A Realistic Approach for Photoelectrochemical Hydrogen Production


 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Photoanode Electrode
2.3. Construction of the Counter Electrode
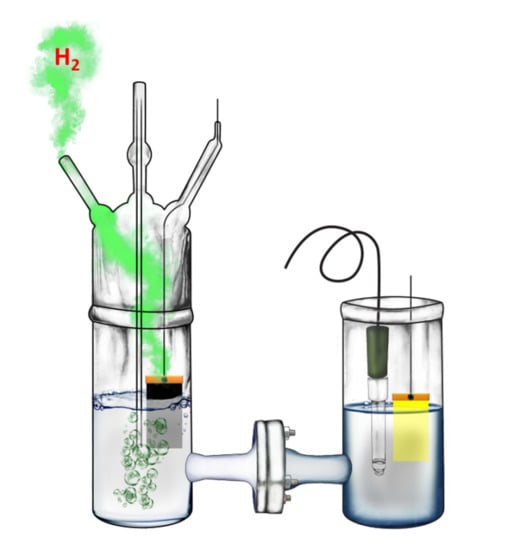
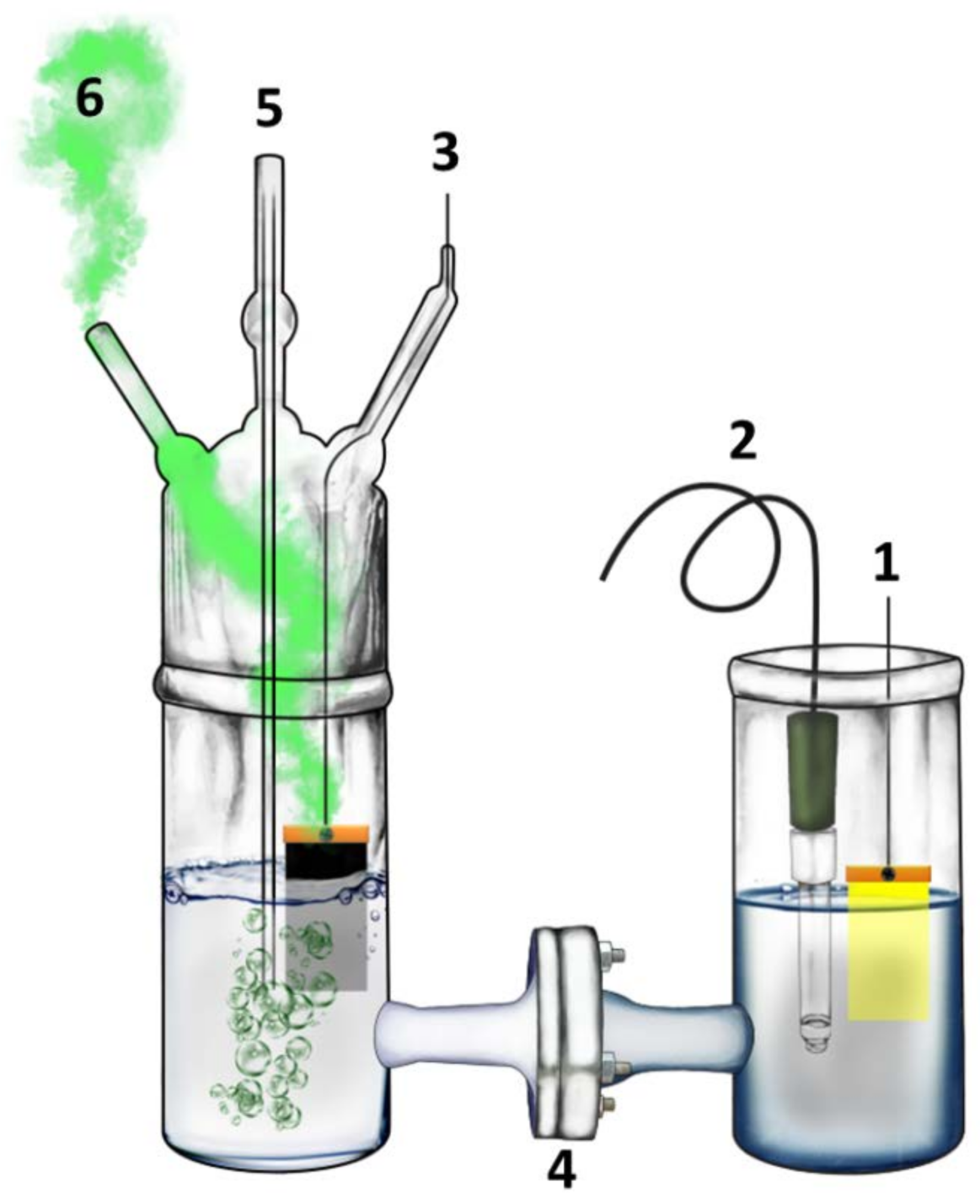
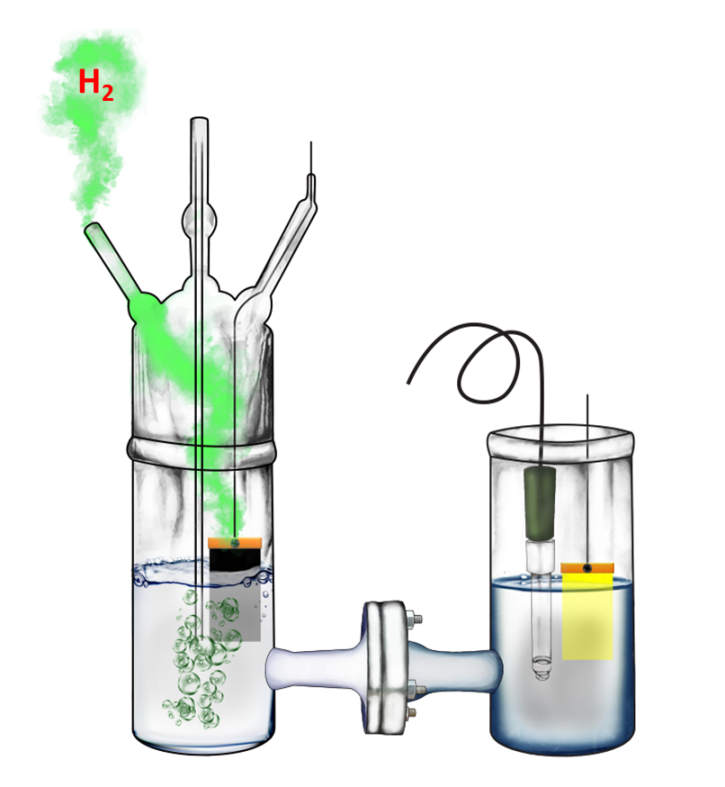
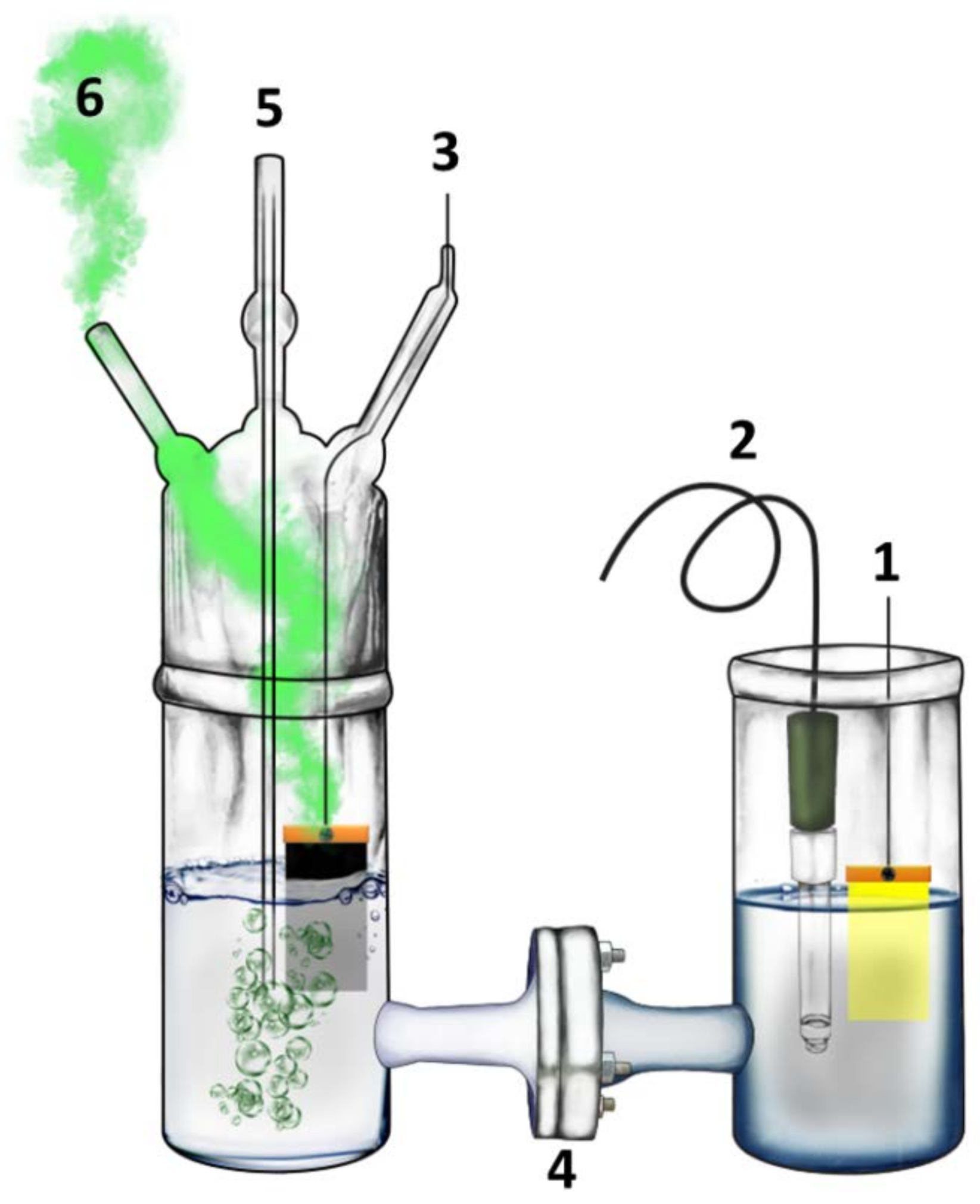
2.4. Description of the Reactor
2.5. Measurements and Characterizations
3. Results and Discussion
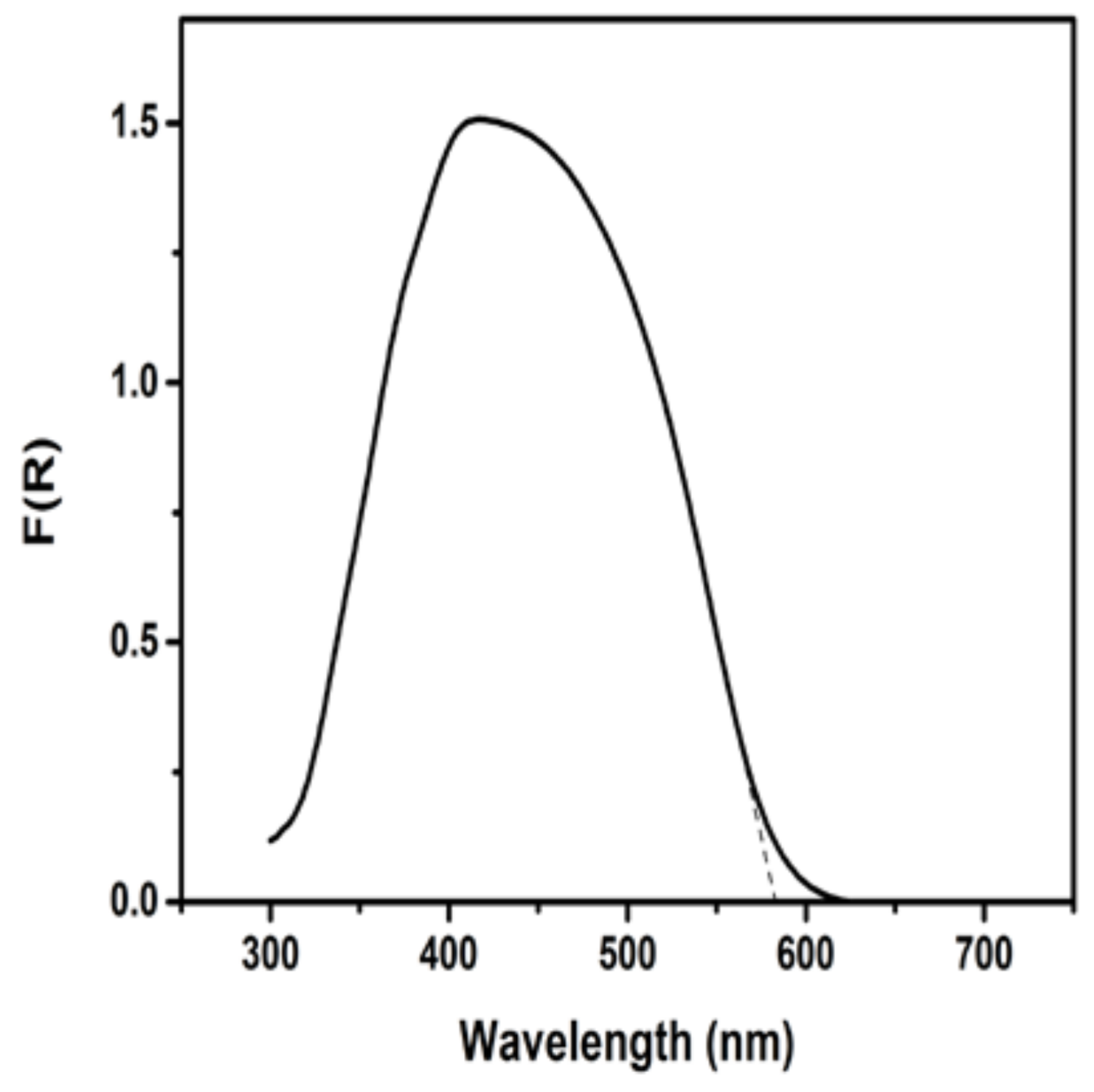
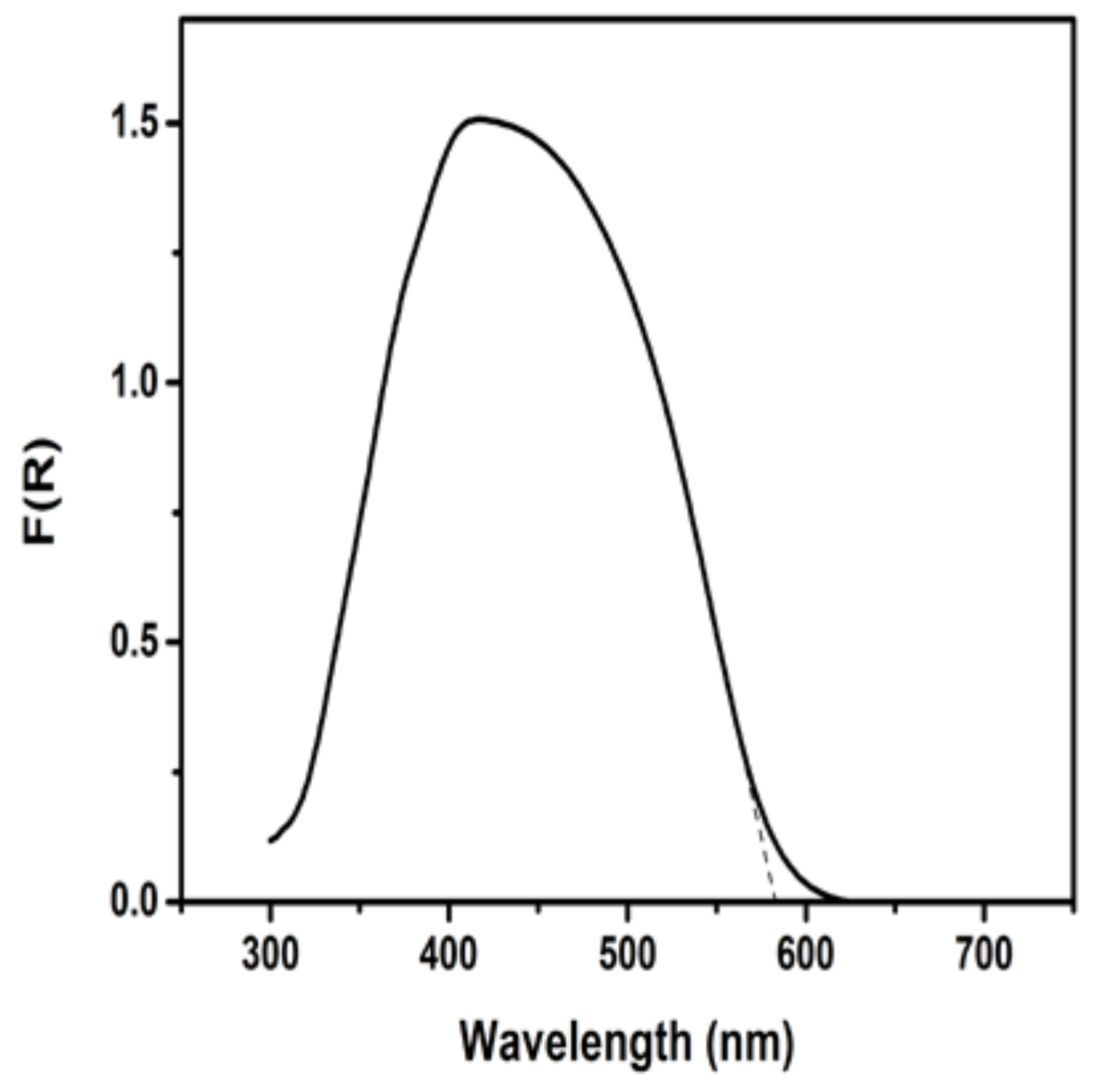
3.1. Materials Characterization
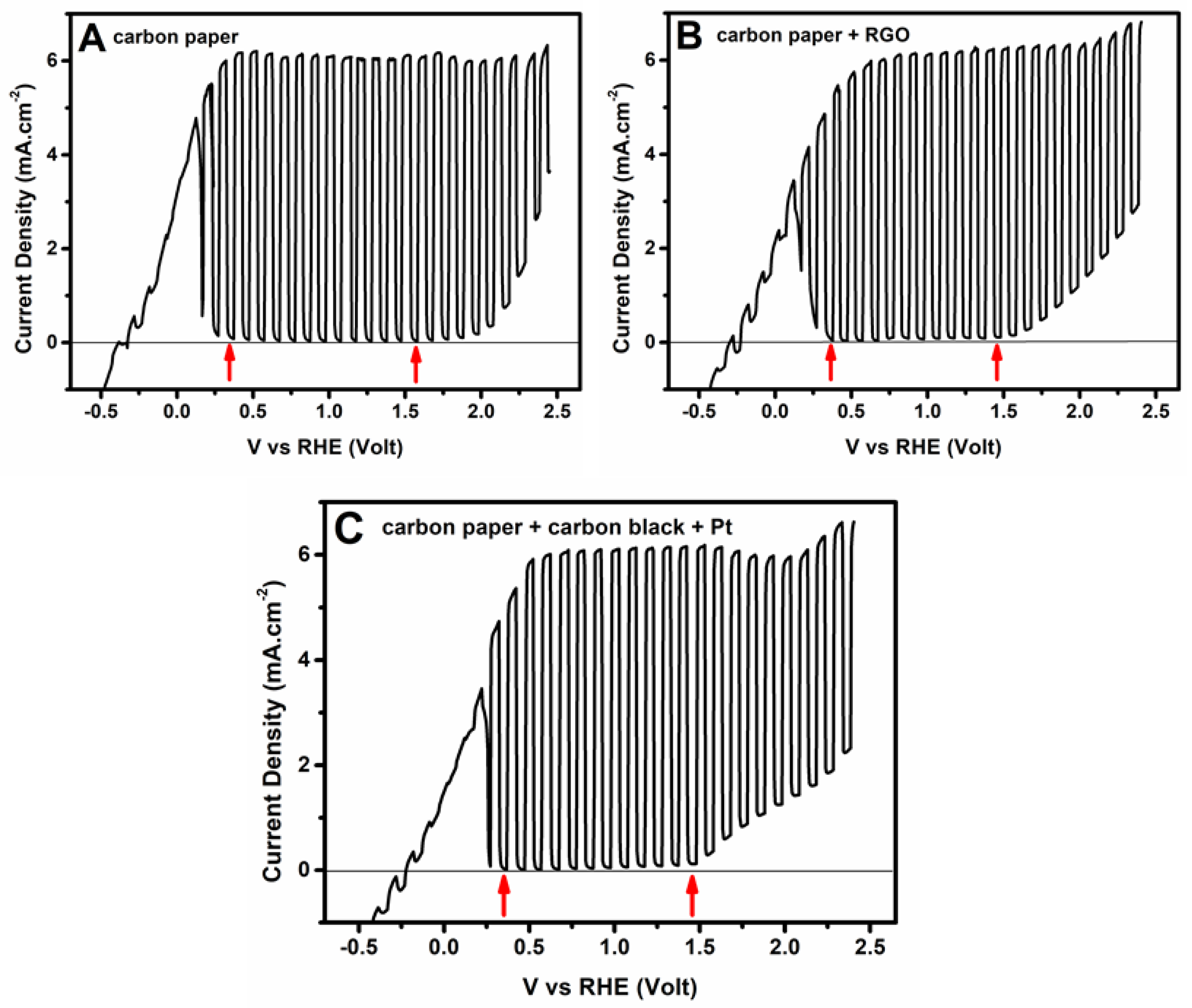
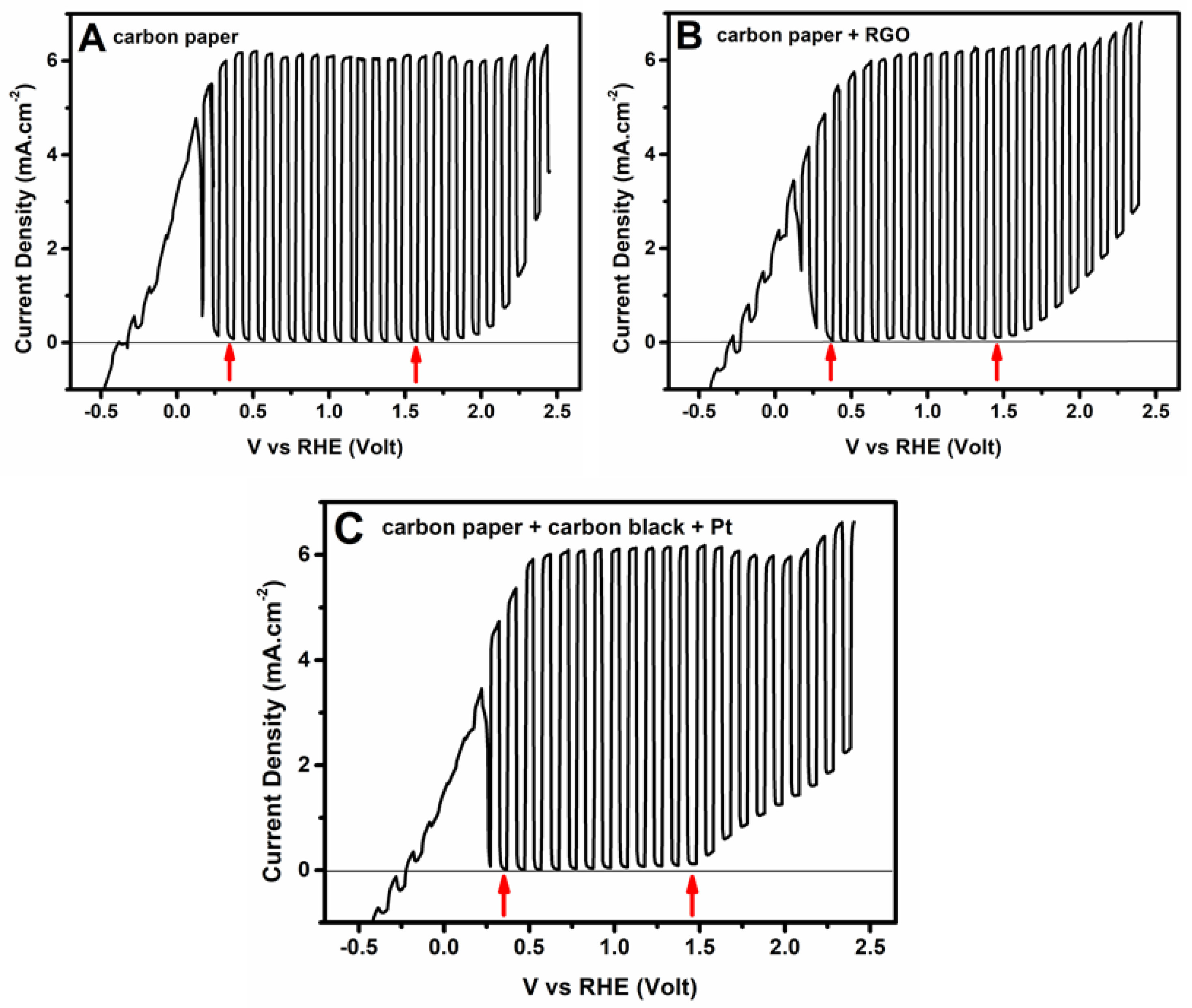
3.2. Current–Voltage Characteristics of the Photoelectrochemical Cell
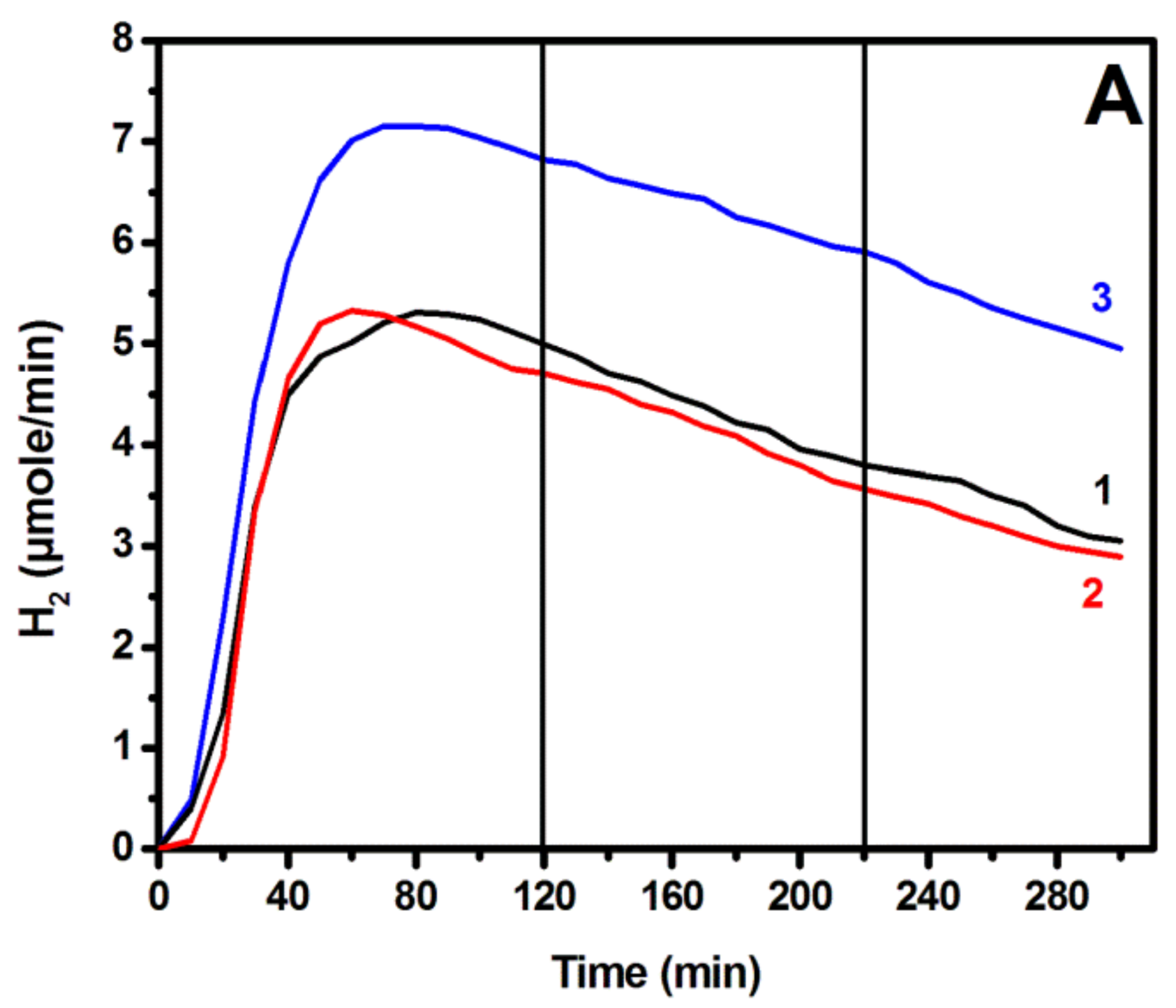
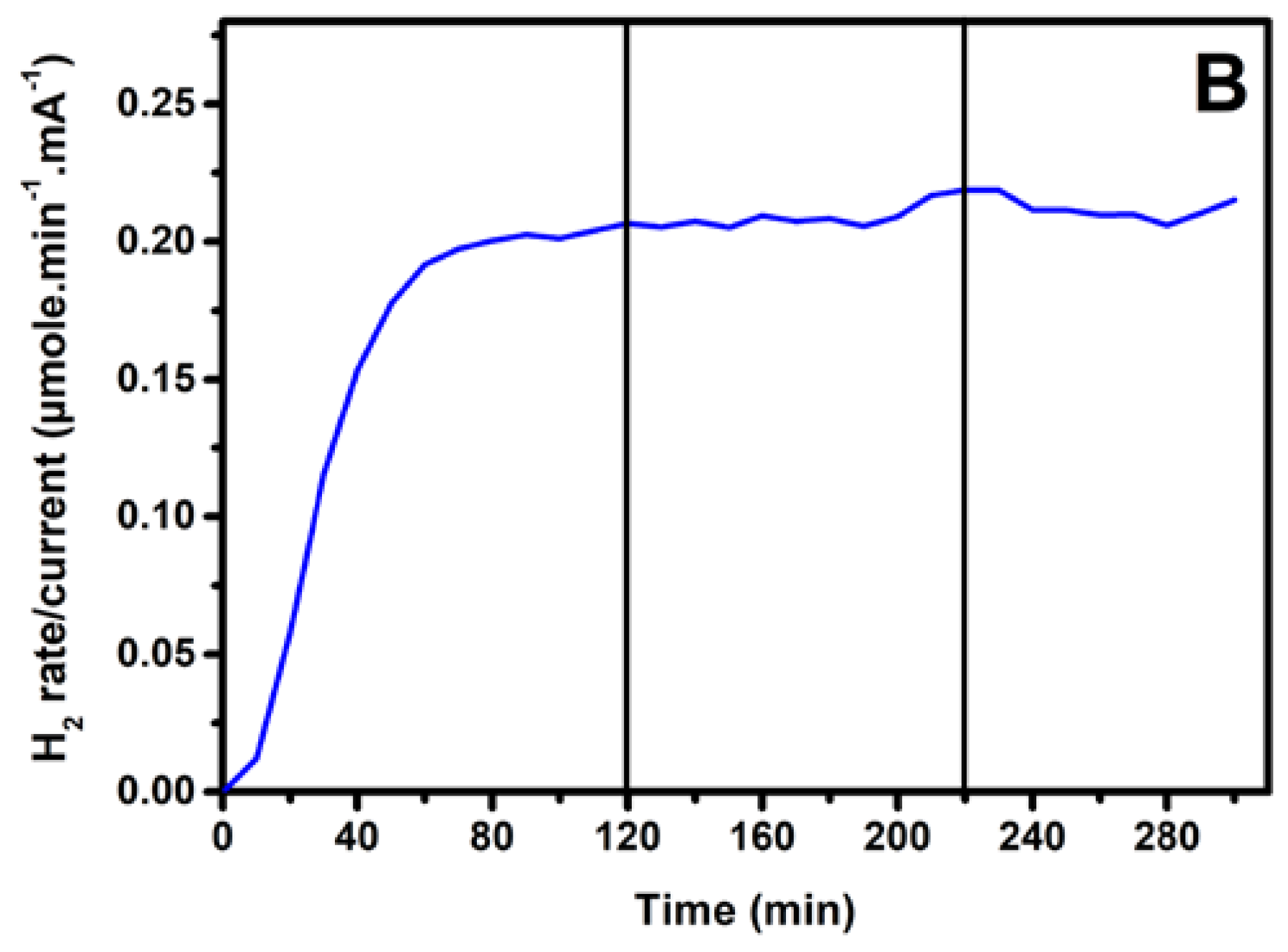
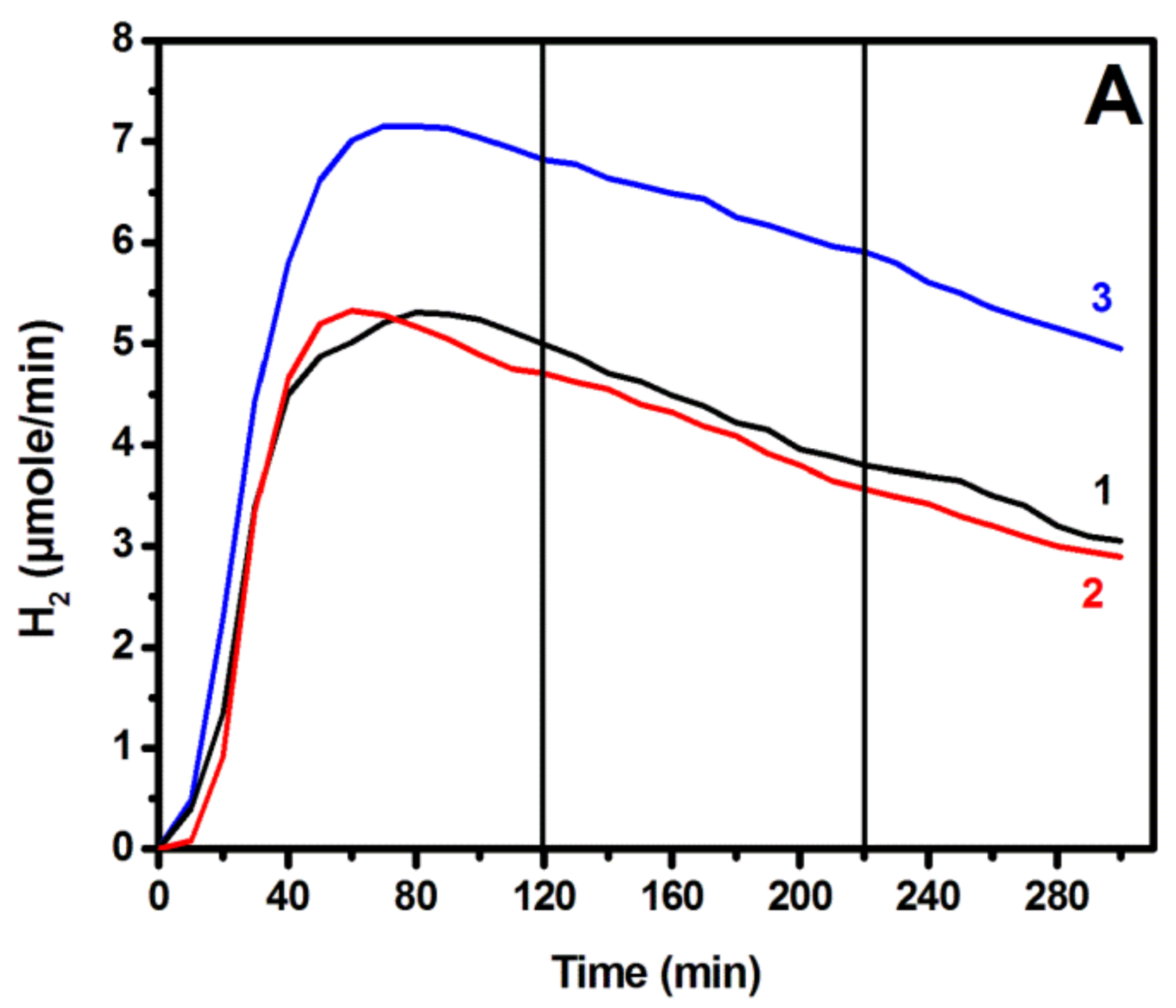
3.3. Photoelectrochemical Hydrogen Production
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Wang, L. Progress in designing effective photoelectrodes for solar water splitting. Chin. J. Catal. 2018, 39, 369–378. