Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Standard Diagnosis and Treatment of Glioblastoma Multiforme
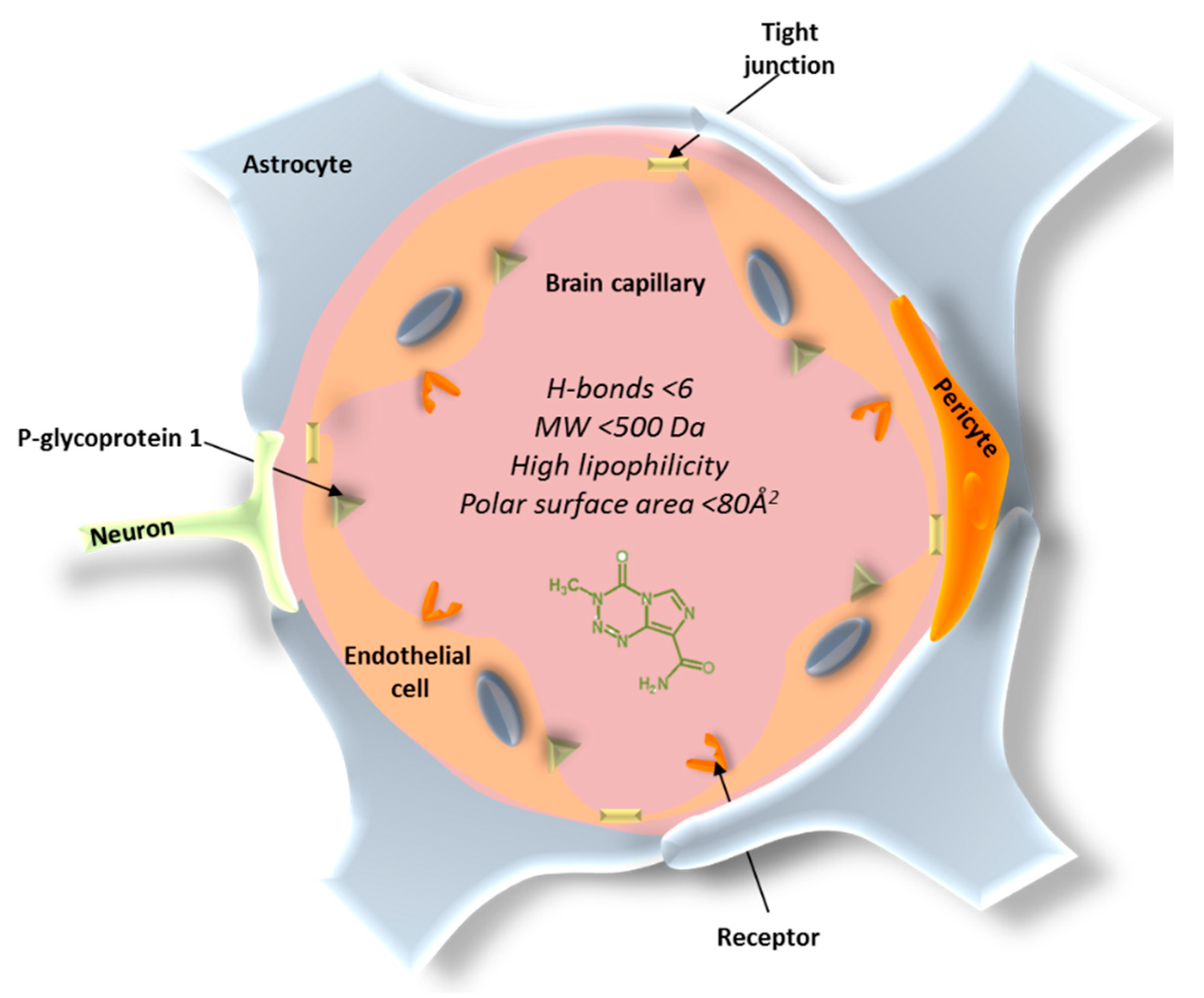
3. Barriers in the Treatment of Glioblastoma
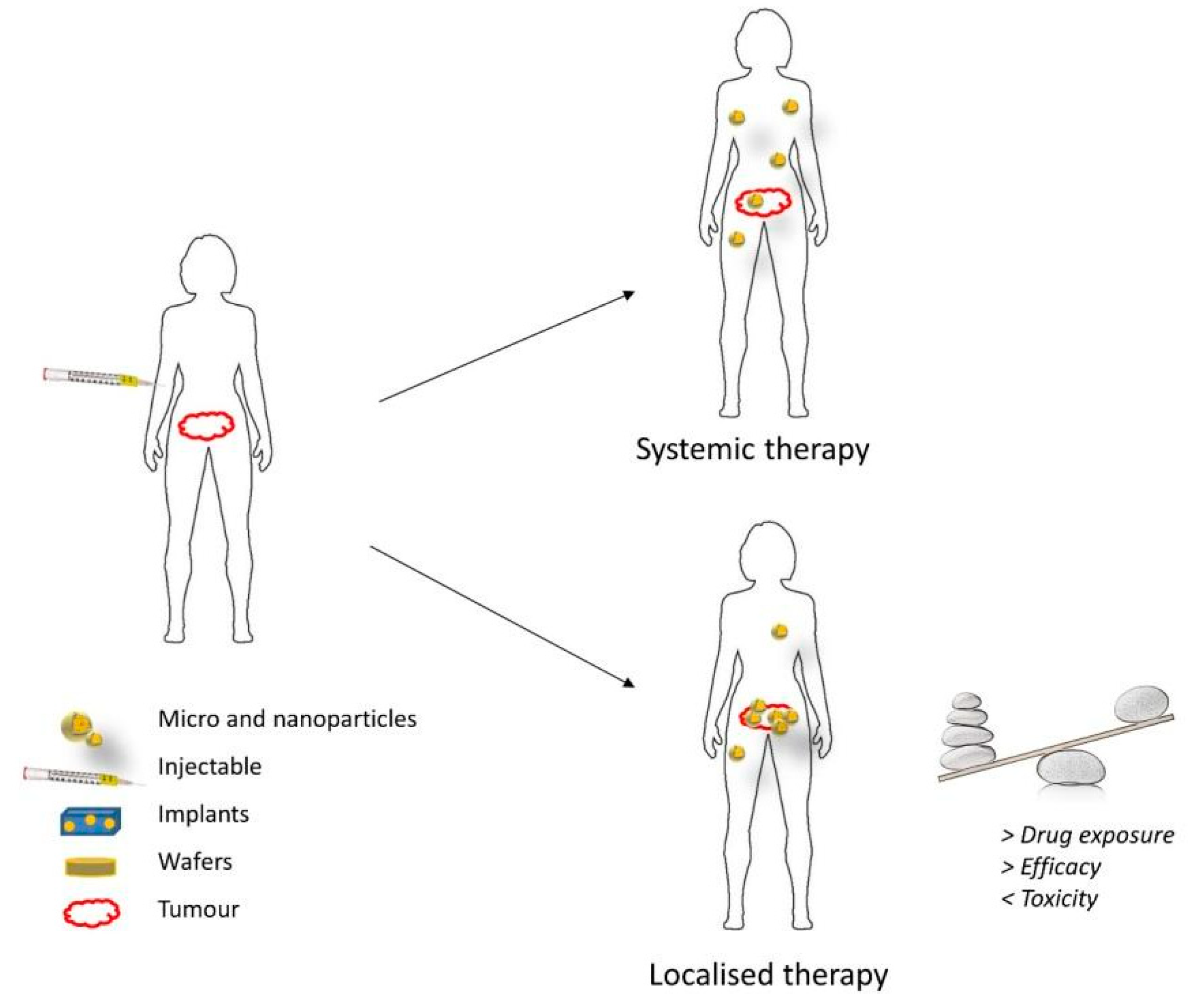
4. Local Treatment of Glioblastoma
4.1. Direct Intratumor Drug Administration
4.2. Wafers and Implants
5. Nanostructures as Alternative Therapeutics for Glioblastoma
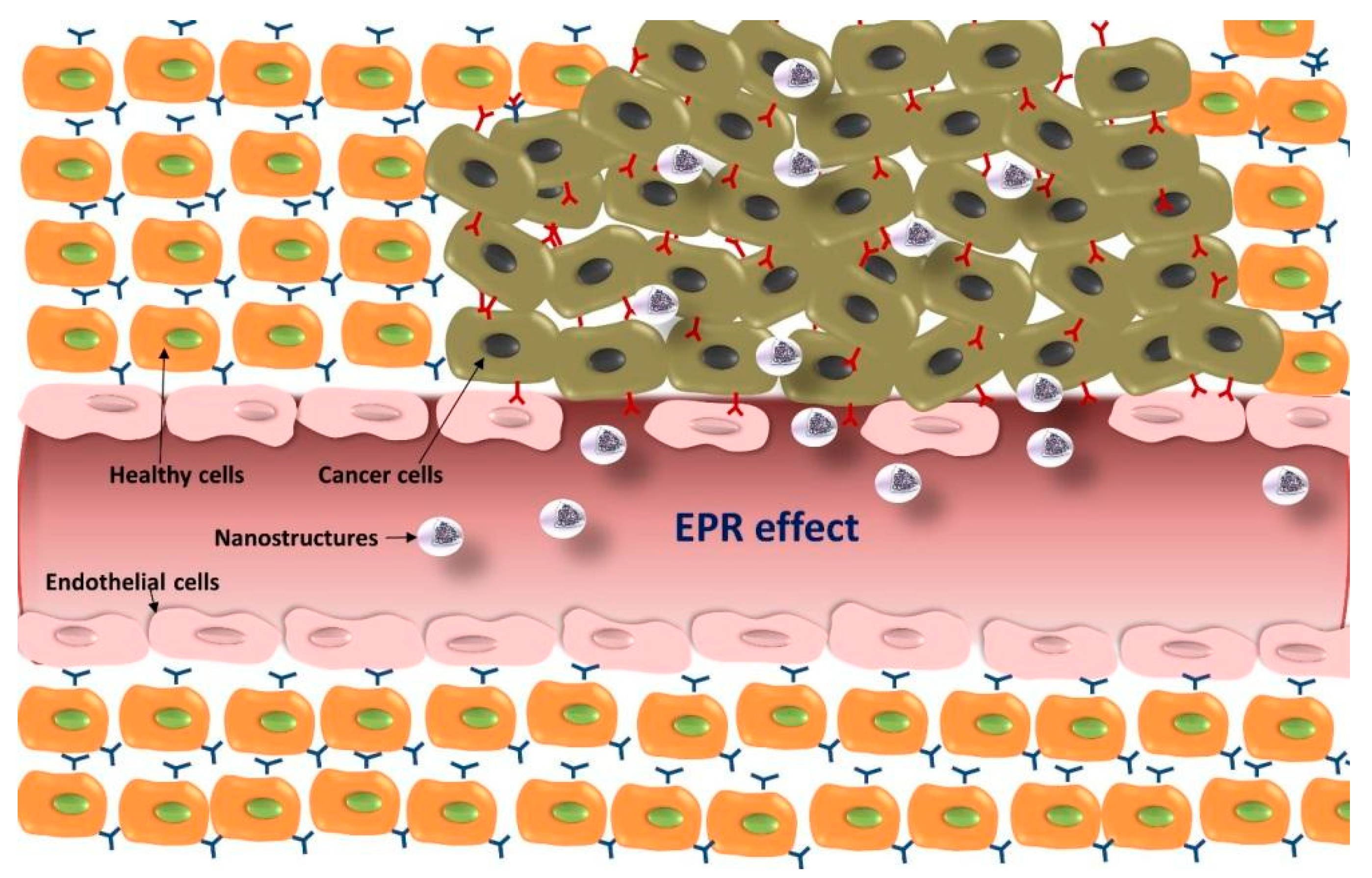
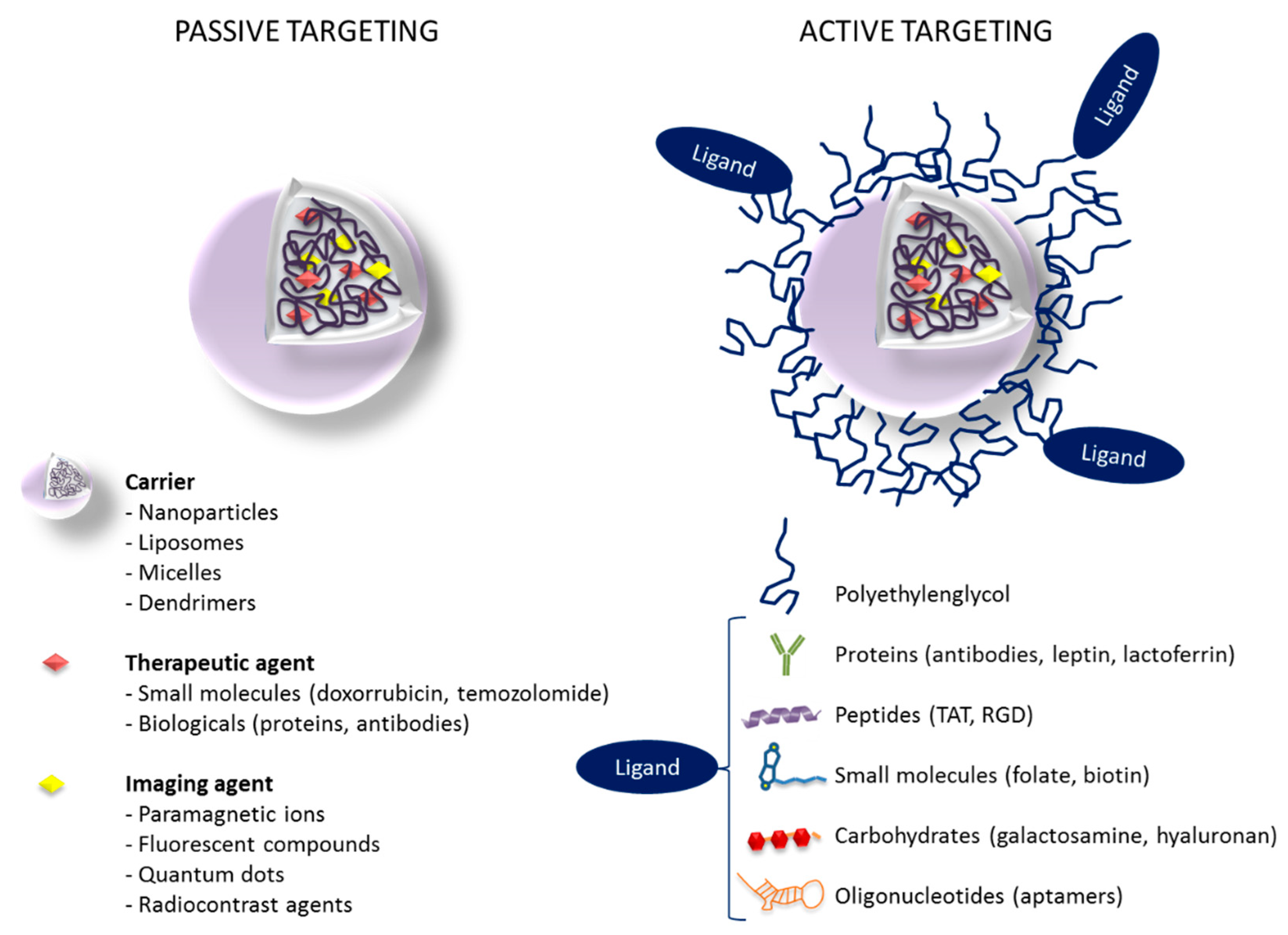
5.1. Passive Targeting Strategies for GBM
5.2. Active Targeting Strategies for GBM
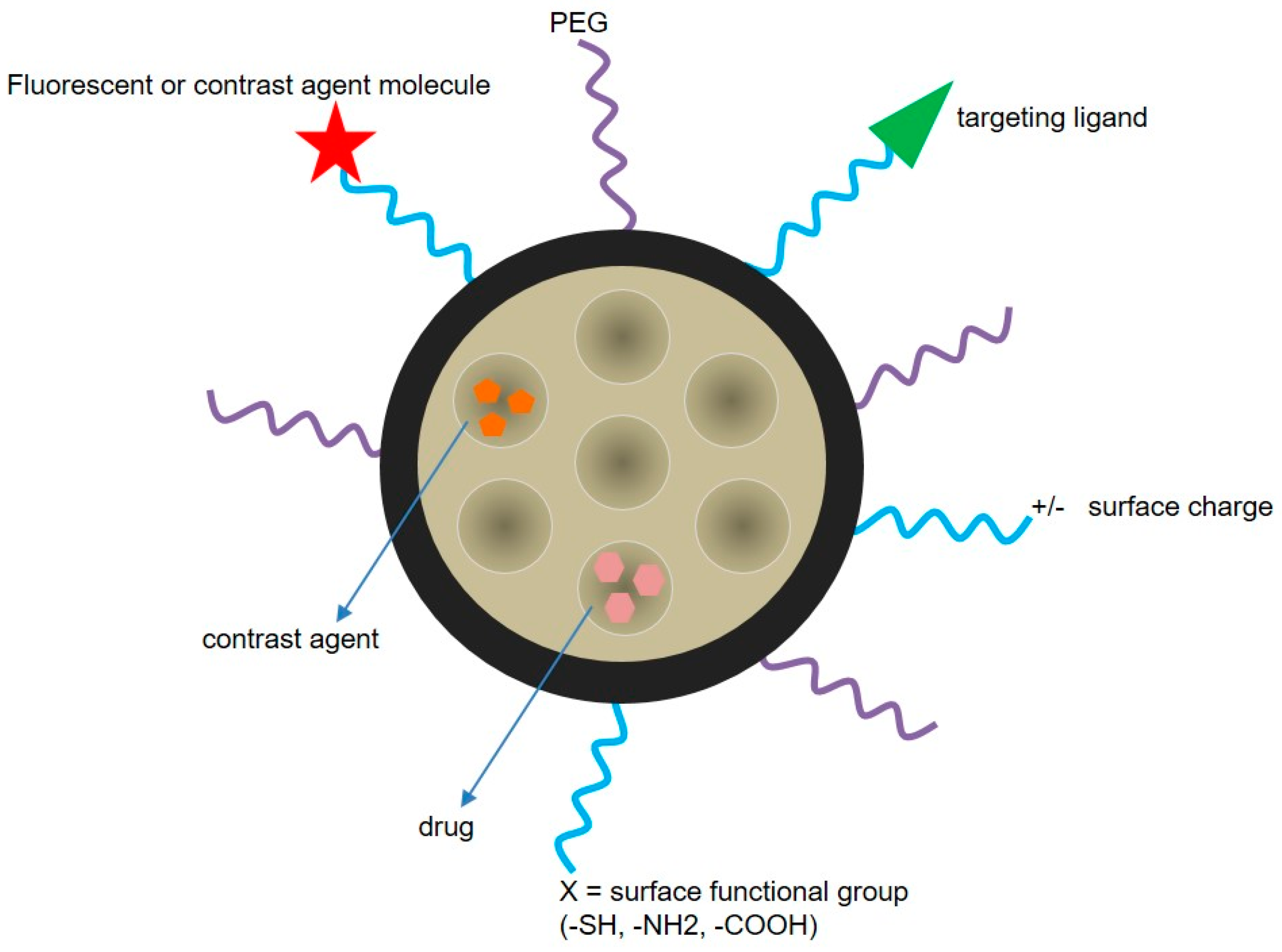
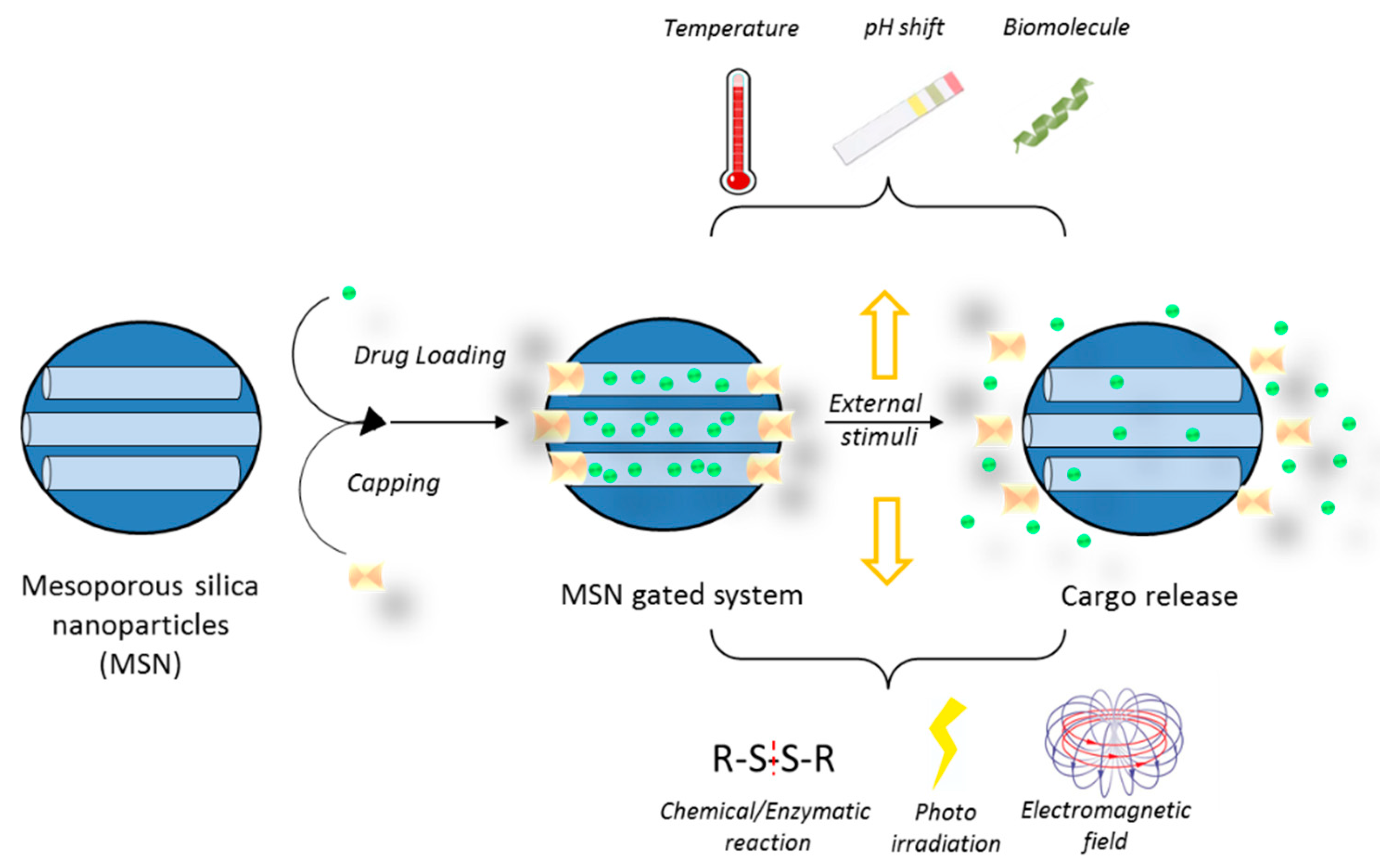
5.3. Drug Delivery Nanosystems Based on Mesoporous Silica Nanoparticles to Treat GBM
5.4. Nanotheranostics
6. Local Treatment with Nanotherapeutics
6.1. Intracranially-Administered Drug-Loaded Nanoparticles
6.2. Gene Delivery
6.3. Thermotherapy
6.4. Theranostics
7. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Gutkin, A.; Cohen, Z.R.; Peer, D. Harnessing nanomedicine for therapeutic intervention in glioblastoma. Expert Opin. Drug Deliv. 2016, 13, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, T. Understanding high grade glioma: Molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013, 331, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-Y.; Gao, P.; Sun, Y.; Duan, Y.-R. Development of targeted therapies in treatment of glioblastoma. Cancer Biol. Med. 2015, 12, 223–237. [Google Scholar] [PubMed]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, F.; Kurisu, K.; Satoh, K.; Arita, K.; Sugiyama, K.; Ohtaki, M.; Takaba, J.; Tominaga, A.; Hanaya, R.; Yoshioka, H. Apparent