Effect of DC Plasma Electrolytic Oxidation on Surface Characteristics and Corrosion Resistance of Zirconium



Abstract
:1. Introduction
2. Materials and Methods
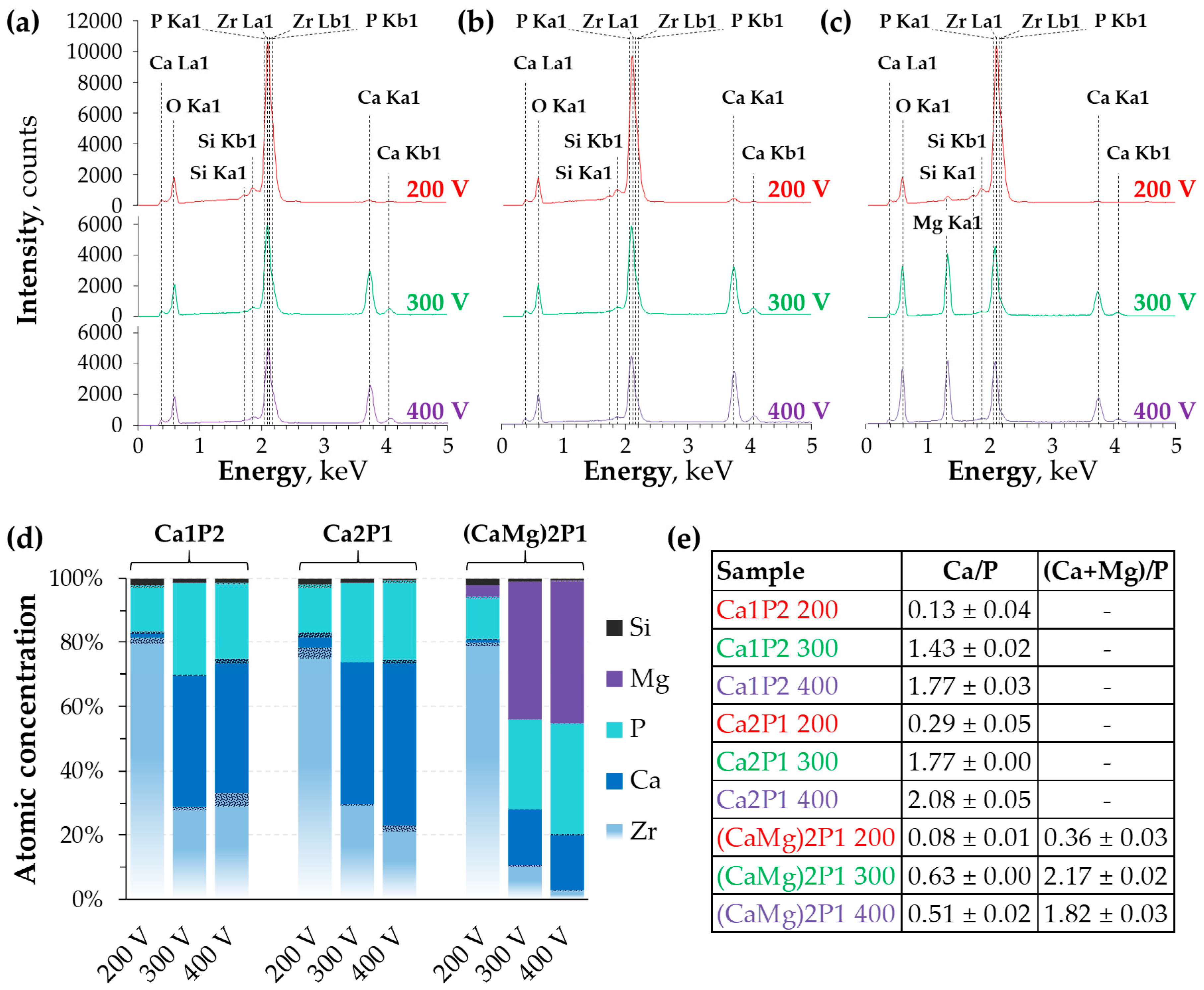
- Ca1P2: 0.5 M Ca(H2PO2)2 solution; Ca/P molar ratio = 0.5;
- Ca2P1: 0.1 M Ca(H2PO2)2 + 0.3 M Ca(HCOO)2 solution; Ca/P molar ratio = 2.0; and,
- (CaMg)2P1: 0.5 M Ca(H2PO2)2 + 1.5 M Mg(CH3COO)2 solution; (Ca+Mg)/P molar ratio = 2.0.
- electrochemical impedance spectroscopy (EIS) at EOC, the amplitude of 10 mV (RMS—root mean square), in the frequency range of 100,000–0.01 Hz and 10 points per decade of frequency. The experiment took approximately 1 h; and,
- potentiodynamic polarization (PDP) scan (0.167 mV/s) from −30 mV vs. EOC to 2000 mV vs. SCE and a reverse scan to −200 mV vs. EOC. If the oxide breakdown was observed in the experiment the reverse scan was commenced after its detection.
3. Results and Discussion
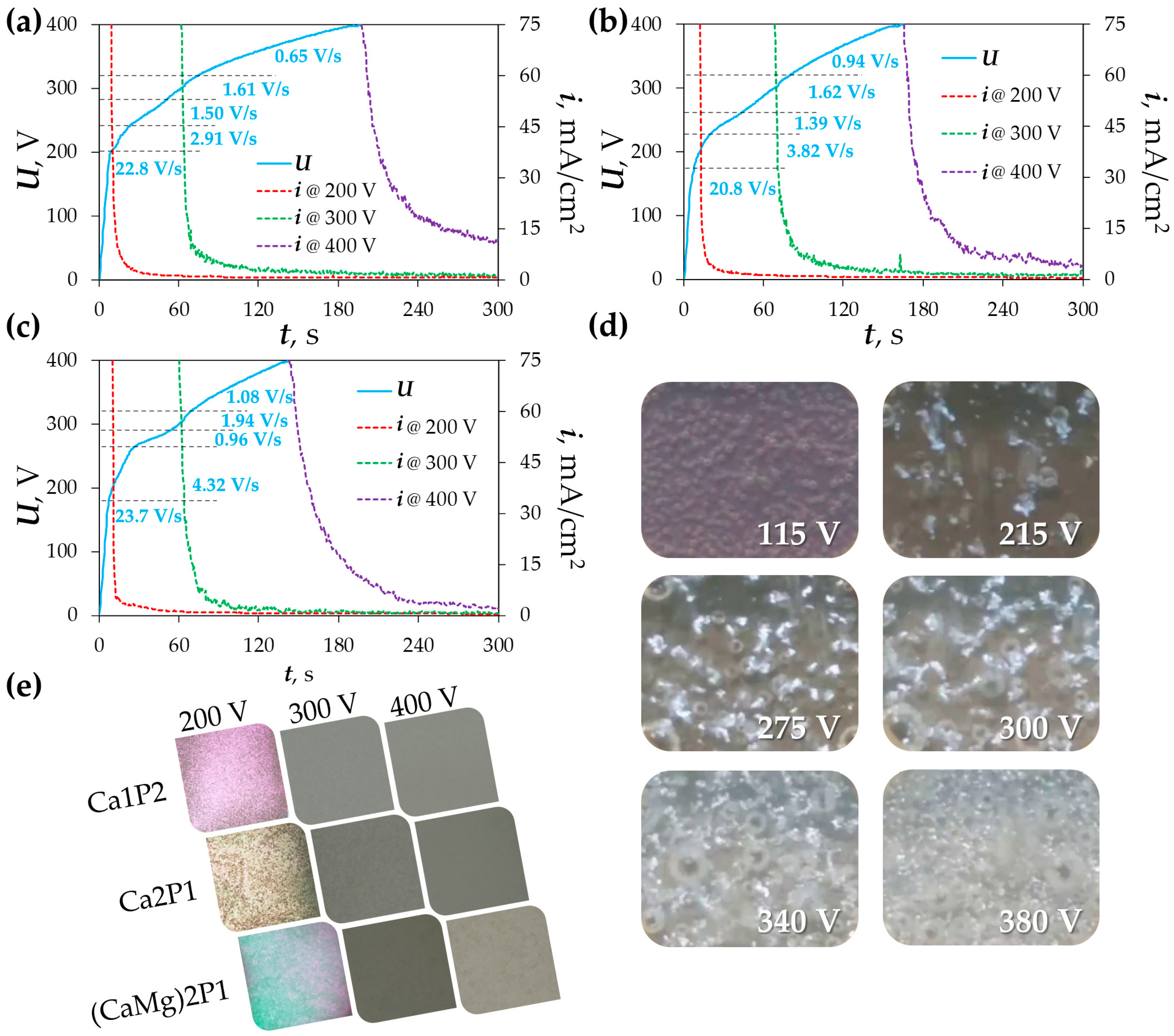
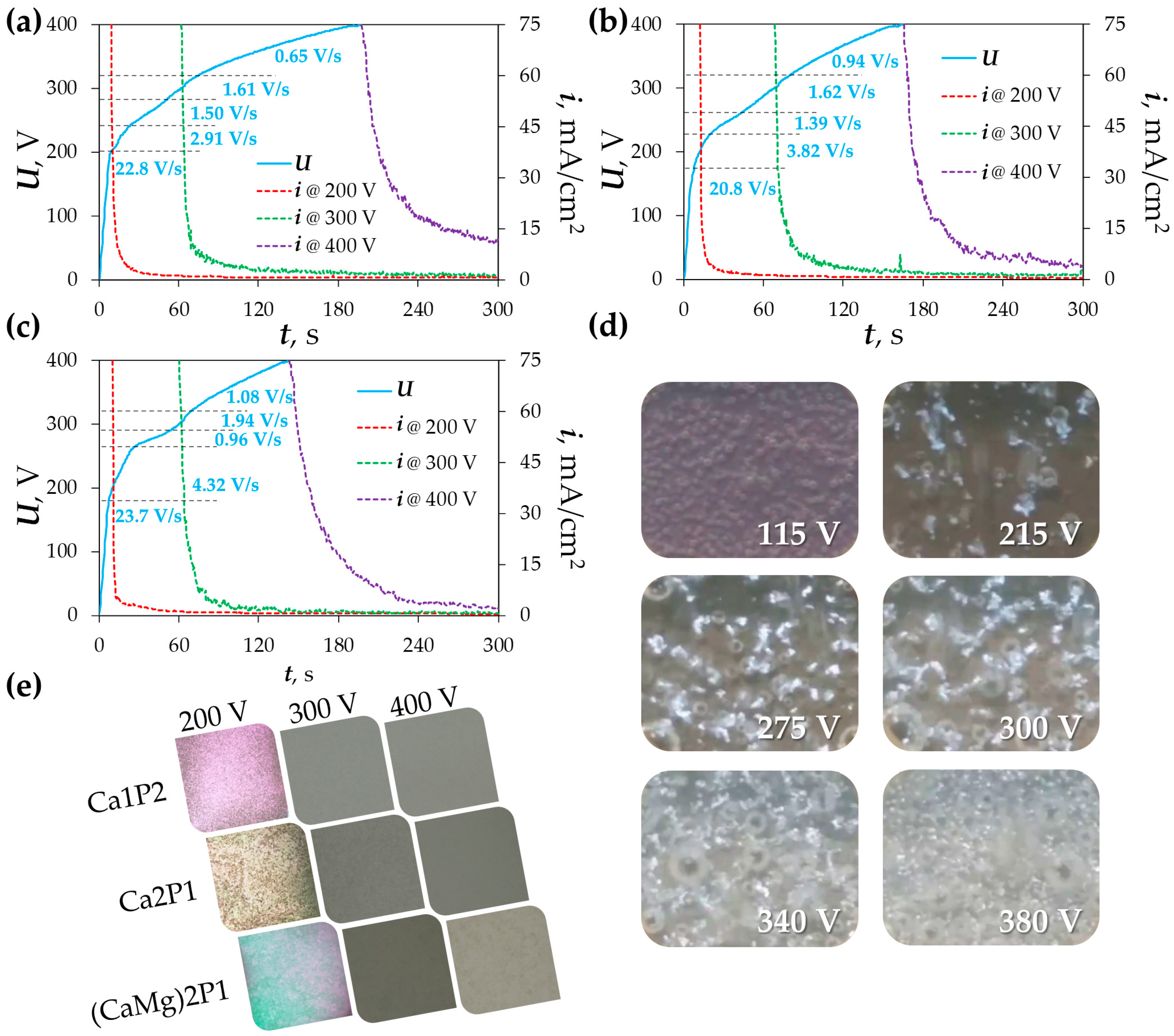
3.1. Plasma Electrolytic Oxidation
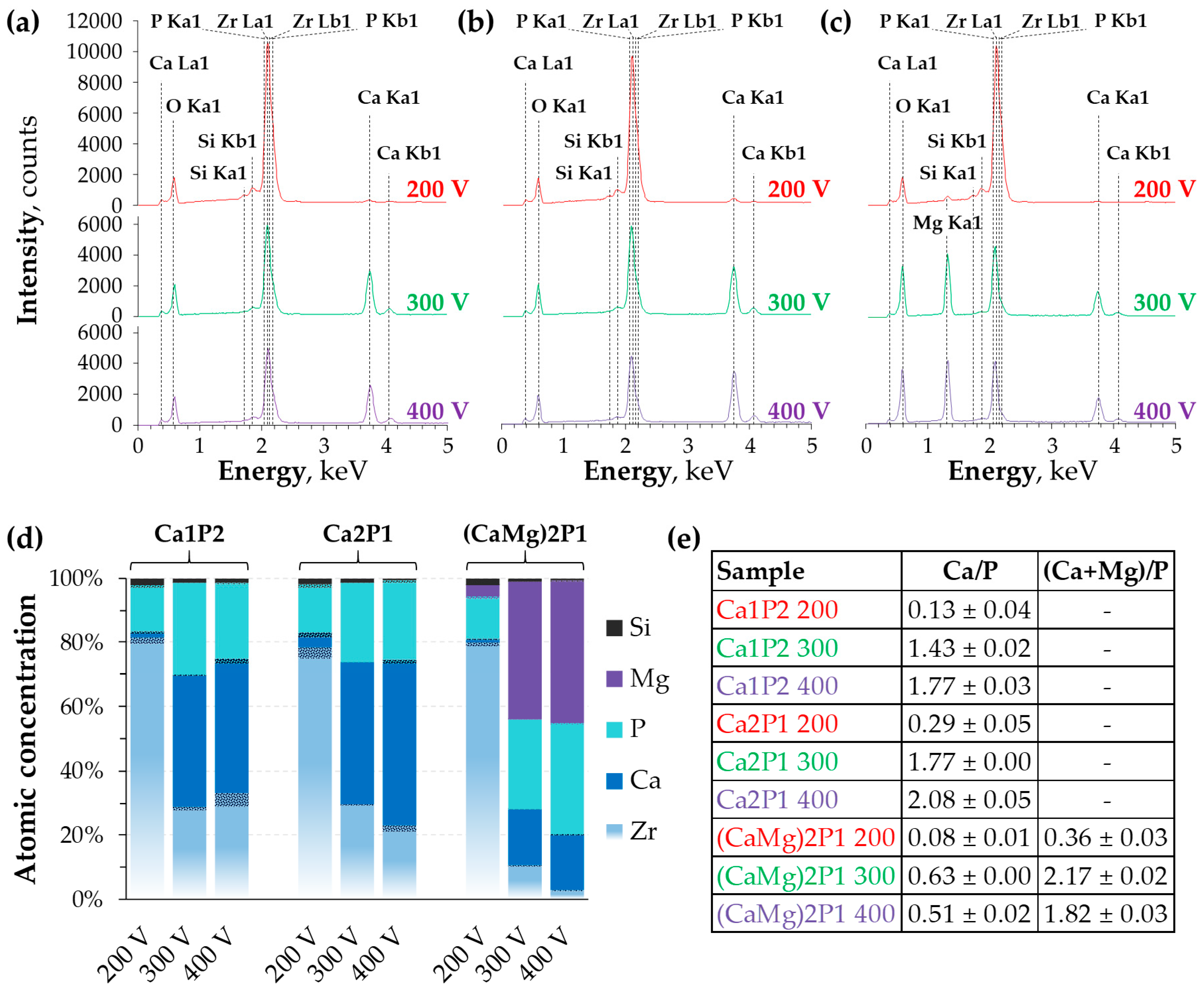
3.2. Surface Morphology and Elemental Composition
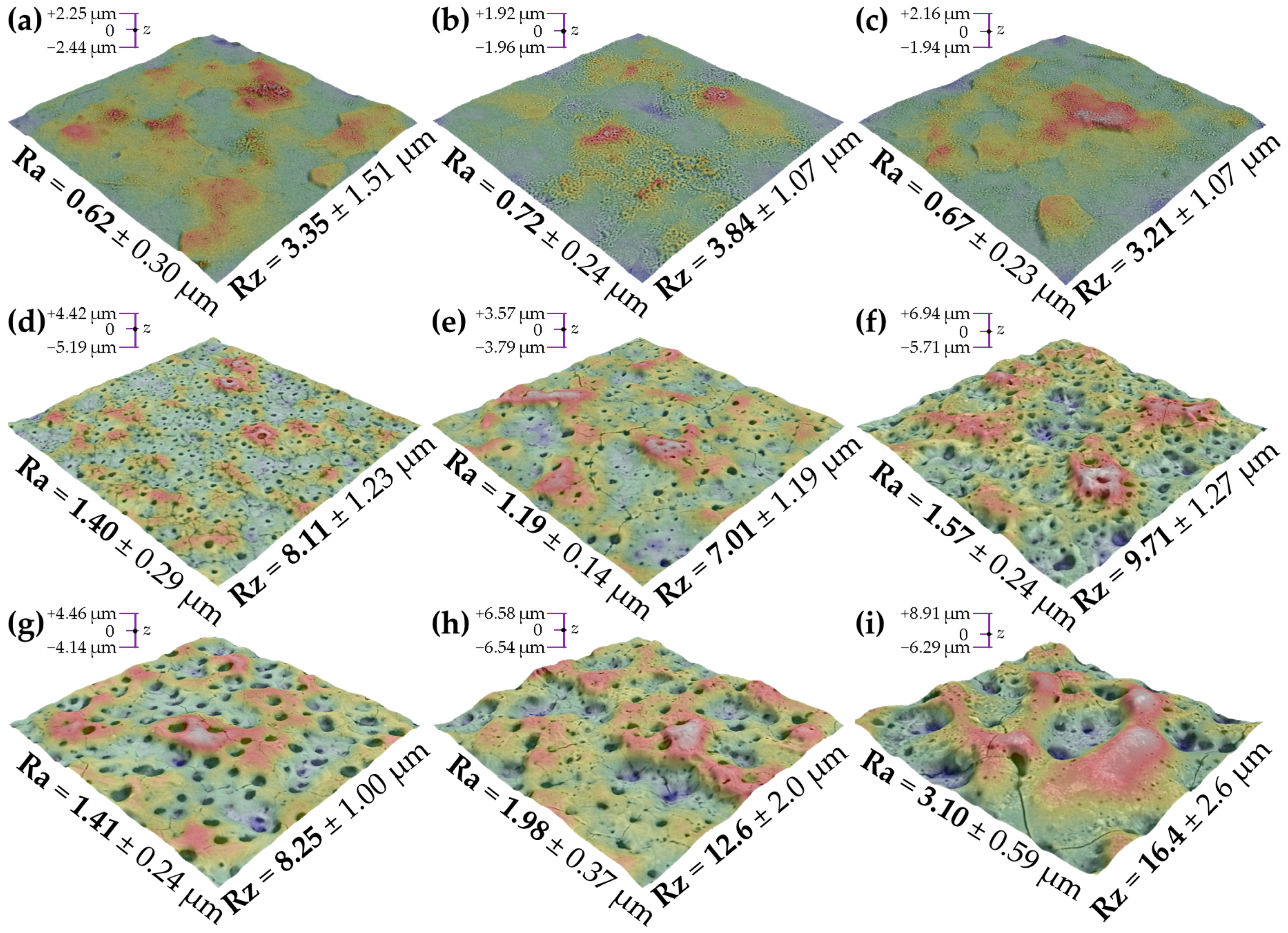
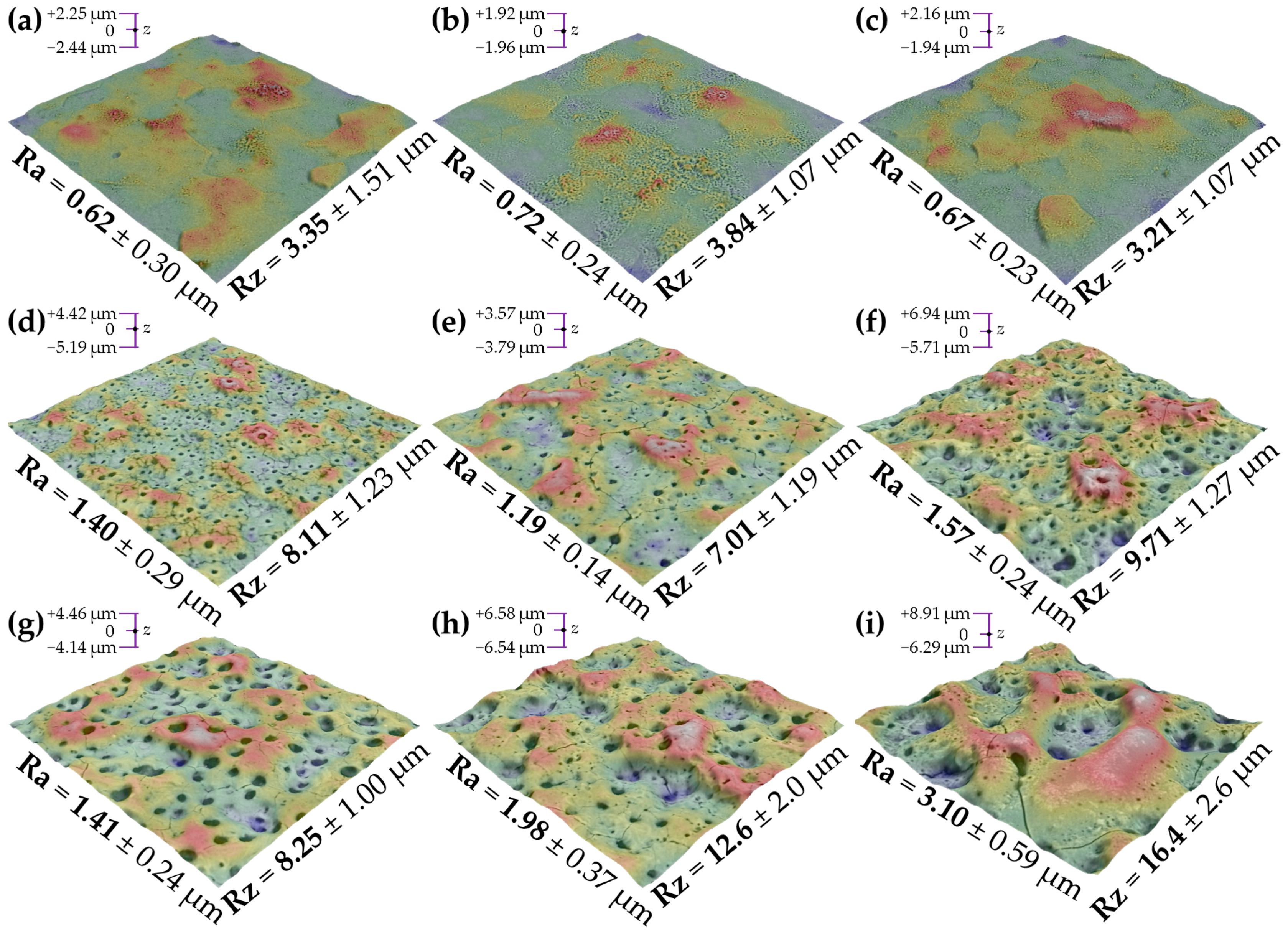
3.3. Coatings’ Thickness and Structure
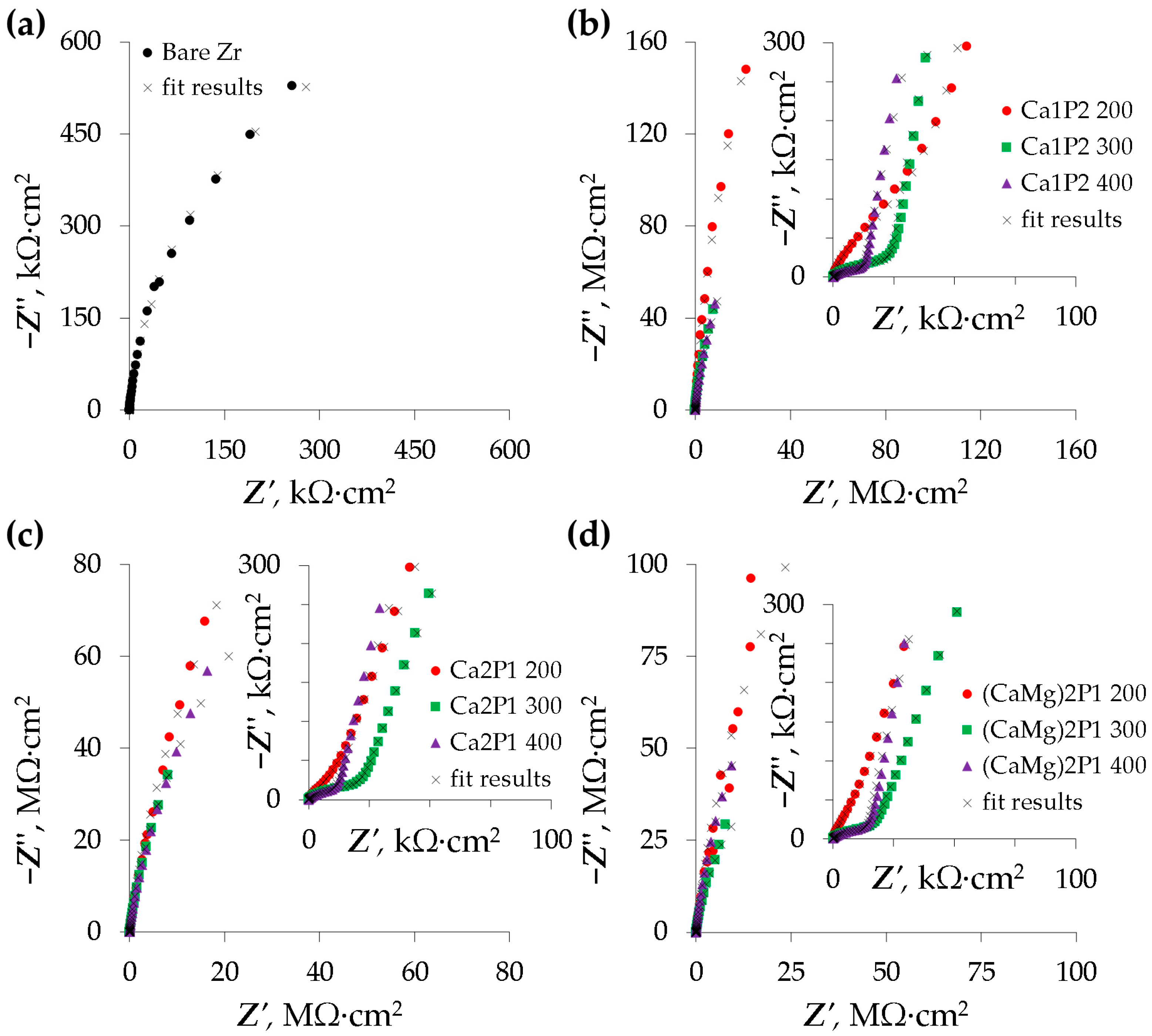
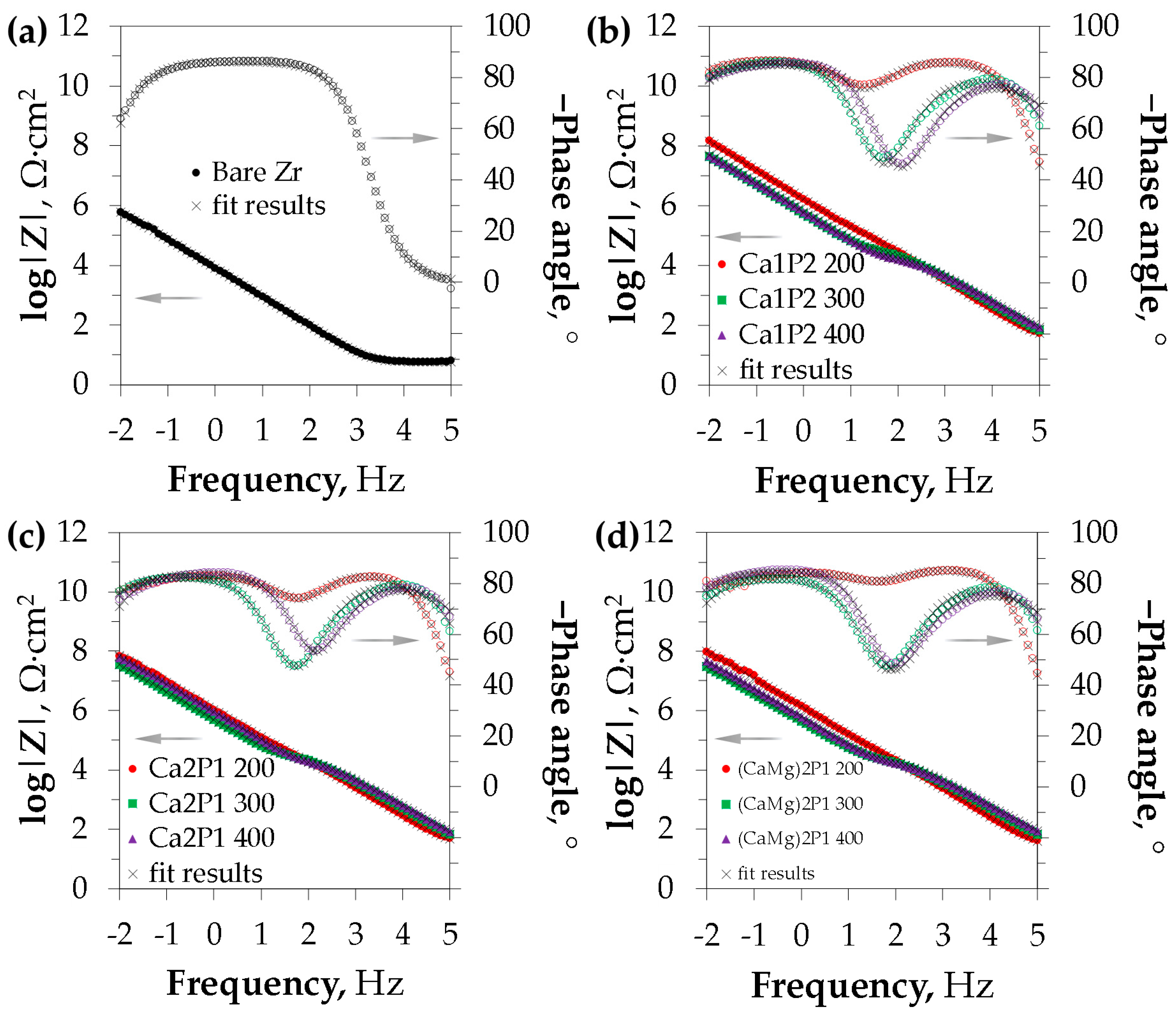
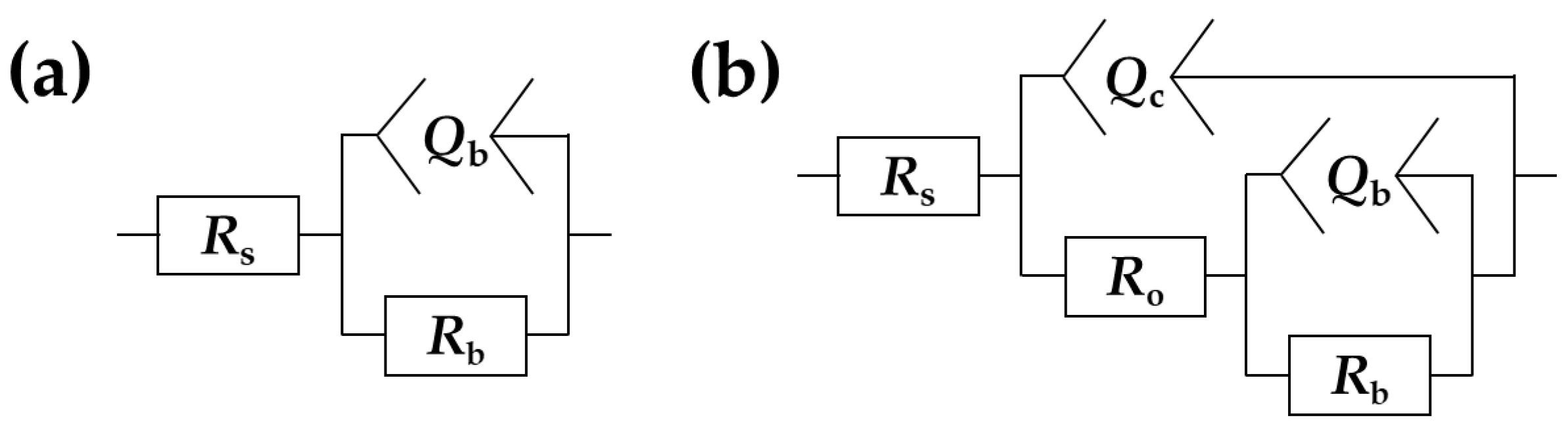
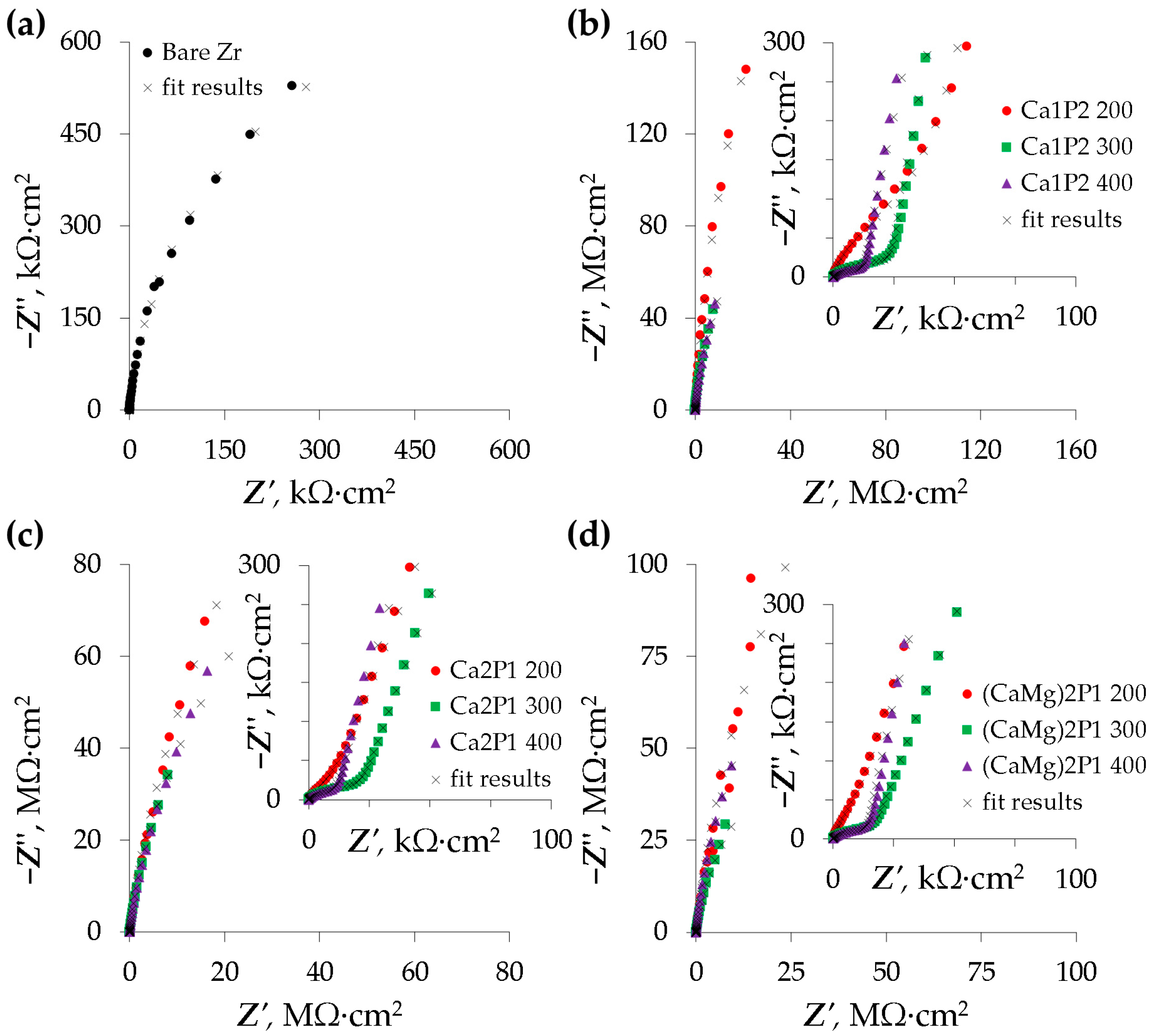
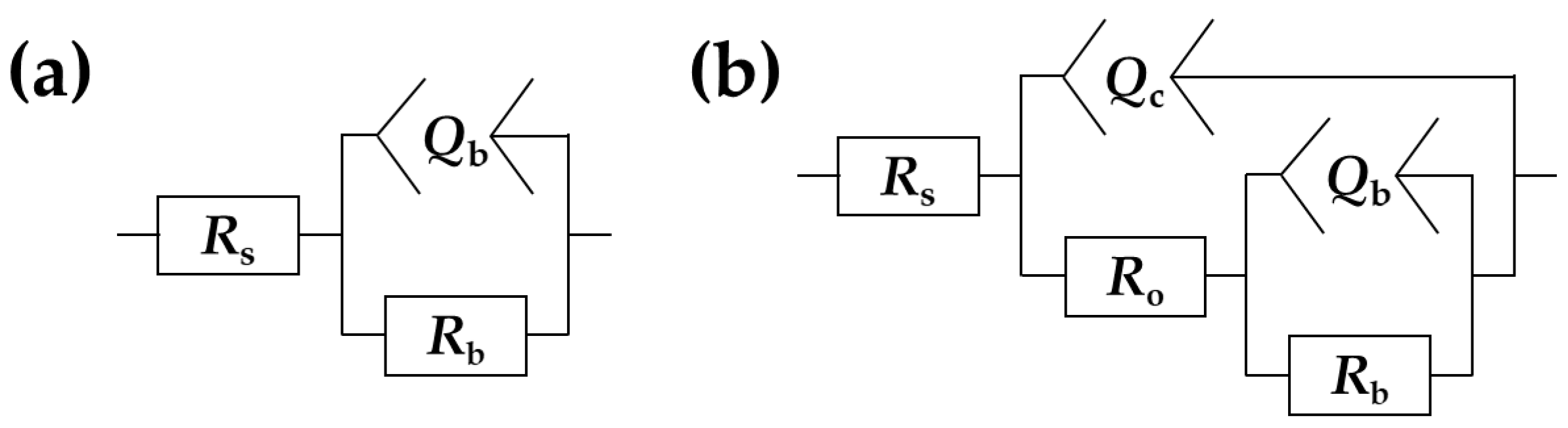
3.4. Electrochemical Impedance Spectroscopy
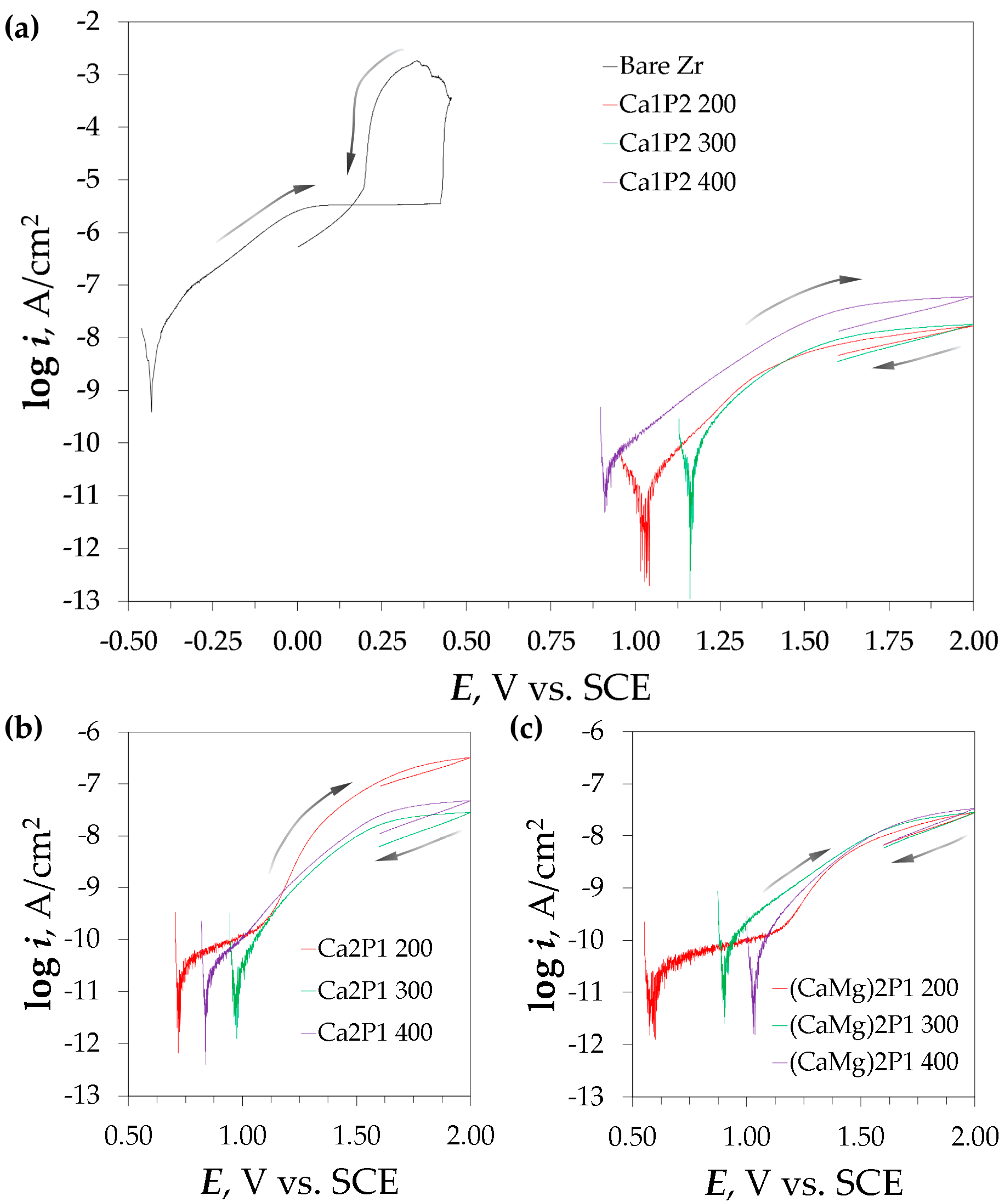
3.5. Polarization Experiments
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shue, L.; Yufeng, Z.; Mony, U. Biomaterials for periodontal regeneration. Biomatter 2012, 2, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.S.; Laurencin, C.T. Polymers as biomaterials for tissue engineering and controlled drug delivery. In Tissue Engineering I; Springer: Berlin Heidelberg, 2005; pp. 47–90. [Google Scholar]
- Brunski, J.B. Metals. In Biomaterials Science: An Introduction to Materials in Medicine; Ratner, B.D., Hoffman, A.S., Schoen, F.J., Lemons, J.E., Eds.; Academic Press: San Diego, CA, USA, 1996; pp. 37–50. [Google Scholar]
- Donato, T.A.G.; de Almeida, L.H.; Nogueira, R.A.; Niemeyer, T.C.; Grandini, C.R.; Caram, R.; Schneider, S.G.; Santos, A.R. Cytotoxicity study of some Ti alloys used as biomaterial. Mater. Sci. Eng. C 2009, 29, 1365–1369. [Google Scholar] [CrossRef]
- Gupta, V.B.; Anitha, S.; Hegde, M.L.; Zecca, L.; Garruto, R.M.; Ravid, R.; Shankar, S.K.; Stein, R.; Shanmugavelu, P.; Jagannatha Rao, K.S. Aluminium in Alzheimer’s disease: Are we still at a crossroad? Cell. Mol. Life Sci. 2005, 62, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Geetha, M.; Singh, A.K.; Asokamani, R.; Gogia, A.K. Ti based biomaterials, the ultimate choice for orthopaedic implants—A review. Prog. Mater. Sci. 2009, 54, 397–425. [Google Scholar] [CrossRef]