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, C.; Chen, B.; Kuhn, A.N.; Ma, D.; Yang, H. Progress in hydrogen production over transition metal carbide catalysts: Challenges and opportunities. Curr. Opin. Chem. Eng. 2018, 20, 68–77. [Google Scholar] [CrossRef]
- Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.; Chen, J.; Starr, J.T.; Baran, P.S. Scalable, electrochemical oxidation of unactivated C−H bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, N.L.; Weinberg, H.R. Electrochemical oxidation of organic compounds. Chem. Rev. 1968, 68, 449–523. [Google Scholar] [CrossRef]
- Lianos, P. Production of electricity and hydrogen by photocatalytic degradation of organic wastes in a photoelectrochemical cell: The concept of the Photofuelcell: A review of a re-emerging research field. J. Hazard. Mater. 2011, 185, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Lianos, P. Review of recent trends in photoelectrocatalytic conversion of solar energy to electricity and hydrogen. Appl. Catal. B Environ. 2017, 210, 235–254. [Google Scholar] [CrossRef]
- Saraswat, S.K.; Rodene, D.D.; Gupta, R.B. Recent advancements in semiconductor materials for photoelectrochemical water splitting for hydrogen production using visible light. Renew. Sustain. Energy Rev. 2018, 89, 228–248. [Google Scholar] [CrossRef]
- Abdi, F.F.; Berglund, S.P. Recent developments in complex metal oxide photoelectrodes. J. Phys. D Appl. Phys. 2017, 50, 193002. [Google Scholar] [CrossRef]
- Tee, S.Y.; Win, K.Y.; Teo, W.S.; Koh, L.D.; Liu, S.; Teng, C.P.; Han, M.Y. Recent progress in energy-driven water splitting. Adv. Sci. 2017, 4, 1600337. [Google Scholar] [CrossRef] [PubMed]
- Antoniadou, M.; Lianos, P. Near ultraviolet and visible light photoelectrochemical degradation of organic substances producing electricity and hydrogen. J. Photochem. Photobiol. A 2009, 204, 69–74. [Google Scholar] [CrossRef]
- Chan, C.H.; Samikkannu, P.; Wang, H.W. Fe2O3/CdS co-sensitized titania nanotube for hydrogen generation from photocatalytic splitting water. Int. J. Hydrogen Energ. 2016, 41, 17818–17825. [Google Scholar] [CrossRef]
- Pareek, A.; Purbia, R.; Paik, P.; Hebalkar, N.Y.; Kim, H.G.; Borse, P.H. Stabilizing effect in nano-titania functionalized CdS photoanode for sustained hydrogen generation. Int. J. Hydrogen Energy 2014, 39, 4170–4180. [Google Scholar] [CrossRef]