Diffusion Coefficient of Human Brain Tumors at MR Imaging 1. Radiology 2005, 235, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Young, R.; Shah, A.; Schweitzer, A.; Graber, J.; Shi, W.; Zhang, Z.; Huse, J.; Omuro, A. Pretreatment dynamic susceptibility contrast MRI perfusion in glioblastoma: Prediction of EGFR gene amplification. Clin. Neuroradiol. 2015, 25, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Drug delivery approaches for the treatment of glioblastoma multiforme. Arti. Cells Nanomed. Biotechnol. 2016, 44, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Štolc, S.; Jakubíková, L.; Kukurová, I. Body distribution of 11C-methionine and 18FDG in rat measured by microPET. Interdiscip. Toxicol. 2011, 4, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Galldiks, N.; Dunkl, V.; Kracht, L.W.; Vollmar, S.; Jacobs, A.H.; Fink, G.R.; Schroeter, M. Volumetry of [(1)(1)C]-methionine positron emission tomographic uptake as a prognostic marker before treatment of patients with malignant glioma. Mol. Imaging 2012, 11, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.; Alvarez-Linera, J.; Carrato, C.; Ley, L.; Luque, R.; Maldonado, X.; Martinez-Aguillo, M.; Navarro, L.M.; Vaz-Salgado, M.A.; Gil-Gil, M. SEOM clinical guidelines for diagnosis and treatment of glioblastoma (2017). Clin. Transl. Oncol. 2018, 20, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.; Karajannis, M.; Harter, D. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Juratli, T.A.; Schackert, G.; Krex, D. Current status of local therapy in malignant gliomas—A clinical review of three selected approaches. Pharmacol. Ther. 2013, 139, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003, 5, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Taillibert, S.; Chamberlain, M.C. The future of high-grade glioma: Where we are and where are we going. Surg. Neurol. Int. 2015, 6, S9–S44. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y. Strategies of temozolomide in future glioblastoma treatment. Oncol. Targets Ther. 2017, 10, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; von Hoff, D.D. Tumor-Treating Fields: A Fourth Modality in Cancer Treatment. Clin. Cancer Res. 2018, 24, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Kesari, S.; Toms, S.A.; Barnett, G.H.; Fink, K.L.; Silvani, A.; Lieberman, F.S.; Zhu, J.-J.; et al. Tumor treating fields (TTFields): A novel treatment modality added to standard chemo- and radiotherapy in newly diagnosed glioblastoma-First report of the full dataset of the EF14 randomized phase III trial. J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Bernard-Arnoux, F.; Lamure, M.; Ducray, F.; Aulagner, G.; Honnorat, J.; Armoiry, X. The cost-effectiveness of tumor-treating fields therapy in patients with newly diagnosed glioblastoma. Neuro Oncol. 2016, 18, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Preusser, M.; de Ribaupierre, S.; Wöhrer, A.; Erridge, S.C.; Hegi, M.; Weller, M.; Stupp, R. Current concepts and management of glioblastoma. Ann. Neurol. 2011, 70, 9–21. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Genentech’s Avastin Full Approval for Most Aggressive Form of Brain Cancer. 2017. Available online: https://www.gene.com/media/press-releases/14695/2017-12-05/fda-grants-genentechs-avastin-full-appro (accessed on 30 April 2018).