- Eisenbarth, E.; Velten, D.; Müller, M.; Thull, R.; Breme, J. Biocompatibility of β-stabilizing elements of titanium alloys. Biomaterials 2004, 25, 5705–5713. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, Z.-M.; Zhao, X.-H.; Gao, Y.; Hu, J.; Gao, B. Improved biological performance of microarc-oxidized low-modulus Ti-24Nb-4Zr-7.9Sn alloy. J. Biomed. Mater. Res. Part B Appl. Biomater. 2010, 92, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Rack, H.J. Titanium alloys in total joint replacement--a materials science perspective. Biomaterials 1998, 19, 1621–1639. [Google Scholar] [CrossRef]
- Niinomi, M. Recent research and development in titanium alloys for biomedical applications and healthcare goods. Sci. Technol. Adv. Mater. 2003, 4, 445–454. [Google Scholar] [CrossRef]
- Hanawa, T. Metal ion release from metal implants. Mater. Sci. Eng. C 2004, 24, 745–752. [Google Scholar] [CrossRef]
- Cremasco, A.; Messias, A.D.; Esposito, A.R.; Duek, E.A.D.R.; Caram, R. Effects of alloying elements on the cytotoxic response of titanium alloys. Mater. Sci. Eng. C 2011, 31, 833–839. [Google Scholar] [CrossRef]
- Chen, J.-S.; Bronson, A.; Knittel, D.R. Pitting corrosion on zirconium in KCl and KCl-H2SO4 solutions. Corrosion 1985, 41, 438–445. [Google Scholar] [CrossRef]
- Piconi, C.; Maccauro, G. Zirconia as a ceramic biomaterial. Biomaterials 1999, 20, 1–25. [Google Scholar] [CrossRef]
- Chevalier, J. What future for zirconia as a biomaterial? Biomaterials 2006, 27, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Hisbergues, M.; Vendeville, S.; Vendeville, P. Zirconia: Established facts and perspectives for a biomaterial in dental implantology. J. Biomed. Mater. Res. Part B Appl. Biomater. 2009, 88B, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Olmedo, D.G.; Tasat, D.R.; Evelson, P.; Rebagliatti, R.; Guglielmotti, M.B.; Cabrini, R.L. In vivo comparative biokinetics and biocompatibility of titanium and zirconium microparticles. J. Biomed. Mater. Res. Part A 2011, 98A, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphate-based bioceramics. Materials 2013, 6, 3840–3942. [Google Scholar] [CrossRef] [PubMed]
- Klinkaewnarong, J.; Sritonwong, P.; Swatsitang, E. Magnesium substitution in hydroxyapatite synthesized by sol-gel method. Adv. Mater. Res. 2012, 506, 150–153. [Google Scholar] [CrossRef]
- Lala, S.; Ghosh, M.; Das, P.K.; Das, D.; Kar, T.; Pradhan, S.K. Magnesium substitution in carbonated hydroxyapatite: Structural and microstructural characterization by Rietveld’s refinement. Mater. Chem. Phys. 2016, 170, 319–329. [Google Scholar] [CrossRef]
- Nabiyouni, M.; Ren, Y.; Bhaduri, S.B. Magnesium substitution in the structure of orthopedic nanoparticles: A comparison between amorphous magnesium phosphates, calcium magnesium phosphates, and hydroxyapatites. Mater. Sci. Eng. C 2015, 52, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Mehrjoo, M.; Javadpour, J.; Shokrgozar, M.A.; Farokhi, M.; Javadian, S.; Bonakdar, S. Effect of magnesium substitution on structural and biological properties of synthetic hydroxyapatite powder. Mater. Express 2015, 5, 41–48. [Google Scholar] [CrossRef]