- Antoniadou, M.; Sfaelou, S.; Dracopoulos, V.; Lianos, P. Platinum-free photoelectrochemical water splitting. Catal. Commun. 2014, 43, 72–74. [Google Scholar] [CrossRef]
- Antoniadou, M.; Sfaelou, S.; Lianos, P. Quantum dot sensitized titania for photo-fuel-cell and for water splitting operation in the presence of sacrificial agents. Chem. Eng. J. 2014, 254, 245–251. [Google Scholar] [CrossRef]
- Kozlova, E.A.; Kozhevnikova, N.S.; Cherepanova, S.V.; Lyubina, T.P.; Gerasimov, E.Y.; Kaichev, V.V.; Vorontsov, A.V.; Tsybulya, S.V.; Rempel, A.A.; Parmon, V.N. Photocatalytic oxidation of ethanol vapors under visible light on CdS–TiO2 nanocatalyst. J. Photochem. Photobiol. A 2012, 250, 103–109. [Google Scholar] [CrossRef]
- Liao, Q.; Li, L.; Chen, R.; Zhu, X.; Wang, H.; Ye, D.; Cheng, X.; Zhang, M.; Zhou, Y. Respective electrode potential characteristics of photocatalytic fuel cell with visible-light responsive photoanode and air-breathing cathode. Int. J. Hydrogen Energy 2015, 40, 16547–16555. [Google Scholar] [CrossRef]