- Wick, W.; Stupp, R.; Gorlia, T.; Bendszus, M.; Sahm, F.; Bromberg, J.E.; Brandes, A.A.; Vos, M.J.; Domont, J.; Idbaih, A.; et al. Phase II part of EORTC study 26101: The sequence of bevacizumab and lomustine in patients with first recurrence of a glioblastoma. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Alyautdin, R.; Khalin, I.; Nafeeza, M.I.; Haron, M.H.; Kuznetsov, D. Nanoscale drug delivery systems and the blood–brain barrier. Int. J. Nanomed. 2014, 9, 795–811. [Google Scholar]
- Liu, W.Y.; Wang, Z.B.; Zhang, L.C.; Wei, X.; Li, L. Tight junction in blood-brain barrier: An overview of structure, regulation, and regulator substances. CNS Neurosci. Ther. 2012, 18, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Targeting blood-brain barrier changes during inflammatory pain: An opportunity for optimizing CNS drug delivery. Ther. Deliv. 2011, 2, 1015–1041. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the Brain—Approaches to Enhance Brain Drug Delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Lalatsa, A.; Schätchlein, A.G.; Uchegbu, I.F. Drug Delivery across the Blood-Brain Barrier. In Comprehensive Biotechnology; Elsevier: Amsterdam, The Netherlands, 2011; pp. 657–668. [Google Scholar]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Serrano Lopez, D.R.; Lalatsa, A. Peptide pills for brain diseases? Reality and future perspectives. Ther. Deliv. 2013, 4, 479–501. [Google Scholar] [PubMed]
- Herve, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.; Wesseling, P.; Wurdinger, T.; De Vries, H. Overcoming the blood–brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [PubMed]
- Laquintana, V.; Trapani, A.; Denora, N.; Wang, F.; Gallo, J.M.; Trapani, G. New strategies to deliver anticancer drugs to brain tumors. Expert Opin. Drug Deliv. 2009, 6, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, M.S.; Upadhyay, U.; Goodwin, R.; Tyler, B.; Brem, H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005, 25, 3825–3831. [Google Scholar] [PubMed]
- Zhan, C.; Gu, B.; Xie, C.; Li, J.; Liu, Y.; Lu, W. Cyclic RGD conjugated poly (ethylene glycol)-co-poly (lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J. Control. Release 2010, 143, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kondo, S.; Tanaka, Y.; Haqqi, T.; Barna, B.P.; Cowell, J.K. Inhibition of telomerase increases the susceptibility of human malignant glioblastoma cells to cisplatin-induced apoptosis. Oncogene 1998, 16, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.P.; Frazier, J.; Brem, H. Local drug delivery to the brain. Adv. Drug Deliv. Rev. 2002, 54, 987–1013. [Google Scholar] [CrossRef]
- De Souza, R.; Zahedi, P.; Allen, C.J.; Piquette-Miller, M. Polymeric drug delivery systems for localized cancer chemotherapy. Drug Deliv. 2010, 17, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. J. Control. Release 2012, 159, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, R.W.; Zhang, P.; Lin, R.; Schiapparelli, P.; Quinones-Hinojosa, A.; Cui, H. Nanotherapeutic systems for local treatment of brain tumors. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017. [CrossRef] [PubMed]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.-K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M. Anti–PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [PubMed]
- Chaichana, K.L.; Pinheiro, L.; Brem, H. Delivery of local therapeutics to the brain: Working toward advancing treatment for malignant gliomas. Ther. Deliv. 2015, 6, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Patchell, R.A.; Regine, W.F.; Ashton, P.; Tibbs, P.A.; Wilson, D.; Shappley, D.; Young, B. A phase I trial of continuously infused intratumoral bleomycin for the treatment of recurrent glioblastoma multiforme. J. Neuro Oncol. 2002, 60, 37–42. [Google Scholar] [CrossRef]
- Hassenbusch, S.J.; Nardone, E.M.; Levin, V.A.; Leeds, N.; Pietronigro, D. Stereotactic injection of DTI-015 into recurrent malignant gliomas: Phase I/II trial. Neoplasia 2003, 5, 9–16. [Google Scholar] [CrossRef]
- Boiardi, A.; Eoli, M.; Salmaggi, A.; Zappacosta, B.; Fariselli, L.; Milanesi, I.; Broggi, G.; Silvani, A. Efficacy of intratumoral delivery of mitoxantrone in recurrent malignant glial tumours. J. Neuro Oncol. 2001, 54, 39–47. [Google Scholar] [CrossRef]
- Oshiro, S.; Tsugu, H.; Komatsu, F.; Ohnishi, H.; Ueno, Y.; Sakamoto, S.; Fukushima, T.; Soma, G.-I. Evaluation of intratumoral administration of tumor necrosis factor-alpha in patients with malignant glioma. Anticancer Res. 2006, 26, 4027–4032. [Google Scholar] [PubMed]
- Lidar, Z.; Mardor, Y.; Jonas, T.; Pfeffer, R.; Faibel, M.; Nass, D.; Hadani, M.; Ram, Z. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: A phase I/II clinical study. J. Neurosurg. 2004, 100, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.N.; Fine, R.L.; Canoll, P.; Yun, J.; Kennedy, B.C.; Rosenfeld, S.S.; Sands, S.A.; Surapaneni, K.; Lai, R.; Yanes, C.L. Regression of recurrent malignant gliomas with convection-enhanced delivery of topotecan. Neurosurgery 2011, 69, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Metellus, P.; Ursu, R.; Zohar, S.; Lafitte, F.; Barrié, M.; Meng, Y.; Richard, M.; Parizot, C.; Laigle-Donadey, F. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: A phase II study. Neuro Oncol. 2010, 12, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.; Mahapatra, A.; Suri, A.A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro Oncol. 2010, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, F.M.; Lamborn, K.R.; Robins, H.I.; Mehta, M.P.; Chang, S.M.; Butowski, N.A.; DeAngelis, L.M.; Abrey, L.E.; Zhang, W.