- Liu, X.; Chu, P.K.; Ding, C. Surface nano-functionalization of biomaterials. Mater. Sci. Eng. R Rep. 2010, 70, 275–302. [Google Scholar] [CrossRef]
- Balamurugan, A.; Rajeswari, S.; Balossier, G.; Rebelo, A.H.S.; Ferreira, J.M.F. Corrosion aspects of metallic implants–An overview. Mater. Corros. 2008, 59, 855–869. [Google Scholar] [CrossRef]
- Yerokhin, A.L.; Nie, X.; Leyland, A.; Matthews, A.; Dowey, S.J. Plasma electrolysis for surface engineering. Surf. Coat. Technol. 1999, 122, 73–93. [Google Scholar] [CrossRef]
- Dzhurinskiy, D.; Gao, Y.; Yeung, W.-K.; Strumban, E.; Leshchinsky, V.; Chu, P.-J.; Matthews, A.; Yerokhin, A.; Maev, R.G. Characterization and corrosion evaluation of TiO2: n-HA coatings on titanium alloy formed by plasma electrolytic oxidation. Surf. Coat. Technol. 2015, 269, 258–265. [Google Scholar] [CrossRef]
- Arrabal, R.; Matykina, E.; Hashimoto, T.; Skeldon, P.; Thompson, G.E. Characterization of AC PEO coatings on magnesium alloys. Surf. Coat. Technol. 2009, 203, 2207–2220. [Google Scholar] [CrossRef]
- Matykina, E.; Berkani, A.; Skeldon, P.; Thompson, G.E. Real-time imaging of coating growth during plasma electrolytic oxidation of titanium. Electrochim. Acta 2007, 53, 1987–1994. [Google Scholar] [CrossRef]
- Krząkała, A.; Kazek-Kęsik, A.; Simka, W. Application of plasma electrolytic oxidation to bioactive surface formation on titanium and its alloys. RSC Adv. 2013, 3, 19725. [Google Scholar] [CrossRef]
- Rokosz, K.; Hryniewicz, T.; Raaen, S.; Chapon, P.; Prima, F. Development of copper-enriched porous coatings on ternary Ti-Nb-Zr alloy by plasma electrolytic oxidation. Int. J. Adv. Manuf. Technol. 2017, 89, 2953–2965. [Google Scholar] [CrossRef]
- Rokosz, K.; Hryniewicz, T. Comparative SEM and EDX analysis of surface coatings created on niobium and titanium alloys after plasma electrolytic oxidation (PEO). Teh. Vjesn. Tech. Gaz. 2017, 24, 465–472. [Google Scholar] [CrossRef]
- Cheng, Y.; Wu, F. Plasma electrolytic oxidation of Zircaloy-4 alloy with DC regime and properties of coatings. Trans. Nonferrous Met. Soc. China 2012, 22, 1638–1646. [Google Scholar] [CrossRef]
- Cheng, Y.; Peng, Z.; Wu, X.; Cao, J.; Skeldon, P.; Thompson, G.E. A comparison of plasma electrolytic oxidation of Ti-6Al-4V and Zircaloy-2 alloys in a silicate-hexametaphosphate electrolyte. Electrochim. Acta 2015, 165, 301–313. [Google Scholar] [CrossRef]
- Matykina, E.; Arrabal, R.; Skeldon, P.; Thompson, G.E.E.; Wang, P.; Wood, P. Plasma electrolytic oxidation of a zirconium alloy under AC conditions. Surf. Coat. Technol. 2010, 204, 2142–2151. [Google Scholar] [CrossRef]