- Li, L.; Xue, S.; Chen, R.; Liao, Q.; Zhu, X.; Wang, Z.; He, X.; Feng, H.; Cheng, X. Performance characteristics of a membraneless solar responsive photocatalytic fuel cell with an air-breathing cathode under different fuels and electrolytes and air conditions. Electrochim. Acta 2015, 182, 280–288. [Google Scholar] [CrossRef]
- Sfaelou, S.; Syggelou, L.; Dracopoulos, V.; Travlos, A.; Lianos, P. Effect of the Nature of Cadmium Salts on the Effectiveness of CdS SILAR Deposition and Its Consequences on the Performance of Sensitized Solar Cells. J. Phys. Chem. B 2014, 118, 22873–22880. [Google Scholar] [CrossRef]
- Kalamaras, E.; Lianos, P. Current Doubling effect revisited: Current multiplication in a PhotoFuelCell. J. Electroanal Chem. 2015, 751, 37–42. [Google Scholar] [CrossRef]
- Nicolau, Y.F. Solution Deposition of Thin Solid Compound Films by a Successive Ionic-Layer Adsorption and Reaction Process. Appl. Surf. Sci. 1985, 22, 1061–1074. [Google Scholar] [CrossRef]
- Staudenmaier, L. Verfahren zur Darstellung der Graphitslure. Berichte der Dtsch. Chem. Gesellschaft 1898, 31, 1481–1487. [Google Scholar] [CrossRef]
