibiofouling polymer interfaces: Poly(ethylene glycol) and other promising candidates. Polym. Chem. 2015, 6, 198–212. [Google Scholar] [CrossRef]
- Campoccia, D.; Montanaro, L.; Arciola, C.R. A review of the biomaterials technologies for infection-resistant surfaces. Biomaterials 2013, 34, 8533–8554. [Google Scholar] [CrossRef] [PubMed]
- Lejars, M.; Margaillan, A.; Bressy, C. Fouling release coatings: A nontoxic alternative to biocidal antifouling coatings. Chem. Rev. 2012, 112, 4347–4390. [Google Scholar] [CrossRef] [PubMed]
- Tiller, J.C.; Liao, C.J.; Lewis, K.; Klibanov, A.M. Designing surfaces that kill bacteria on contact. Proc. Natl. Acad. Sci. USA 2001, 98, 5981–5985. [Google Scholar] [CrossRef] [PubMed]
- Kugler, R.; Bouloussa, O.; Rondelez, F. Evidence of a charge-density threshold for optimum efficiency of biocidal cationic surfaces. Microbiology 2005, 151, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Asri, L.A.T.W.; Crismaru, M.; Roest, S.; Chen, Y.; Ivashenko, O.; Rudolf, P.; Tiller, J.C.; van der Mei, H.C.; Loontjens, T.J.A.; Busscher, H.J. A shape-adaptive, antibacterial-coating of immobilized quaternary-ammonium compounds tethered on hyperbranched polyurea and its mechanism of action. Adv. Funct. Mater. 2014, 24, 346–355. [Google Scholar] [CrossRef]
- Riga, E.K.; Vöhringer, M.; Widyaya, V.T.; Lienkamp, K. Polymer-based surfaces designed to reduce biofilm formation: From antimicrobial polymers to strategies for long-term applications. Macromol. Rapid Commun. 2017, 38, 1700216. [Google Scholar] [CrossRef] [PubMed]
- Hartleb, W.; Saar, J.S.; Zou, P.; Lienkamp, K. Just antimicrobial is not enough: Toward bifunctional polymer surfaces with dual antimicrobial and protein-repellent functionality. Macromol. Chem. Phys. 2016, 217, 225–231. [Google Scholar] [CrossRef]
- Kurowska, M.; Eickenscheidt, A.; Guevara-Solarte, D.L.; Widyaya, V.T.; Marx, F.; Al-Ahmad, A.; Lienkamp, K. A simultaneously antimicrobial, protein-repellent and cell-compatible polyzwitterion network. Biomacromolecules 2017, 18, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wu, Z.; Chen, H. Dual-function antibacterial surfaces for biomedical applications. Acta Biomater. 2015, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.B.; Seyer, D.; Jouenne, T.; Thébault, P. Elaboration of antibacterial plastic surfaces by a combination of antiadhesive and biocidal coatings of natural products. Colloids Surf. B 2017, 156, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.J.; Cai, T.; Neoh, K.-G.; Kang, E.T.; Dickinson, G.H.; Teo, S.L.M.; Rittschof, D. Biomimetic anchors for antifouling and antibacterial polymer brushes on stainless steel. Langmuir 2011, 27, 7065–7076. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Lee, J.; Perreault, F.; Elimelech, M. Controlled architecture of dual-functional block copolymer brushes on thin-film composite membranes for integrated “defending” and “attacking” strategies against biofouling. Appl. Mater. Interf. 2015, 7, 23069–23079. [Google Scholar] [CrossRef] [PubMed]
- Charnley, M.; Textor, M.; Acikgoz, C. Designed polymer structures with antifouling–antimicrobial properties. React. Funct. Polym. 2011, 71, 329–334. [Google Scholar] [CrossRef]
- Zou, P.; Hartleb, W.; Lienkamp, K. It takes walls and knights to defend a castle—synthesis of surface coatings from antimicrobial and antibiofouling polymers. J. Mater. Chem. 2012, 22, 19579–19589. [Google Scholar] [CrossRef]