- Xue, W.; Zhu, Q.; Jin, Q.; Hua, M. Characterization of ceramic coatings fabricated on zirconium alloy by plasma electrolytic oxidation in silicate electrolyte. Mater. Chem. Phys. 2010, 120, 656–660. [Google Scholar] [CrossRef]
- Farrakhov, R.G.; Mukaeva, V.R.; Fatkullin, A.R.; Gorbatkov, M.V.; Tarasov, P.V.; Lazarev, D.M.; Ramesh Babu, N.; Parfenov, E.V. Plasma electrolytic oxidation treatment mode influence on corrosion properties of coatings obtained on Zr-1Nb alloy in silicate-phosphate electrolyte. IOP Conf. Ser. Mater. Sci. Eng. 2018, 292, 12006. [Google Scholar] [CrossRef]
- Yan, Y.; Han, Y. Structure and bioactivity of micro-arc oxidized zirconia films. Surf. Coat. Technol. 2007, 201, 5692–5695. [Google Scholar] [CrossRef]
- Stojadinović, S.; Vasilić, R.; Petković, M.; Belča, I.; Kasalica, B.; Perić, M.; Zeković, L. Luminescence during the anodization of zirconium. Electrochim. Acta 2012, 79, 133–140. [Google Scholar] [CrossRef]
- Stojadinović, S.; Tadić, N.; Vasilić, R. Down-conversion photoluminescence of ZrO2: Er3+ coatings formed by plasma electrolytic oxidation. Mater. Lett. 2018, 219, 251–255. [Google Scholar] [CrossRef]
- Trivinho-Strixino, F.; da Silva, D.X.; Paiva-Santos, C.O.; Pereira, E.C. Tetragonal to monoclinic phase transition observed during Zr anodisation. J. Solid State Electrochem. 2013, 17, 191–199. [Google Scholar] [CrossRef]
- Sandhyarani, M.; Rameshbabu, N.; Venkateswarlu, K.; Sreekanth, D.; Subrahmanyam, C. Surface morphology, corrosion resistance and in vitro bioactivity of P containing ZrO2 films formed on Zr by plasma electrolytic oxidation. J. Alloys Compd. 2013, 553, 324–332. [Google Scholar] [CrossRef]
- Cengiz, S.; Uzunoglu, A.; Stanciu, L.; Tarakci, M.; Gencer, Y. Direct fabrication of crystalline hydroxyapatite coating on zirconium by single-step plasma electrolytic oxidation process. Surf. Coat. Technol. 2016, 301, 74–79. [Google Scholar] [CrossRef]
- Fidan, S.; Muhaffel, F.; Riool, M.; Cempura, G.; de Boer, L.; Zaat, S.A.J.; Filemonowicz, A.C.; Cimenoglu, H. Fabrication of oxide layer on zirconium by micro-arc oxidation: Structural and antimicrobial characteristics. Mater. Sci. Eng. C 2017, 71, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, S.; Gencer, Y. The characterization of the oxide based coating synthesized on pure zirconium by plasma electrolytic oxidation. Surf. Coat. Technol. 2014, 242, 132–140. [Google Scholar] [CrossRef]
- Cengiz, S.; Azakli, Y.; Tarakci, M.; Stanciu, L.; Gencer, Y. Microarc oxidation discharge types and bio properties of the coating synthesized on zirconium. Mater. Sci. Eng. C 2017, 77, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-Y.Y.; Tsutsumi, Y.; Doi, H.; Nomura, N.; Kim, K.-H.H.; Hanawa, T. Enhancement of calcium phosphate formation on zirconium by micro-arc oxidation and chemical treatments. Surf. Coat. Technol. 2011, 205, 4948–4955. [Google Scholar] [CrossRef]