-T.; Prados, M.D.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Brem, S.; Tyler, B.; Li, K.; Pradilla, G.; Legnani, F.; Caplan, J.; Brem, H. Local delivery of temozolomide by biodegradable polymers is superior to oral administration in a rodent glioma model. Cancer Chemother. Pharmacol. 2007, 60, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Recinos, V.R.; Tyler, B.M.; Bekelis, K.; Sarah Brem Sunshine, B.; Vellimana, A.; Li, K.W.; Brem, H. Combination of intracranial temozolomide with intracranial carmustine improves survival when compared with either treatment alone in a rodent glioma model. Neurosurgery 2010, 66, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Storm, P.B.; Moriarity, J.L.; Tyler, B.; Burger, P.C.; Brem, H.; Weingart, J. Polymer delivery of camptothecin against 9L gliosarcoma: Release, distribution, and efficacy. J. Neuro Oncol. 2002, 56, 209–217. [Google Scholar] [CrossRef]
- Scott, A.W.; Tyler, B.M.; Masi, B.C.; Upadhyay, U.M.; Patta, Y.R.; Grossman, R.; Basaldella, L.; Langer, R.S.; Brem, H.; Cima, M.J. Intracranial microcapsule drug delivery device for the treatment of an experimental gliosarcoma model. Biomaterials 2011, 32, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Tyler, B.M.; Tupper, M.M.; Karp, J.M.; Langer, R.S.; Brem, H.; Cima, M.J. Resorbable polymer microchips releasing BCNU inhibit tumor growth in the rat 9L flank model. J. Control. Release 2007, 123, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Masi, B.C.; Tyler, B.M.; Bow, H.; Wicks, R.T.; Xue, Y.; Brem, H.; Langer, R.; Cima, M.J. Intracranial MEMS based temozolomide delivery in a 9L rat gliosarcoma model. Biomaterials 2012, 33, 5768–5775. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tsibouklis, J.; Weng, T.; Zhang, B.; Yin, G.; Feng, G.; Cui, Y.; Savina, I.N.; Mikhalovska, L.I.; Sandeman, S.R. Nano carriers for drug transport across the blood–brain barrier. J. Drug Target. 2017, 25, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Mangraviti, A.; Gullotti, D.; Tyler, B.; Brem, H. Nanobiotechnology-based delivery strategies: New frontiers in brain tumor targeted therapies. J. Control. Release 2016, 240, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.R.; Lalatsa, A. Active targeting. In Fundamentals of Pharmaceutical Nanoscience; Springer: New York, NY, USA, 2013; p. 337. [Google Scholar]
- Torchilin, V.P. Passive and active drug targeting: Drug delivery to tumors as an example. Handb. Exp. Pharmacol. 2010, 197, 3–53. [Google Scholar]
- Rippe, B.; Rosengren, B.I.; Carlsson, O.; Venturoli, D. Transendothelial transport: The vesicle controversy. J. Vasc. Res. 2002, 39, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Swami, A.; Shi, J.J.; Gadde, S.; Votruba, A.R.; Kolishetti, N.; Farokhzad, O.C. Nanoparticles for Targeted and Temporally Controlled Drug Delivery. In Multifunctional Nanoparticles for Drug Delivery Applications: Imaging, Targeting, and Delivery; Svenson, S., Prudhomme, R.K., Eds.; Springer: New York, NY, USA, 2012; pp. 9–29. [Google Scholar]
- Chouly, C.; Pouliquen, D.; Lucet, I.; Jeune, J.J.; Jallet, P. Development of superparamagnetic nanoparticles for MRI: Effect of particle size, charge and surface nature on biodistribution. J. Microencapsul. 1996, 13, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Morales, C.; Zhang, L.; Langer, R.; Farokhzad, O.C. Immunocompatibility properties of lipid-polymer hybrid nanoparticles with heterogeneous surface functional groups. Biomaterials 2009, 30, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Lu, W. The blood-brain/tumor barriers: Challenges and chances for malignant gliomas targeted drug delivery. Curr. Pharm. Biotechnol. 2012, 13, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Leite, D.M.; Pilkington, G.J. Nanomedicines and the future of glioma. Oncol. News 2015, 10, 51–57. [Google Scholar]
- Steiniger, S.C.; Kreuter, J.; Khalansky, A.S.; Skidan, I.N.; Bobruskin, A.I.; Smirnova, Z.S.; Severin, S.E.; Uhl, R.; Kock, M.; Geiger, K.D. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int. J. Cancer 2004, 109, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Khalansky, A.S.; Bernreuther, C.; Michaelis, M.; Cinatl, J.; Glatzel, M.; Kreuter, J. Treatment of glioblastoma with poly (isohexyl cyanoacrylate) nanoparticles. Int. J. Pharm. 2011, 415, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.H.; Lin, X.N.; Wei, F.; Feng, W.; Huang, Z.C.; Wang, P.; Ren, L.; Diao, Y. Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int. J. Nanomed. 2011, 6, 445–452. [Google Scholar]
- Zanotto-Filho, A.; Coradini, K.; Braganhol, E.; Schroder, R.; de Oliveira, C.M.; Simoes-Pires, A.; Battastini, A.M.; Pohlmann, A.R.; Guterres, S.S.; Forcelini, C.M.; et al. Curcumin-loaded lipid-core nanocapsules as a strategy to improve pharmacological efficacy of curcumin in glioma treatment. Eur. J. Pharm. Biopharm. 2013, 83, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Gao, H. Perspectives on Dual Targeting Delivery Systems for Brain Tumors. Int. J. Neuroimmune Pharmacol. 2017, 12, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.P.; Arce, M.; Yameen, B.; Vilos, C. Targeted brain delivery nanoparticles for malignant gliomas. Nanomedicine 2017, 12, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wang, K.; Stephen, Z.R.; Mu, Q.; Kievit, F.M.; Chiu, D.T.; Press, O.W.; Zhang, M. Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appl. Mater. Interfaces 2015, 7, 6674. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.; Shao, K.; Huang, R.; Han, L.; Liu, Y.; Li, J.; Kuang, Y.; Ye, L.; Lou, J.; Jiang, C. Gene delivery targeted to the brain using an Angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials 2009, 30, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