- Vöhringer, M.; Hartleb, W.; Lienkamp, K. Surface structuring meets orthogonal chemical modifications: Toward a technology platform for site-selectively functionalized polymer surfaces and biomems. Biomater. Sci. Eng. 2017, 3, 909–921. [Google Scholar] [CrossRef]
- Zou, P.; Laird, D.; Riga, E.K.; Deng, Z.; Perez-Hernandez, H.R.; Guevara-Solarte, D.L.; Steinberg, T.; Al-Ahmad, A.; Lienkamp, K. Antimicrobial and cell-compatible surface-attached polymer networks—how the correlation of chemical structure to physical and biological data leads to a modified mechanism of action. J. Mater. Chem. B 2015, 3, 6224–6238. [Google Scholar] [CrossRef]
- Nurioglu, A.G.; Esteves, A.C.C.; de With, G. Non-toxic, non-biocide-release antifouling coatings based on molecular structure design for marine applications. J. Mater. Chem. 2015, 3, 6547–6570. [Google Scholar] [CrossRef]
- Al-Ahmad, A.; Zou, P.; Guevara-Solarte, D.L.; Hellwig, E.; Steinberg, T.; Lienkamp, K. Development of a standardized and safe airborne antibacterial assay, and its evaluation on antibacterial biomimetic model surfaces. PLoS One 2014, e111357. [Google Scholar] [CrossRef] [PubMed]
- Alfred, S.F.; Al-Badri, Z.M.; Madkour, A.E.; Lienkamp, K.; Tew, G.N. Water soluble poly(ethylene oxide) functionalized norbornene polymers. J. Ploym. Sci. Part A Ploym. Chem. 2008, 46, 2640–2648. [Google Scholar] [CrossRef]
- Al-Ahmad, A.; Laird, D.; Zou, P.; Tomakidi, P.; Steinberg, T.; Lienkamp, K. Nature-inspired antimicrobial polymers – assessment of their potential for biomedical applications. PLoS ONE 2013, 8, e73812. [Google Scholar] [CrossRef] [PubMed]
- Lienkamp, K.; Madkour, A.E.; Musante, A.; Nelson, C.F.; Nüsslein, K.; Tew, G.N. Antimicrobial polymers prepared by romp with unprecedented selectivity: A molecular construction kit approach. J. Am. Chem. Soc. 2008, 130, 9836–9843. [Google Scholar] [CrossRef] [PubMed]
- Riga, E.K.; Rühe, J.; Lienkamp, K. Non-delaminating Polymer Hydrogel Coatings via C,H Insertion Crosslinking (CHic)—A Case Study of Poly(oxanorbornenes). Chem. Eur. J. 2018, submitted. [Google Scholar]
- Sharma, S.; Johnson, R.W.; Desai, T.A. XPS and AFM analysis of antifouling peg interfaces for microfabricated silicon biosensors. Biosens. Bioelectron. 2004, 20, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Sun, W. Functionalization of surfaces with branched polymers. RSC Adv. 2016, 6, 42089–42108. [Google Scholar] [CrossRef]
- Dickson, J.; Koohmaraie, M. Cellsurface charge characteristics and their relationship to bacterial attachment tomeatsurfaces. Appl. Environ. Microbiol. 1989, 55, 832–836. [Google Scholar] [PubMed]
- Guo, S.; Jańczewski, D.; Zhu, X.; Quintana, R.; He, T.; Neoh, K.G. Surface charge control for zwitterionic polymer brushes: Tailoring surface properties to antifouling applications. J. Colloid Interf. Sci. 2015, 452, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Jiang, Y.; Nüsslein, K.; Rotello, V.M.; Santore, M.M. Antimicrobial surfaces containing cationic nanoparticles: How immobilized, clustered, and protruding cationic charge presentation affects killing activity and kinetics. Colloids Surf. B 2015, 125, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, A.; Mierczynska, A.; Barton, M.; Majewski, P.; Vasilev, K. Influence of immobilized quaternary ammonium group surface density on antimicrobial efficacy and cytotoxicity. Biofouling 2016, 32, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Gottenbos, B.; Grijpma, D.W.; van der Mei, H.C.; Feijen, J.; Busscher, H.J. Antimicrobial effects of positively charged surfaces on adhering gram-positive and gram-negative bacteria. J. Antimicrob. Chemother. 2001, 48, 7–13. [Google Scholar] [CrossRef]