- Sandhyarani, M.; Rameshbabu, N.; Venkateswarlu, K.; Rama Krishna, L. Fabrication, characterization and in-vitro evaluation of nanostructured zirconia/hydroxyapatite composite film on zirconium. Surf. Coat. Technol. 2014, 238, 58–67. [Google Scholar] [CrossRef]
- Sandhyarani, M.; Prasadrao, T.; Rameshbabu, N. Role of electrolyte composition on structural, morphological and in-vitro biological properties of plasma electrolytic oxidation films formed on zirconium. Appl. Surf. Sci. 2014, 317, 198–209. [Google Scholar] [CrossRef]
- Apelfeld, A.V.; Betsofen, S.Y.; Borisov, A.M.; Vladimirov, B.V.; Savushkina, S.V.; Knyazev, E.V. Stabilization of the high-temperature phases in ceramic coatings on zirconium alloy produced by plasma electrolytic oxidation. J. Phys. Conf. Ser. 2016, 748, 12019. [Google Scholar] [CrossRef]
- Apelfeld, A.V.; Ashmarin, A.A.; Borisov, A.M.; Vinogradov, A.V.; Savushkina, S.V.; Shmytkova, E.A. Formation of zirconia tetragonal phase by plasma electrolytic oxidation of zirconium alloy in electrolyte comprising additives of yttria nanopowder. Surf. Coat. Technol. 2017, 328, 513–517. [Google Scholar] [CrossRef]
- Savushkina, S.V.; Ashmarin, A.A.; Apelfeld, A.V.; Borisov, A.M.; Vinogradov, A.V.; Polyansky, M.N.; Bogdashkina, N.L. Investigation of zirconia tetragonal phase coatings formed by plasma electrolytic oxidation. J. Phys. Conf. Ser. 2017, 857, 12037. [Google Scholar] [CrossRef]
- Cheng, Y.; Wu, F.; Matykina, E.; Skeldon, P.; Thompson, G.E. The influences of microdischarge types and silicate on the morphologies and phase compositions of plasma electrolytic oxidation coatings on Zircaloy-2. Corros. Sci. 2012, 59, 307–315. [Google Scholar] [CrossRef]
- Legostaeva, E.V.; Kulyashova, K.S.; Komarova, E.G.; Epple, M.; Sharkeev, Y.P.; Khlusov, I.A. Physical, chemical and biological properties of micro-arc deposited calcium phosphate coatings on titanium and zirconium-niobium alloy. Materwiss. Werksttech. 2013, 44, 188–197. [Google Scholar] [CrossRef]
- Simka, W.; Sowa, M.; Socha, R.P.; Maciej, A.; Michalska, J. Anodic oxidation of zirconium in silicate solutions. Electrochim. Acta 2013. [Google Scholar] [CrossRef]
- Sowa, M.; Dercz, G.; Suchanek, K.; Simka, W. Investigation of anodic oxide coatings on zirconium after heat treatment. Appl. Surf. Sci. 2015, 346, 534–542. [Google Scholar] [CrossRef]
- Sowa, M.; Simka, W. Electrochemical behavior of plasma electrolytically oxidized niobium in simulated physiological environment. Surf. Coat. Technol. 2018, 344. [Google Scholar] [CrossRef]
- Sowa, M.; Simka, W. Electrochemical impedance and polarization corrosion studies of tantalum surface modified by DC plasma electrolytic oxidation. Materials 2018, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.; Woszczak, M.; Kazek-Kęsik, A.; Dercz, G.; Korotin, D.M.; Zhidkov, I.S.; Kurmaev, E.Z.; Cholakh, S.O.; Basiaga, M.; Simka, W. Influence of process parameters on plasma electrolytic surface treatment of tantalum for biomedical applications. Appl. Surf. Sci. 2017, 407, 52–63. [Google Scholar] [CrossRef]