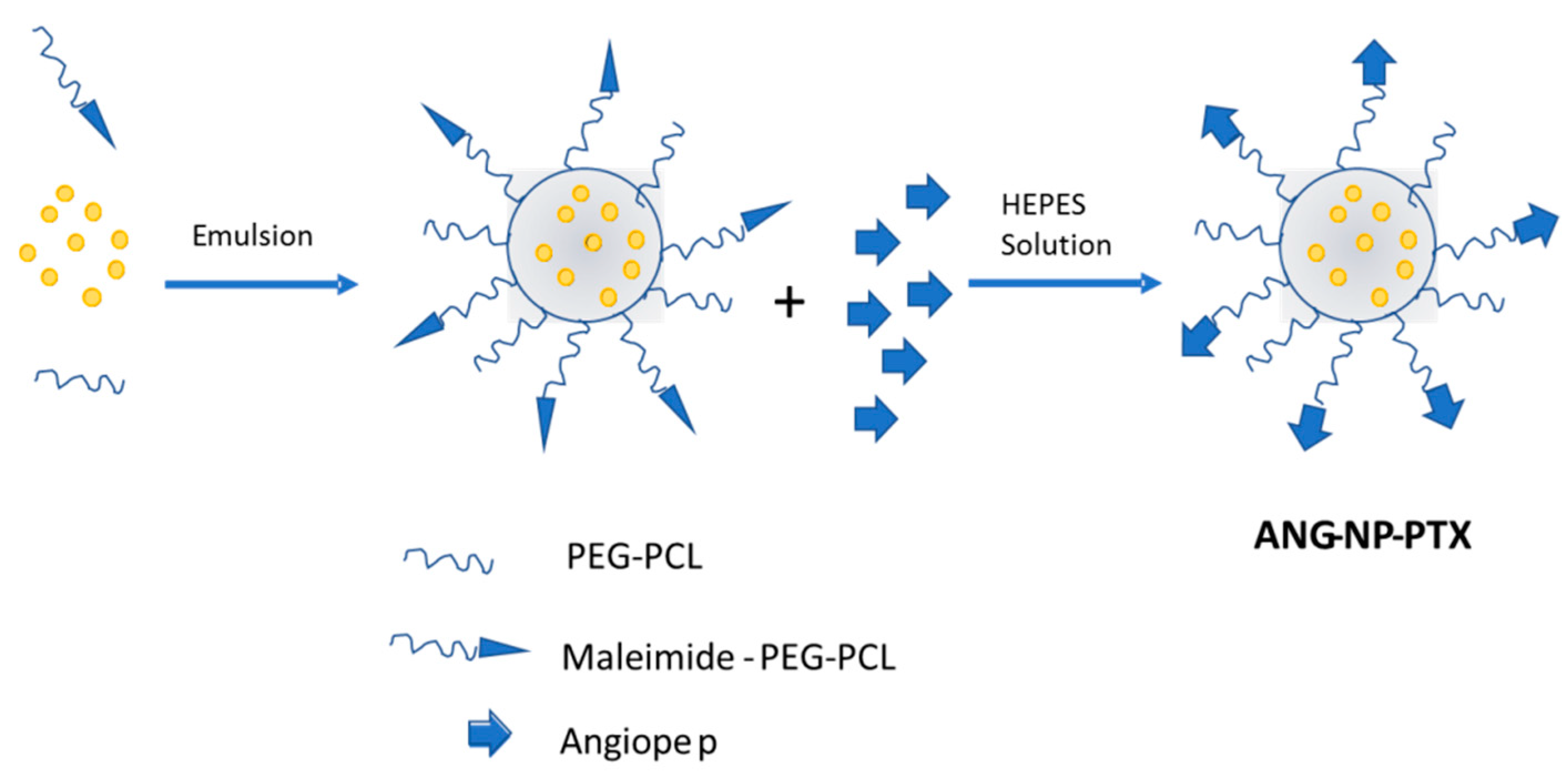
- Xin, H.; Jiang, X.; Gu, J.; Sha, X.; Chen, L.; Law, K.; Chen, Y.; Wang, X.; Jiang, Y.; Fang, X. Angiopep-conjugated poly(ethylene glycol)-co-poly(epsilon-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials 2011, 32, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, H.; Liao, Z.; Wang, Y.; Hu, Y.; Yang, J.; Shen, S.; Chen, J.; Mei, H.; Shi, W.; et al. EGFP–EGF1-conjugated nanoparticles for targeting both neovascular and glioma cells in therapy of brain glioma. Biomaterials 2014, 35, 4133–4145. [Google Scholar] [CrossRef] [PubMed]
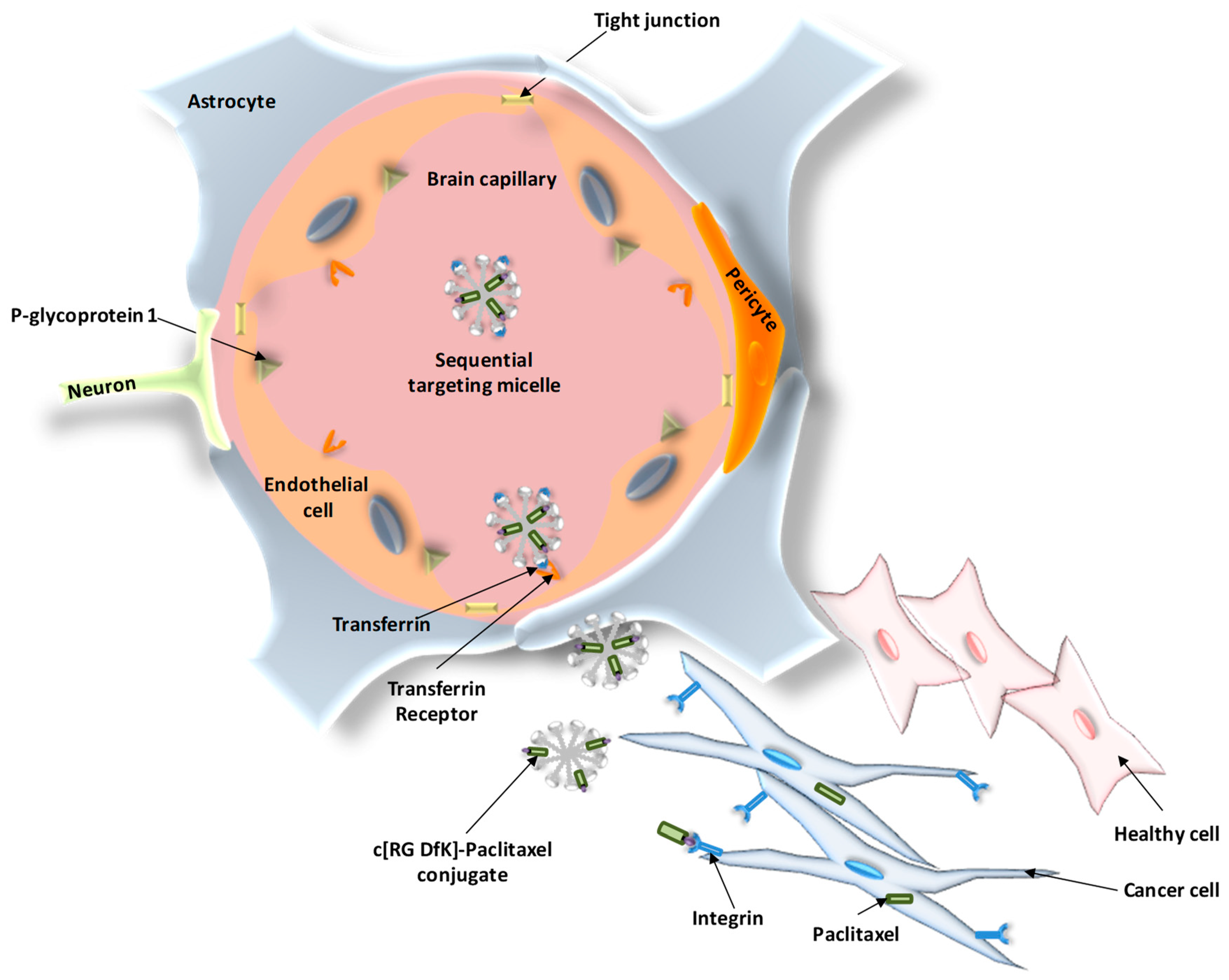
- Zhang, P.; Hu, L.; Yin, Q.; Feng, L.; Li, Y. Transferrin-modified c[RGDfK]-paclitaxel loaded hybrid micelle for sequential blood-brain barrier penetration and glioma targeting therapy. Mol. Pharm. 2012, 9, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Ma, D. Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale 2014, 6, 6415–6425. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.S.; Kwon, Y.J. Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications. Adv. Drug Deliv. Rev. 2012, 64, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Zarebkohan, A.; Najafi, F.; Moghimi, H.R.; Hemmati, M.; Deevband, M.R.; Kazemi, B. Synthesis and characterization of a PAMAM dendrimer nanocarrier functionalized by SRL peptide for targeted gene delivery to the brain. Eur. J. Pharm. Sci. 2015, 78, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Nance, E.; Timbie, K.; Miller, G.W.; Song, J.; Louttit, C.; Klibanov, A.L.; Shih, T.Y.; Swaminathan, G.; Tamargo, R.J.; Woodworth, G.F.; et al. Non-invasive delivery of stealth, brain-penetrating nanoparticles across the blood-brain barrier using MRI-guided focused ultrasound. J. Control. Release 2014, 189, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.P.; Mastorakos, P.; Suk, J.S.; Klibanov, A.L.; Hanes, J.; Price, R.J. Targeted gene transfer to the brain via the delivery of brain-penetrating DNA nanoparticles with focused ultrasound. J. Control. Release 2016, 223, 109–117. [Google Scholar] [CrossRef] [PubMed]
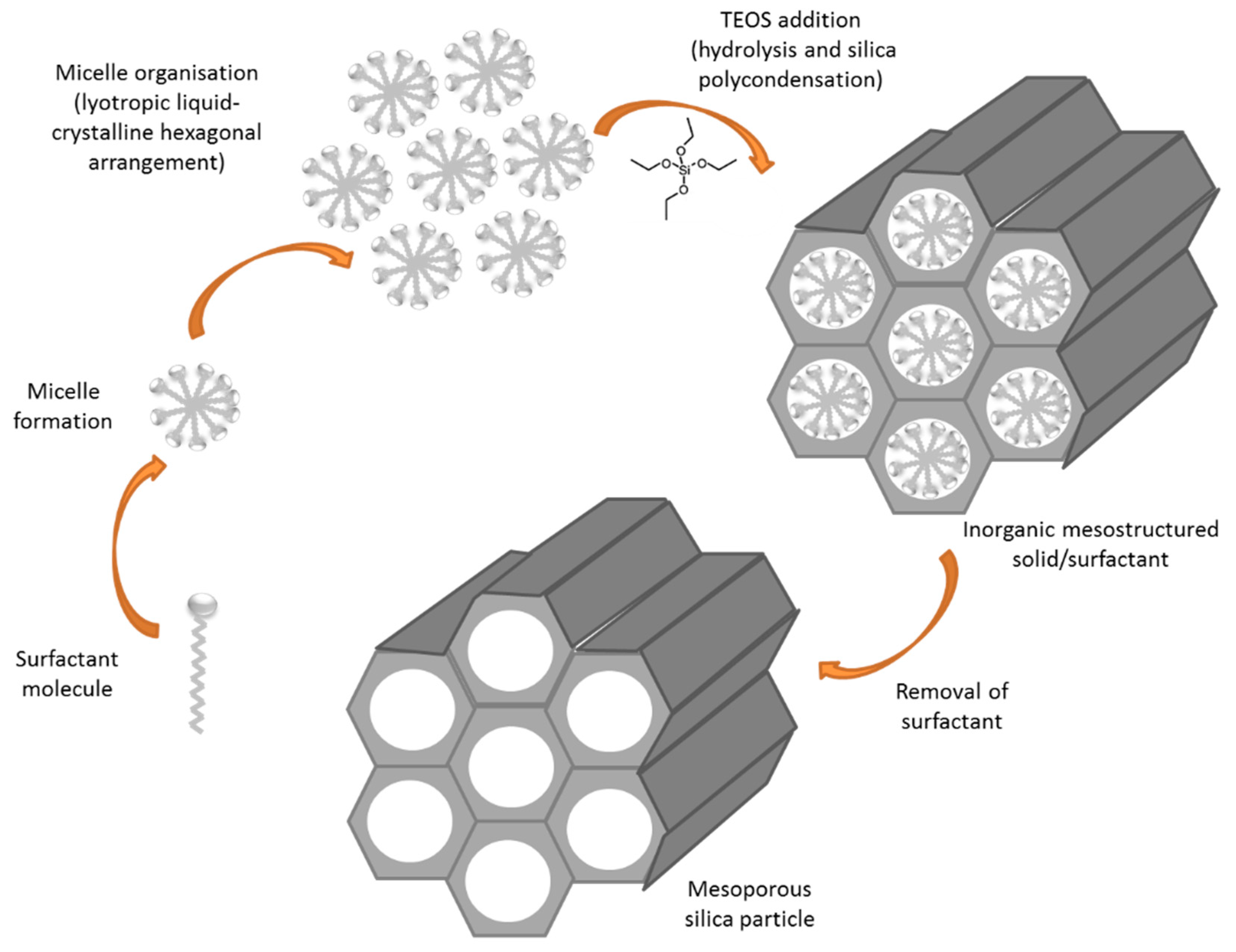
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A New Family of Mesoporous Molecular Sieves Prepared with Liquid Crystal Templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Raman, N.K.; Anderson, M.T.; Brinker, C.J. Template-based approaches to the preparation of amorphous, nanoporous silicas. Chem. Mater. 1996, 8, 1682–1701. [Google Scholar] [CrossRef]
- Mo, J.; He, L.; Ma, B.; Chen, T. Tailoring Particle Size of Mesoporous Silica Nanosystem To Antagonize Glioblastoma and Overcome Blood-Brain Barrier. ACS Appl. Mater. Interfaces 2016, 8, 6811–6825. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Liu, Y.; Wang, X.Q.; Liu, L.H.; Hu, J.J.; Luo, G.F.; Chen, W.H.; Rong, L.; Zhang, X.Z. One-pot construction of functional mesoporous silica nanoparticles for the tumor-acidity-activated synergistic chemotherapy of glioblastoma. ACS Appl. Mater. Interfaces 2013, 5, 7995–8001. [Google Scholar] [CrossRef] [PubMed]
- Kresge, C.T.; Roth, W.J. The discovery of mesoporous molecular sieves from the twenty year perspective. Chem. Soc. Rev. 2013, 42, 3663–3670. [Google Scholar] [CrossRef] [PubMed]
- Aznar, E.; Mondragon, L.; Ros-Lis, J.V.; Sancenon, F.; Marcos, M.D.; Martinez-Manez, R.; Soto, J.; Perez-Paya, E.; Amoros, P. Finely tuned temperature-controlled cargo release using paraffin-capped mesoporous silica nanoparticles. Angew. Chem. Int. Ed. Engl. 2011, 50, 11172–11175. [Google Scholar] [CrossRef] [PubMed]
- Bernardos, A.; Mondragón, L.; Aznar, E.; Marcos, M.D.; Martínez-Máñez, R.; Sancenón, F.; Soto, J.; Barat, J.M.; Pérez-Payá, E.; Guillem, C.; et al. Enzyme-responsive intracellular controlled release using nanometric silica mesoporous supports capped with “saccharides”. ACS Nano 2010, 4, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, C.; Agostini, A.; Mondragon, L.; Orzaez, M.; Sancenon, F.; Martinez-Manez, R.; Marcos, M.D.; Amoros, P.; Perez-Paya, E. Temperature-controlled release by changes in the secondary structure of peptides anchored onto mesoporous silica supports. Chem. Commun. 2014, 50, 3184–3186. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, K.; Gupta, S.; Gnanadhas, D.P.; Ramamurthy, P.C.; Chakravortty, D.; Raichur, A.M. Protamine-Capped Mesoporous Silica Nanoparticles for Biologically Triggered Drug Release. Part. Part. Syst. Charact. 2014, 31, 449–458. [Google Scholar] [CrossRef]
- Tarn, D.; Xue, M.; Zink, J.I. pH-responsive dual cargo delivery from mesoporous silica nanoparticles with a metal-latched nanogate. Inorg. Chem. 2013, 52, 2044–2049. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi Nasab, N.; Hassani Kumleh, H.; Beygzadeh, M.; Teimourian, S.; Kazemzad, M. Delivery of curcumin by a pH-responsive chitosan mesoporous silica nanoparticles for cancer treatment. Artif. Cells Nanomed. Biotechnol. 2018, 46, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, F.; Wang, H.; Niu, G.; Choi, K.Y.; Swierczewska, M.; Zhang, G.; Gao, H.; Wang, Z.; Zhu, L.; et al. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials 2013, 34, 1772–1780. [Google Scholar] [CrossRef] [PubMed]
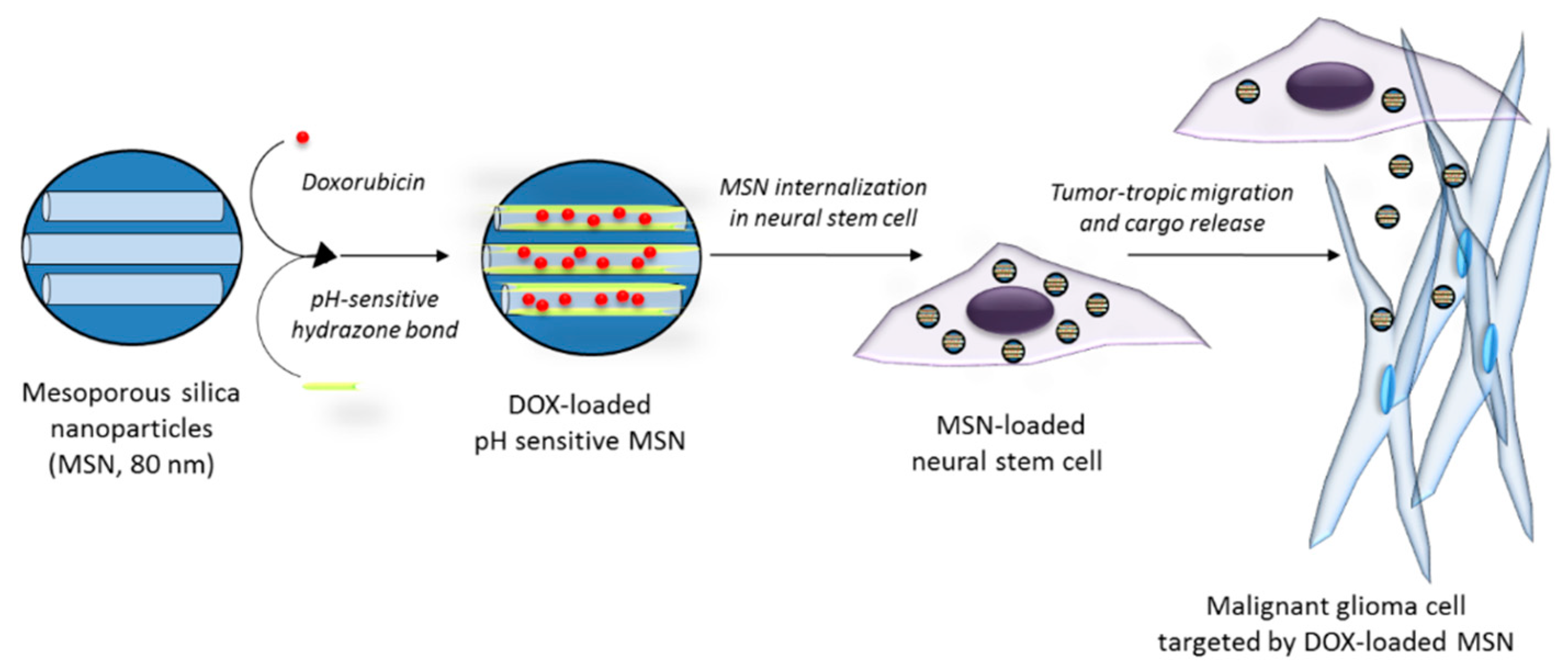