- Kurowska, M.; Eickenscheidt, A.; Al-Ahmad, A.; Lienkamp, K. Simultaneously antimicrobial, protein-repellent and cell-compatible polyzwitterion networks: More insight on bioactivity and physical properties. Appl. Bio Mater. 2018. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Monomer M | Monomer P | Catalyst G3 | Solvent | |||
|---|---|---|---|---|---|---|---|
| n/mmol | m/mg | n/mmol | m/mg | n/mmol | m/mg | mL | |
| SMAMP*-co-5%PEG | 0.07 | 116 | 1.36 | 500 | 5.7·10−3 | 4.2 | 7 |
| SMAMP*-co-10%PEG | 0.15 | 244 | 1.36 | 500 | 6.5·10−3 | 4.7 | 7 |
| SMAMP*-co-25%PEG | 0.45 | 733 | 1.36 | 500 | 9.5·10−3 | 6.9 | 8 |
| Copolymer | SMAMP to PEG Ratio | PEG Content | Mn/kg mol−1 | Mw/Mn | |||
|---|---|---|---|---|---|---|---|
| mol % | mass % | ||||||
| calc. | NMR | calc. | NMR | ||||
| SMAMP-co-5%PEG | 95:5 | 5 | 4.5 | 18.8 | 17.3 | 66 | 1.3 |
| SMAMP-co-10%PEG | 90:10 | 10 | 8.1 | 32.8 | 28.0 | 63 | 1.6 |
| SMAMP-co-25%PEG | 75:25 | 25 | 22.1 | 59.5 | 55.6 | 50 | 1.2 |
| SMAMP | SMAMP* | SMAMP-co-5% PEG | SMAMP*-co-5% PEG | SMAMP-co-10% PEG | SMAMP*-co-10% PEG | SMAMP-co-25% PEG | SMAMP*-co-25% PEG | |
|---|---|---|---|---|---|---|---|---|
| Thickness/nm | 62 ± 2 | 53 ± 4 | 86 ± 2 | 79 ± 1 | 101 ± 2 | 93 ± 1 | 79 ± 2 | 71 ± 3 |
| /° | 82 ± 2 | 51 ± 1 | 89 ± 1 | 52 ± 2 | 85 ± 3 | 51 ± 2 | 79 ± 1 | 46 ± 2 |
| /° | 91 ± 4 | 56 ± 2 | 90 ± 2 | 61 ± 2 | 87 ± 1 | 58 ± 3 | 85 ± 2 | 52 ± 1 |
| /° | 43 ± 4 | 26 ± 2 | 42 ± 3 | 24 ± 2 | 41 ± 1 | 27 ± 1 | 37 ± 3 | 22 ± 2 |
| Roughness/nm | 2.1 | 1.1 | 1.9 | 0.6 |
| Polymer | Elemental Composition/% | Ratio XPS/calc. | |||||||
|---|---|---|---|---|---|---|---|---|---|
| XPS | calc. | ||||||||
| C 1s | N 1s | O 1s | C | N | O | C | N | O | |
| SMAMP* | 71.2 | 3.2 | 25.6 | 68.4 | 5.3 | 26.3 | 1.04 | 0.61 | 1.02 |
| SMAMP*-co-5%PEG | 70.6 | 3.1 | 26.3 | 68.3 | 5.0 | 26.7 | 1.03 | 0.62 | 1.02 |
| SMAMP*-co-10%PEG | 70.1 | 2.8 | 27.1 | 68.2 | 4.7 | 27.0 | 1.03 | 0.38 | 1.03 |
| SMAMP*-co-25% PEG | 70.5 | 1.3 | 28.2 | 68.0 | 3.9 | 28.1 | 1.04 | 0.33 | 1.04 |
| Polymer | Protein Adhesion/ng mm−2 | Antimicrobial Activity/% growth | |
|---|---|---|---|
| E. coli | S. aureus | ||
| SMAMP* | 11.3 | - | - |
| SMAMP*-co-5%PEG | 9.8 | 0.1 | 11 |
| SMAMP*-co-10%PEG | 8.4 | 0.1 | 18.2 |
| SMAMP*-co-25% PEG | 0.99 | 0 | 47.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurowska, M.; Widyaya, V.T.; Al-Ahmad, A.; Lienkamp, K. Surface-Attached Poly(oxanorbornene) Hydrogels with Antimicrobial and Protein-Repellent Moieties: The Quest for Simultaneous Dual Activity. Materials 2018, 11, 1411. https://doi.org/10.3390/ma11081411
Kurowska M, Widyaya VT, Al-Ahmad A, Lienkamp K. Surface-Attached Poly(oxanorbornene) Hydrogels with Antimicrobial and Protein-Repellent Moieties: The Quest for Simultaneous Dual Activity. Materials. 2018; 11(8):1411. https://doi.org/10.3390/ma11081411
Chicago/Turabian StyleKurowska, Monika, Vania Tanda Widyaya, Ali Al-Ahmad, and Karen Lienkamp. 2018. "Surface-Attached Poly(oxanorbornene) Hydrogels with Antimicrobial and Protein-Repellent Moieties: The Quest for Simultaneous Dual Activity" Materials 11, no. 8: 1411. https://doi.org/10.3390/ma11081411
APA StyleKurowska, M., Widyaya, V. T., Al-Ahmad, A., & Lienkamp, K. (2018). Surface-Attached Poly(oxanorbornene) Hydrogels with Antimicrobial and Protein-Repellent Moieties: The Quest for Simultaneous Dual Activity. Materials, 11(8), 1411. https://doi.org/10.3390/ma11081411