- Geometrical Product Specifications (GPS)—Surface Texture: Profile Method—Terms, Definitions and Surface Texture Parameters; ISO 4287:1997; International Organization for Standardization: Geneva, Switzerland, 1997.
- Gomes, M.A.B.; Onofre, S.; Juanto, S.; Bulhões, L.O.S. Anodization of niobium in sulphuric acid media. J. Appl. Electrochem. 1991, 21, 1023–1026. [Google Scholar] [CrossRef]
- Dorri, M.; Turgeon, S.; Brodusch, N.; Cloutier, M.; Chevallier, P.; Gauvin, R.; Mantovani, D. Characterization of amorphous oxide nano-thick layers on 316L stainless steel by electron channeling contrast imaging and electron backscatter diffraction. Microsc. Microanal. 2016, 22, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Lagar, J.H.; Raborar, M.G.C. Material contrast identification and compositional contrast mapping using backscattered electron imaging. In Proceedings of the 20th IEEE International Symposium on the Physical and Failure Analysis of Integrated Circuits (IPFA), Suzhou, China, 15–19 July 2013; pp. 464–469. [Google Scholar]
- Rokosz, K.; Hryniewicz, T.; Raaen, S.; Chapon, P. Investigation of porous coatings obtained on Ti-Nb-Zr-Sn alloy biomaterial by plasma electrolytic oxidation: Characterisation and modelling. Int. J. Adv. Manuf. Technol. 2016, 87, 3497–3512. [Google Scholar] [CrossRef]
- Dang, X.-D.; Plieth, W.; Richter, S.; Plötner, M.; Fischer, W.-J. Aluminum oxide film as gate dielectric for organic FETs: Anodization and characterization. Phys. Status Solidi 2008, 205, 626–632. [Google Scholar] [CrossRef]
- Liu, J.; Wu, G.; Li, S.; Yu, M.; Yi, J.; Wu, L. Surface analysis of chemical stripping titanium alloy oxide films. J. Wuhan Univ. Technol. Sci. Ed. 2012, 27, 399–404. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Shin, S.H.; Lim, H.-T.; Koo, B.H. Transformation of plasma electrolytic oxidation coatings from crater to cluster–based structure with increase in DC voltage and the role of ZrO2 nanoparticles. Surf. Coat. Technol. 2017, 311, 383–390. [Google Scholar] [CrossRef]
- Frankel, G.S.; Papavinasam, S.; Berke, N.; Brossia, S.; Dean, S.W. Electrochemical techniques in corrosion: Status, limitations, and needs. J. ASTM Int. 2008, 5, 101241. [Google Scholar] [CrossRef]
- Babaei, M.; Dehghanian, C.; Taheri, P.; Babaei, M. Effect of duty cycle and electrolyte additive on photocatalytic performance of TiO2-ZrO2 composite layers prepared on CP Ti by micro arc oxidation method. Surf. Coat. Technol. 2016, 307, 554–564. [Google Scholar] [CrossRef]
- Rosalbino, F.; Macciò, D.; Scavino, G.; Saccone, A. Corrosion behavior of new ternary zirconium alloys as alternative materials for biomedical applications. Mater. Corros. 2015, 66, 1125–1132. [Google Scholar] [CrossRef]
