Regulating the Optoelectronic Properties of Nickel Dithiolene by the Substituents: A Theoretical Study



Abstract
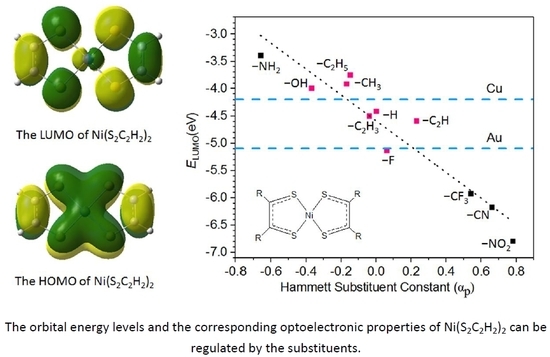
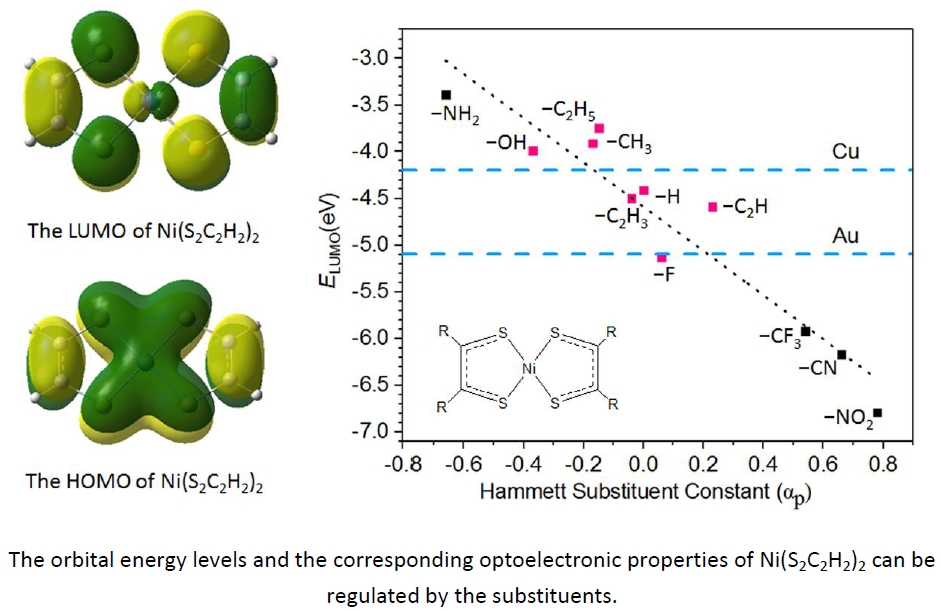
:
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Verification of Computational Method