- Raptis, D.; Dracopoulos, V.; Lianos, P. Renewable energy production by photoelectrochemical oxidation of organic wastes using WO3 photoanodes. J. Hazard. Mater. 2017, 333, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Sfaelou, S.; Antoniadou, M.; Dracopoulos, V.; Bourikas, K.; Kondarides, D.I.; Lianos, P. Quantum dot sensitized titania as visible-light photocatalyst for solar operation of photofuel cells. J. Adv. Oxid. Technol. 2014, 17, 59–65. [Google Scholar] [CrossRef]
- Sfaelou, S.; Raptis, D.; Dracopoulos, V.; Lianos, P. BiOI solar cells. RSC Adv. 2015, 5, 95813–95816. [Google Scholar] [CrossRef]
- Pop, L.C.; Sfaelou, S.; Lianos, P. Cation adsorption by mesoporous titania photoanodes and its effect on the current-voltage characteristics of photoelectrochemical cells. Electrochim. Acta 2015, 156, 223–227. [Google Scholar] [CrossRef]
- Li, Z.; Luo, W.; Zhang, M.; Feng, J.; Zou, Z. Photoelectrochemical cells for solar hydrogen production: current state of promising photoelectrodes, methods to improve their properties, and outlook. Energy Environ. Sci. 2013, 6, 347–370. [Google Scholar] [CrossRef]