- Rosalbino, F.; Macciò, D.; Giannoni, P.; Quarto, R.; Saccone, A. Study of the in vitro corrosion behavior and biocompatibility of Zr-2.5Nb and Zr-1.5Nb-1Ta (at%) crystalline alloys. J. Mater. Sci. Mater. Med. 2011, 22, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Wenger, F.; Ponthiaux, P.; Celis, J.-P.; Pinto, A.M.; Rocha, L.A.; Fernandes, J.C.S. Corrosion mechanisms in titanium oxide-based films produced by anodic treatment. Electrochim. Acta 2017, 234, 16–27. [Google Scholar] [CrossRef]
- Orazem, M.E.; Tribollet, B. Electrochemical Impedance Spectroscopy; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Babaei, M.M.; Dehghanian, C.; Babaei, M.M. Electrochemical assessment of characteristics and corrosion behavior of Zr-containing coatings formed on titanium by plasma electrolytic oxidation. Surf. Coat. Technol. 2015, 279, 79–91. [Google Scholar] [CrossRef]
- Mareci, D.; Trincă, L.C.; Căilean, D.; Souto, R.M. Corrosion resistance of ZrTi alloys with hydroxyapatite-zirconia-silver layer in simulated physiological solution containing proteins for biomaterial applications. Appl. Surf. Sci. 2016, 389, 1069–1075. [Google Scholar] [CrossRef]
- Huang, C.-H.; Chen, R.-S.; Yoshimura, M. Direct bioactive ceramics coating via reactive growing integration layer method on α-Ti-alloy. Mater. Sci. Eng. C 2017, 76, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Yeum, B. EChem Software Tech Note 24—Pseudocapacitance Associated with CPE; EChem Software: Ann Arbor, MI, USA, 2002. [Google Scholar]
- Frangini, S.; De Cristofaro, N. Analysis of the galvanostatic polarization method for determining reliable pitting potentials on stainless steels in crevice-free conditions. Corros. Sci. 2003, 45, 2769–2786. [Google Scholar] [CrossRef]
- Galvele, J.R. Tafel’s law in pitting corrosion and crevice corrosion susceptibility. Corros. Sci. 2005, 47, 3053–3067. [Google Scholar] [CrossRef]
- Yu, S.Y.; Scully, J.R. Corrosion and passivity of Ti-13% Nb-13% Zr in comparison to other biomedical implant alloys. Corrosion 1997, 53, 965–976. [Google Scholar] [CrossRef]
- Assis, S.L.; Wolynec, S.; Costa, I. The electrochemical behaviour of Ti-13Nb-13Zr alloy in various solutions. Mater. Corros. 2008, 59, 739–743. [Google Scholar] [CrossRef]
- Milošev, I.; Žerjav, G.; Calderon Moreno, J.M.; Popa, M. Electrochemical properties, chemical composition and thickness of passive film formed on novel Ti–20Nb–10Zr–5Ta alloy. Electrochim. Acta 2013, 99, 176–189. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
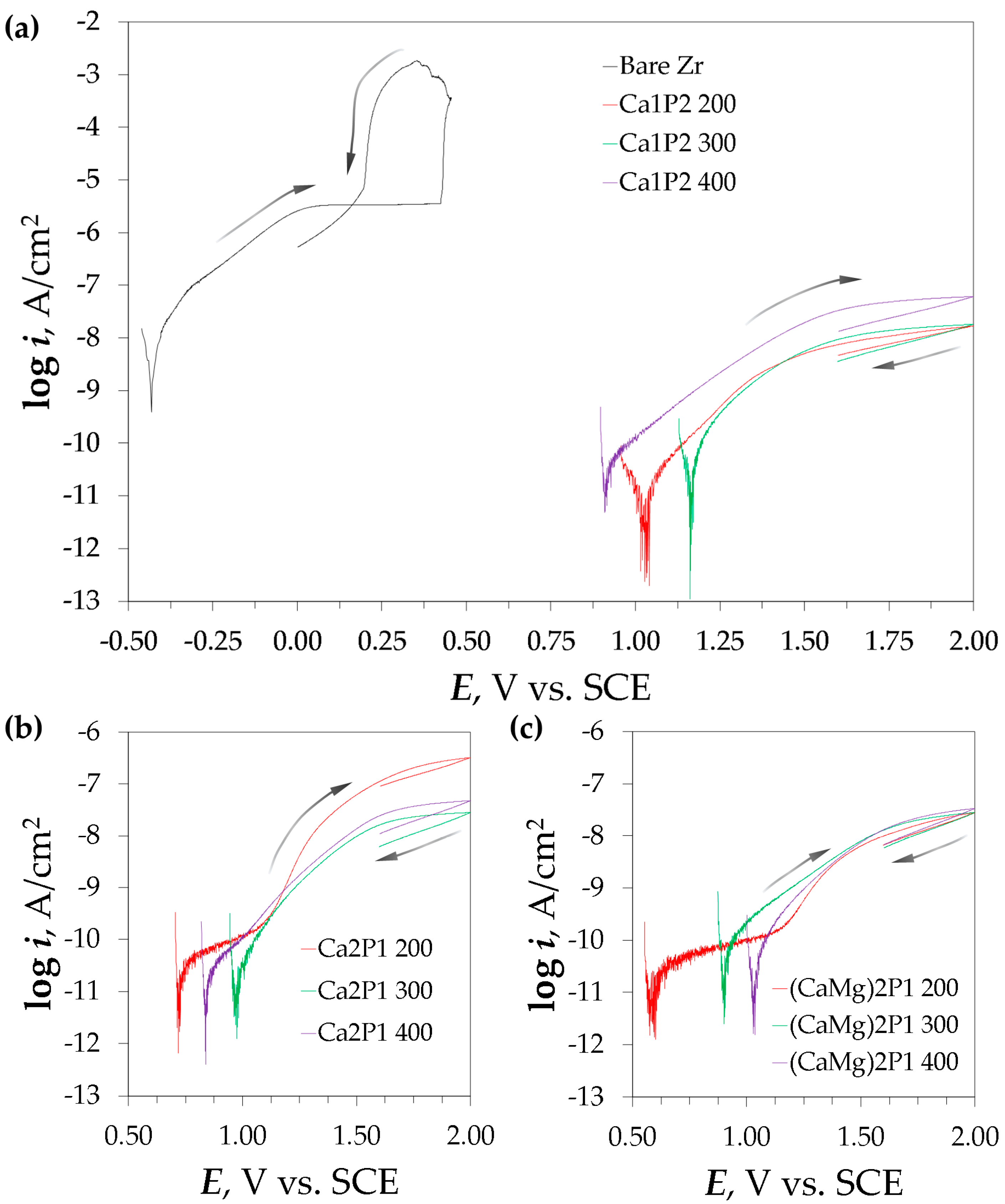{kind=link}
| Sample | Rs Ω·cm2 | Qc × 108 sn/(Ω·cm2) | nc | Cc × 108 F/cm2 | Ro × 10−4 Ω·cm2 | Qb×108 sn/(Ω·cm2) | nb | Rb × 10−8 Ω·cm2 | χ2 × 104 |
|---|---|---|---|---|---|---|---|---|---|
| Bare Zr | 5.83 ± 0.13 | - | - | - | - | 2111 ± 45 | 0.96 ± 0.00 | 0.0135 ± 0.0020 | <3.72 |
| Ca1P2 200 | 35.4 ± 9.4 | 6.59 ± 0.04 | 0.97 ± 0.00 | 7.79 ± 0.18 | 34.6 ± 16.3 | 3.63 ± 1.83 | 0.97 ± 0.03 | 19.7 ± 5.5 | <9.92 |