- Cheng, Y.; Morshed, R.; Cheng, S.H.; Tobias, A.; Auffinger, B.; Wainwright, D.A.; Zhang, L.; Yunis, C.; Han, Y.; Chen, C.T.; et al. Nanoparticle-programmed self-destructive neural stem cells for glioblastoma targeting and therapy. Small 2013, 9, 4123–4129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, W.; Zhou, Y.; Jiang, Y.; Li, S. Dual Functional Mesoporous Silicon Nanoparticles Enhance the Radiosensitivity of VPA in Glioblastoma. Transl. Oncol. 2017, 10, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Funkhouser, J. Reinventing pharma: The theranostic revolution. Curr. Drug Discov. 2002, 2, 17–19. [Google Scholar]
- Chowdhury, M.R.; Schumann, C.; Bhakta-Guha, D.; Guha, G. Cancer nanotheranostics: Strategies, promises and impediments. Biomed. Pharmacother. 2016, 84, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, P.; Kumar, S.U.; Matai, I.; Bhushan, B.; Malwal, D.; Sachdev, A.; Dubey, P. Cancer nanotheranostics. In Cancer Nanotheranostics; Springer: New York, NY, USA, 2015; pp. 1–93. [Google Scholar]
- Lammers, T.; Aime, S.; Hennink, W.E.; Storm, G.; Kiessling, F. Theranostic nanomedicine. Acc. Chem. Res. 2011, 44, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Blau, R.; Krivitsky, A.; Epshtein, Y.; Satchi-Fainaro, R. Are nanotheranostics and nanodiagnostics-guided drug delivery stepping stones towards precision medicine? Drug Resist. Updates 2016, 27, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Chen, F.; Hong, H.; Valdovinos, H.F.; Hernandez, R.; Shi, S.; Barnhart, T.E.; Cai, W. VEGF(1)(2)(1)-conjugated mesoporous silica nanoparticle: A tumor targeted drug delivery system. ACS Appl. Mater. Interfaces 2014, 6, 21677–21685. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Yu, D.; Tsai, H.M.; Morshed, R.A.; Kanojia, D.; Lo, L.W.; Leoni, L.; Govind, Y.; Zhang, L.; Aboody, K.S.; et al. Dynamic In Vivo SPECT Imaging of Neural Stem Cells Functionalized with Radiolabeled Nanoparticles for Tracking of Glioblastoma. J. Nucl. Med. 2016, 57, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-L.; Mao, K.-L.; Huang, Y.-P.; Yang, J.-J.; Xu, J.; Chen, P.-P.; Fan, Z.-L.; Zou, S.; Gao, Z.-Z.; Yin, J.-Y. Glioma-targeted superparamagnetic iron oxide nanoparticles as drug-carrying vehicles for theranostic effects. Nanoscale 2016, 8, 14222–14236. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Ifediba, M.A.; Ghosh, S.; Medarova, Z.; Moore, A. Combination treatment with theranostic nanoparticles for glioblastoma sensitization to TMZ. Mol. Imaging Biol. 2014, 16, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Wei, K.; Zou, F.; Zhong, S. Temozolomide loaded PLGA-based superparamagnetic nanoparticles for magnetic resonance imaging and treatment of malignant glioma. Int. J. Pharm. 2012, 430, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Le Fevre, R.; Durand-Dubief, M.; Chebbi, I.; Mandawala, C.; Lagroix, F.; Valet, J.P.; Idbaih, A.; Adam, C.; Delattre, J.Y.; Schmitt, C.; et al. Enhanced antitumor efficacy of biocompatible magnetosomes for the magnetic hyperthermia treatment of glioblastoma. Theranostics 2017, 7, 4618–4631. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, M.; Umemura, M.; Sato, I.; Akimoto, T.; Oda, K.; Nagasako, A.; Kim, J.H.; Fujita, T.; Yokoyama, U.; Nakayama, T.; et al. Hyperthermia and chemotherapy using Fe(Salen) nanoparticles might impact glioblastoma treatment. Sci. Rep. 2017, 7, 42783. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Joh, D.Y.; Al-Zaki, A.; Stangl, M.; Murty, S.; Davis, J.J.; Baumann, B.C.; Alonso-Basanta, M.; Kao, G.D.; Tsourkas, A.; et al. Theranostic application of mixed gold and superparamagnetic iron oxide nanoparticle micelles in glioblastoma multiforme. J. Biomed. Nanotechnol. 2016, 12, 347–356. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zeng, L.; Mai, X.; Shi, C.; Luo, L.; Chen, T. Nucleolin-targeted selenium nanocomposites with enhanced theranostic efficacy to antagonize glioblastoma. J. Mater. Chem. B 2017, 5, 3024–3034. [Google Scholar] [CrossRef]
- Runge, V.M.; Muroff, L.R.; Jinkins, J.R. Central nervous system: Review of clinical use of contrast media. Top. Magn. Reson. Imaging 2001, 12, 231–263. [Google Scholar] [CrossRef] [PubMed]
- Stefancikova, L.; Lacombe, S.; Salado, D.; Porcel, E.; Pagacova, E.; Tillement, O.; Lux, F.; Depes, D.; Kozubek, S.; Falk, M. Effect of gadolinium-based nanoparticles on nuclear DNA damage and repair in glioblastoma tumor cells. J. Nanobiotechnol. 2016, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Patel, T.R.; Sirianni, R.W.; Strohbehn, G.; Zheng, M.-Q.; Duong, N.; Schafbauer, T.; Huttner, A.J.; Huang, Y.; Carson, R.E. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc. Natl. Acad. Sci. USA 2013, 110, 11751–11756. [Google Scholar] [CrossRef] [PubMed]
- Çırpanlı, Y.; Allard, E.; Passirani, C.; Bilensoy, E.; Lemaire, L.; Çalış, S.; Benoit, J.-P. Antitumoral activity of camptothecin-loaded nanoparticles in 9L rat glioma model. Int. J. Pharm. 2011, 403, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Nance, E.A.; Mastorakos, P.; Chisholm, J.; Berry, S.; Eberhart, C.; Tyler, B.; Brem, H.; Suk, J.S.; Hanes, J. Convection enhanced delivery of cisplatin-loaded brain penetrating nanoparticles cures malignant glioma in rats. J. Control. Release 2017, 263, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Huang, W.T.; Liu, D.M.; Losic, D. Local co-administration of gene-silencing RNA and drugs in cancer therapy: State-of-the art and therapeutic potential. Cancer Treat. Rev. 2017, 55, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, D.-Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Mangraviti, A.; Tzeng, S.Y.; Kozielski, K.L.; Wang, Y.; Jin, Y.; Gullotti, D.; Pedone, M.; Buaron, N.; Liu, A.; Wilson, D.R. Polymeric nanoparticles for nonviral gene therapy extend brain tumor survival in vivo. ACS Nano 2015, 9, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Khan, O.F.; Suva, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Scholz, R.; Maier-Hauff, K.; van Landeghem, F.K.; Waldoefner, N.; Teichgraeber, U.; Pinkernelle, J.; Bruhn, H.; Neumann, F.; Thiesen, B. The effect of thermotherapy using magnetic nanoparticles on rat malignant glioma. J. Neuro Oncol. 2006, 78, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Orawa, H.; Budach, V.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neuro Oncol. 2011, 103, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Krauze, M.T.; Bringas, J.R.; Noble, C.; McKnight, T.R.; Jackson, P.; Wendland, M.F.; Mamot, C.; Drummond, D.C.; Kirpotin, D.B. Gadolinium-loaded liposomes allow for real-time magnetic resonance imaging of convection-enhanced delivery in the primate brain. Exp. Neurol. 2005, 196, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Strohbehn, G.; Coman, D.; Han, L.; Ragheb, R.R.; Fahmy, T.M.; Huttner, A.J.; Hyder, F.; Piepmeier, J.M.; Saltzman, W.M.; Zhou, J. Imaging the delivery of brain-penetrating PLGA nanoparticles in the brain using magnetic resonance. J. Neuro Oncol. 2015, 121, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Bernal, G.M.; LaRiviere, M.J.; Mansour, N.; Pytel, P.; Cahill, K.E.; Voce, D.J.; Kang, S.; Spretz, R.; Welp, U.; Noriega, S.E. Convection-enhanced delivery and in vivo imaging of polymeric nanoparticles for the treatment of malignant glioma. Nanomedicine 2014, 10, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Archer, G.; Pedain, C.; Wembacher-Schröder, E.; Westphal, M.; Kunwar, S.; Vogelbaum, M.A.; Coan, A.; Herndon, J.E.; Raghavan, R. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J. Neurosurg. 2010, 113, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, P.A.; Kievit, F.M.; Zhang, M.; Ellenbogen, R.G. Bionanotechnology and the future of glioma. Surg. Neurol. Int. 2015, 6, S45–S58. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeted Receptor/Stimuli | Ligand/Responsive Entity | Carrier (Size) | Drug | Beneficial Outcome | Reference |
|---|---|---|---|---|---|
| Chloride channel and MMP 2 | CTX peptide | CS nanoparticles (<100 nm) | TMZ | Higher uptake (2–6-fold) and IC50 reduction (50–90%) in glioma cell lines (U188, SF767, and GBM6) compared to CS nanoparticles without CTX and free TMZ | [79] |
| LDL | Angiopep-2 | PEG–PCL nanoparticles (<100 nm) | PTX | Improved transport across BBB (2-fold higher than Taxol) | [81] |
| Serine-arginine-leucine (SRL) peptide | Poly(amidoamine) (PAMAM) dendrimer | Plasmid pEGFP | Increased uptake and accumulation of DNA–PAMAM–SRL system in the brain compared with nontargeted systems | [86] | |
| Tissue factor | EGFP–EGF1 fusion protein | PEG–PLA nanoparticles (<150 nm) | PTX | Longer survival time of glioma-bearing mice (27 days) compared to saline group, Taxol group and nontargeted particles (14, 13, 21 days, respectively) | [82] |
| Transferrin receptor (Tfr1, also known as CD17) | Transferrin + modified c[RGDfK] | Micelle (98 nm) | PTX | Longer survival time of mice bearing intracranial U87 MG glioma (39.5 days) compared to PTX-loaded micelle (34.8 days), Taxol (33.6 days), and saline solution (34.5 days) | [83] |
| Acidic pH | Hydrazone bond | MSN | DOX + CPT | Increased drug release at pH 6.5 when compared to pH 7.4, improving the chemotherapeutic effect | [99] |
| Acidic pH | Hydrazone bond | MSN (80 nm) incorporated into neural stem cells | DOX | Tumortropic migration of neural stem cells carrying DOX-loaded MSN in an intracranial U87 xenograft mouse model, resulting in the induction of apoptosis and improvements in survival (41–42 days) compared to PBS (34 days) | [102] |
| Nanoparticle Type | Cargo | Surface Functionalization | Contrast Agent | Detection Method | Combined Therapy | Reference |
|---|---|---|---|---|---|---|
| MSN | Sunitinib | VEGF121 and 64Cu | 64Cu | PET | - | [117] |
| MSN | - | In111 | In111 | SPECT and fluorescence microscopy | - | [118] |
| SPION | DOX | - | Iron oxide | MRI | Magnetic hyperthermia | [119] |
| SPION | TMZ and siRNA of the MGMT gene | - | Iron oxide | MRI | - | [120] |
| PLGA-SPION | TMZ | PLGA coating | Iron oxide | MRI | - | [121] |
| SPION | - | Lysine coating | Iron oxide | MRI | Magnetic hyperthermia | [122] |
| Fe(Salen) nanoparticles | - | - | Iron oxide | MRI | Magnetic hyperthermia | [123] |
| Micelles | SPION and Au nanoparticles | PEG-PCL coating | Iron oxide | MRI | Radiotherapy | [124] |
| Selenium nanoparticles | CdTe/ZnS quantum dots and ruthenium complexes | - | Quantum dots | Fluorescence | - | [125] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, L.; Coll, C.; Erthal, L.C.S.; De la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.J.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779. https://doi.org/10.3390/ma11050779
Nam L, Coll C, Erthal LCS, De la Torre C, Serrano D, Martínez-Máñez R, Santos-Martínez MJ, Ruiz-Hernández E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials. 2018; 11(5):779. https://doi.org/10.3390/ma11050779
Chicago/Turabian StyleNam, L., C. Coll, L. C. S. Erthal, C. De la Torre, D. Serrano, R. Martínez-Máñez, M. J. Santos-Martínez, and E. Ruiz-Hernández. 2018. "Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme" Materials 11, no. 5: 779. https://doi.org/10.3390/ma11050779
APA StyleNam, L., Coll, C., Erthal, L. C. S., De la Torre, C., Serrano, D., Martínez-Máñez, R., Santos-Martínez, M. J., & Ruiz-Hernández, E. (2018). Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials, 11(5), 779. https://doi.org/10.3390/ma11050779