- Sfaelou, S.; Pop, L.C.; Monfort, O.; Dracopoulos, V.; Lianos, P. Mesoporous WO3 photoanodes for hydrogen production by water splitting and PhotoFuelCell operation. Int. J. Hydrogen Energy 2016, 41, 5902–5907. [Google Scholar] [CrossRef]
- Karuppuchamy, S.; Iwasaki, M.; Minoura, H. Electrochemical properties of electrosynthesized TiO2 thin films. Appl. Surf. Sci. 2006, 253, 2924–2929. [Google Scholar] [CrossRef]
- Wang, C.-M.; Lin, S.-Y.; Chen, Y.-C. Electrochromic properties of TiO2 thin films prepared by chemical solution deposition method. J. Phys. Chem. Solids 2008, 69, 451–455. [Google Scholar] [CrossRef]
- Khalifa, Z.S. Electronic structure changes of TiO2 thin films due to electrochromism. Sol. Energy Mater. Sol. Cell 2014, 124, 186–191. [Google Scholar] [CrossRef]
- Kanai, F.; Kurita, S.; Sugioka, S.; Li, M.; Mita, Y. Optical characteristics of WO3 electrochromic cells under heavy Li ion injection. J. Electrochem. Soc. 1982, 129, 2633–2635. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Counter Electrode | Type of Electrolyte in the Counter Electrode Compartment | Faradaic Efficiency of Hydrogen Production |
|---|---|---|