| Ca1P2 300 | 19.6± 4.0 | 10.1 ± 0.3 | 0.89 ± 0.00 | 15.9 ± 1.4 | 3.99 ± 0.46 | 21.2 ± 3.8 | 0.99 ± 0.01 | 4.64 ± 1.61 | <16.9 |
| Ca1P2 400 | 15.0 ± 1.2 | 9.83 ± 0.69 | 0.88 ± 0.01 | 16.9 ± 1.6 | 2.06 ± 0.32 | 19.0 ± 4.7 | 0.99 ± 0.00 | 4.96 ± 1.48 | <9.89 |
| Ca2P1 200 | 31.4 ± 6.0 | 10.3 ± 1.8 | 0.94 ± 0.01 | 13.3 ± 3.1 | 7.05 ± 1.92 | 7.69 ± 3.19 | 0.89 ± 0.03 | 6.14 ± 1.76 | <9.88 |
| Ca2P1 300 | 20.1 ± 2.9 | 9.47 ± 1.02 | 0.90 ± 0.01 | 13.7 ± 2.0 | 3.46 ± 0.19 | 27.2 ± 5.3 | 0.94 ± 0.05 | 3.12 ± 0.41 | <11.2 |
| Ca2P1 400 | 16.2 ± 2.3 | 8.55 ± 0.13 | 0.89 ± 0.00 | 12.6 ± 0.3 | 2.47 ± 0.10 | 11.3 ± 1.3 | 0.97 ± 0.01 | 2.87 ± 0.50 | <11.2 |
| (CaMg)2P1 200 | 28.0± 1.1 | 8.99 ± 0.87 | 0.96± 0.01 | 10.6 ± 1.5 | 17.0 ± 9.8 | 3.76 ± 0.81 | 0.88 ± 0.06 | 8.71 ± 3.20 | <59.2 |
| (CaMg)2P1 300 | 18.4 ± 1.5 | 10.8 ± 0.4 | 0.89 ± 0.00 | 15.4 ± 0.8 | 2.64 ± 0.17 | 30.7 ± 5.9 | 0.93 ± 0.03 | 1.71 ± 0.34 | <13.1 |
| (CaMg)2P1 400 | 14.5 ± 2.2 | 11.2 ± 0.9 | 0.87 ± 0.01 | 20.8 ± 2.3 | 2.50 ± 0.39 | 17.7 ± 3.4 | 0.99 ± 0.01 | 4.73 ± 0.81 | <13.6 |
| Sample | Ecor mV vs. SCE | Rp GΩ·cm2 | Rp,EIS GΩ·cm2 | Ebreak mV vs. SCE | Eprot mV vs. SCE | ipas nA/cm2 |
|---|---|---|---|---|---|---|
| Bare Zr | −446 ± 28 | 0.00250 ± 0.00018 | 0.00135 ± 0.00020 | 412 ± 39 | 173 ± 8 | 3609 ± 100 |
| Ca1P2 200 | 996 ± 28 | 2.92 ± 1.05 | 1.98 ± 0.55 | - | - | 13.8 ± 4.4 |
| Ca1P2 300 | 1042 ± 117 | 0.490 ± 0.210 | 0.464 ± 0.161 | - | - | 16.1 ± 2.3 |
| Ca1P2 400 | 950 ± 52 | 0.673 ± 0.073 | 0.496 ± 0.148 | - | - | 65.2 ± 12.6 |
| Ca2P1 200 | 856 ± 163 | 0.714 ± 0.288 | 0.614 ± 0.176 | - | - | 440 ± 173 |
| Ca2P1 300 | 941 ± 132 | 0.734 ± 0.231 | 0.312 ± 0.041 | - | - | 28.0 ± 3.9 |
| Ca2P1 400 | 889 ± 100 | 0.610 ± 0.075 | 0.287 ± 0.050 | - | - | 59.0 ± 8.2 |
| (CaMg)2P1 200 | 644 ± 70 | 1.40 ± 0.47 | 0.871 ± 0.320 | - | - | 33.3 ± 9.3 |
| (CaMg)2P1 300 | 758 ± 199 | 0.374 ± 0.055 | 0.171 ± 0.034 | - | - | 31.9 ± 3.4 |
| (CaMg)2P1 400 | 886 ± 228 | 0.617 ± 0.167 | 0.473 ± 0.081 | - | - | 40.3 ± 6.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sowa, M.; Simka, W. Effect of DC Plasma Electrolytic Oxidation on Surface Characteristics and Corrosion Resistance of Zirconium. Materials 2018, 11, 723. https://doi.org/10.3390/ma11050723
Sowa M, Simka W. Effect of DC Plasma Electrolytic Oxidation on Surface Characteristics and Corrosion Resistance of Zirconium. Materials. 2018; 11(5):723. https://doi.org/10.3390/ma11050723
Chicago/Turabian StyleSowa, Maciej, and Wojciech Simka. 2018. "Effect of DC Plasma Electrolytic Oxidation on Surface Characteristics and Corrosion Resistance of Zirconium" Materials 11, no. 5: 723. https://doi.org/10.3390/ma11050723
APA StyleSowa, M., & Simka, W. (2018). Effect of DC Plasma Electrolytic Oxidation on Surface Characteristics and Corrosion Resistance of Zirconium. Materials, 11(5), 723. https://doi.org/10.3390/ma11050723