| Carbon paper | NaOH | 51% |
| Carbon paper + RGO | NaOH | 52% |
| Carbon paper + Carbon Black | NaOH | 53% |
| Carbon paper + Carbon Black + Pt | NaOH | 67% |
| Carbon paper | H2SO4 | 57% |
| Carbon paper + Carbon Black + Pt | H2SO4 | 60% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doukas, E.; Balta, P.; Raptis, D.; Avgouropoulos, G.; Lianos, P. A Realistic Approach for Photoelectrochemical Hydrogen Production. Materials 2018, 11, 1269. https://doi.org/10.3390/ma11081269
Doukas E, Balta P, Raptis D, Avgouropoulos G, Lianos P. A Realistic Approach for Photoelectrochemical Hydrogen Production. Materials. 2018; 11(8):1269. https://doi.org/10.3390/ma11081269
Chicago/Turabian StyleDoukas, Elias, Paraskevi Balta, Dimitrios Raptis, George Avgouropoulos, and Panagiotis Lianos. 2018. "A Realistic Approach for Photoelectrochemical Hydrogen Production" Materials 11, no. 8: 1269. https://doi.org/10.3390/ma11081269
APA StyleDoukas, E., Balta, P., Raptis, D., Avgouropoulos, G., & Lianos, P. (2018). A Realistic Approach for Photoelectrochemical Hydrogen Production. Materials, 11(8), 1269. https://doi.org/10.3390/ma11081269