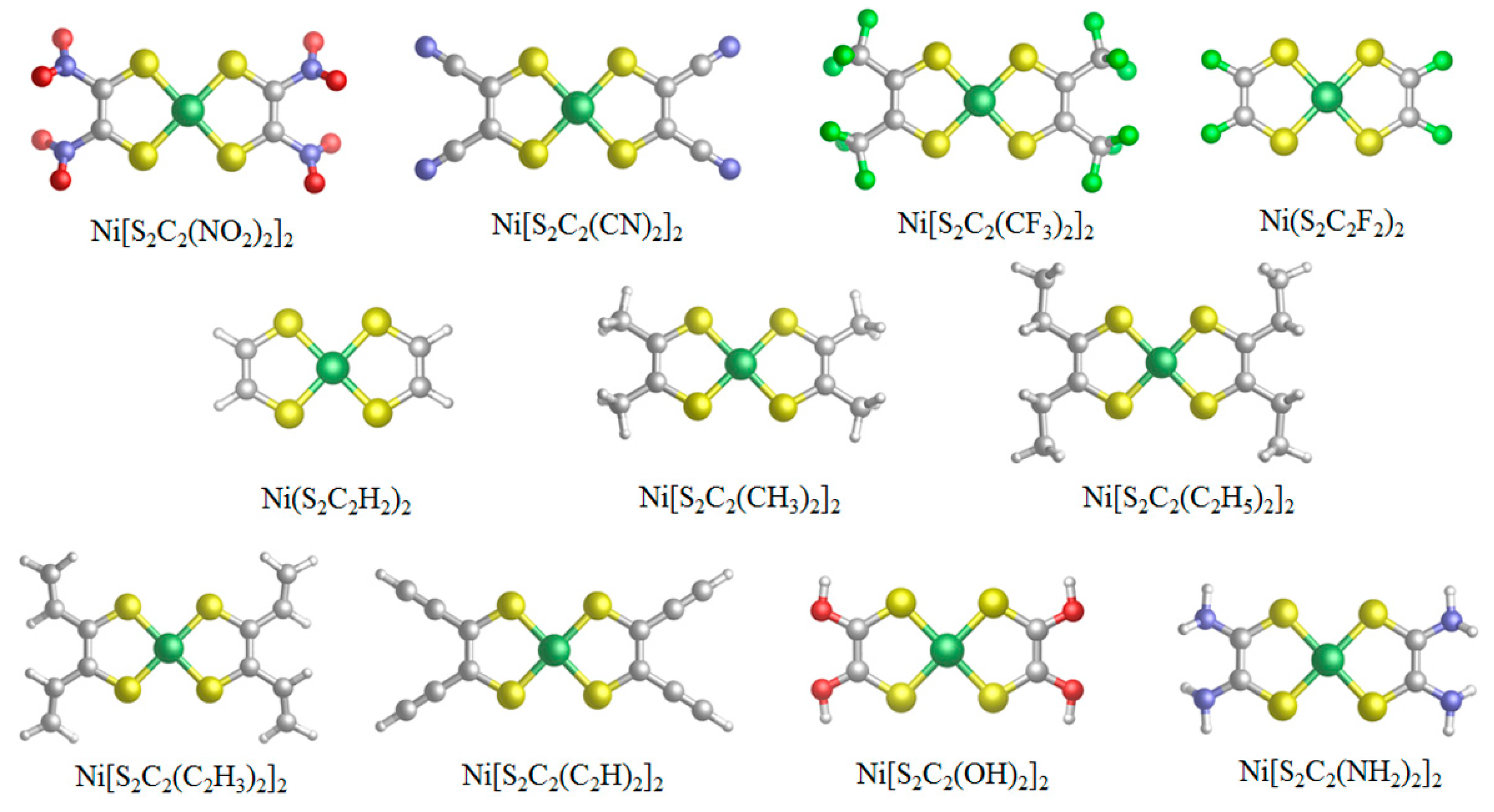
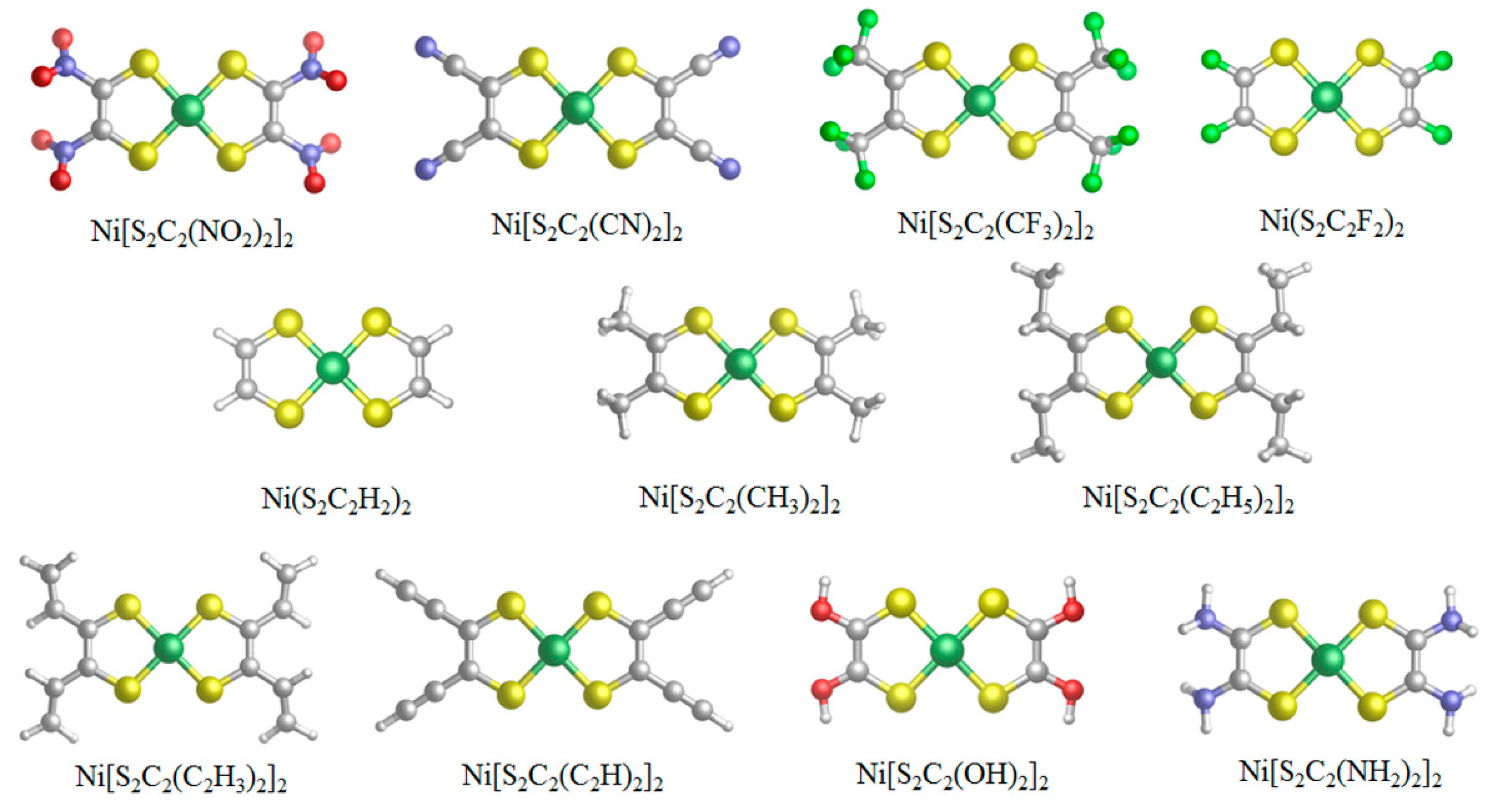
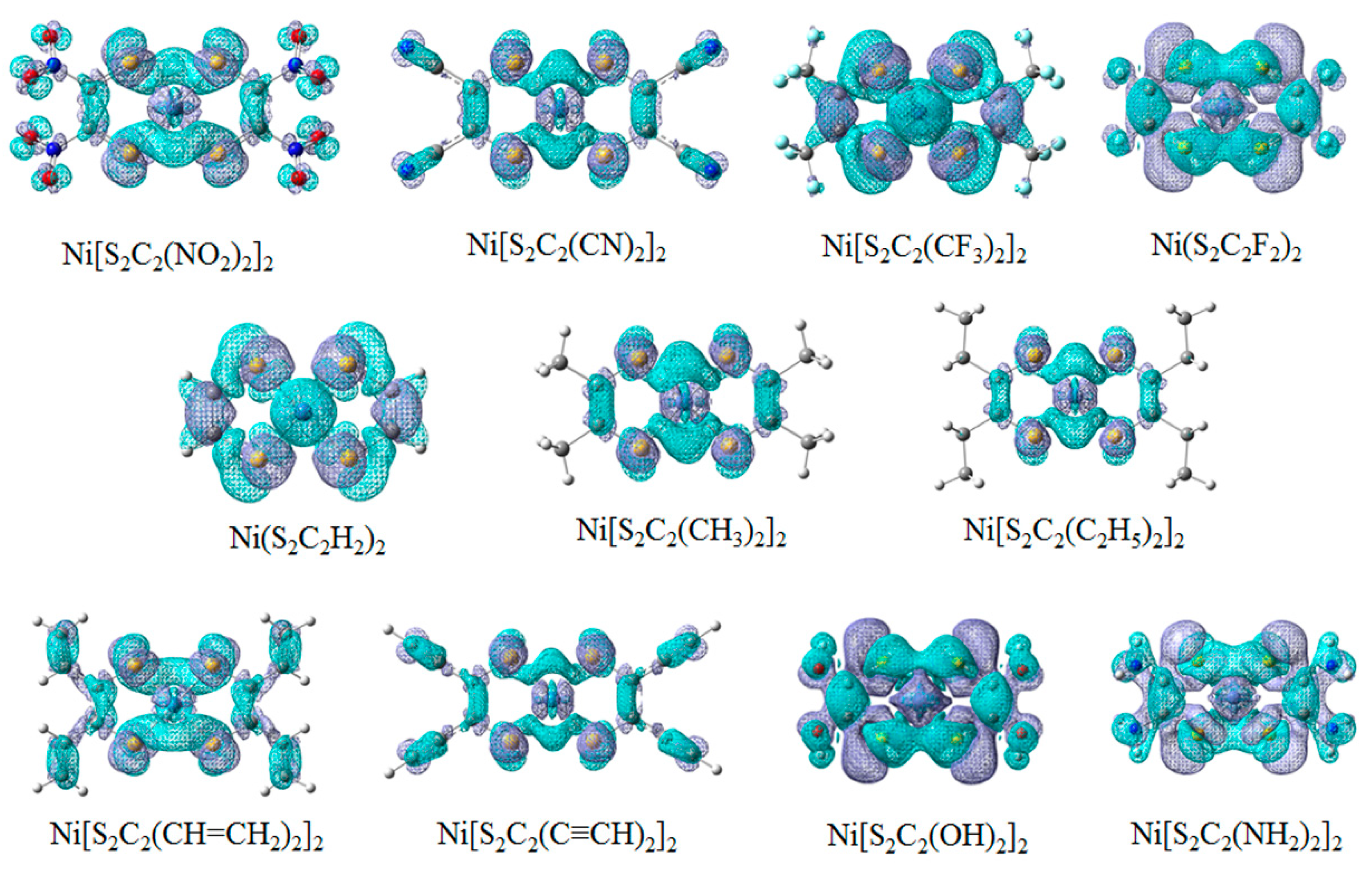
3.2. Geometry and Frontier Molecular Orbitals
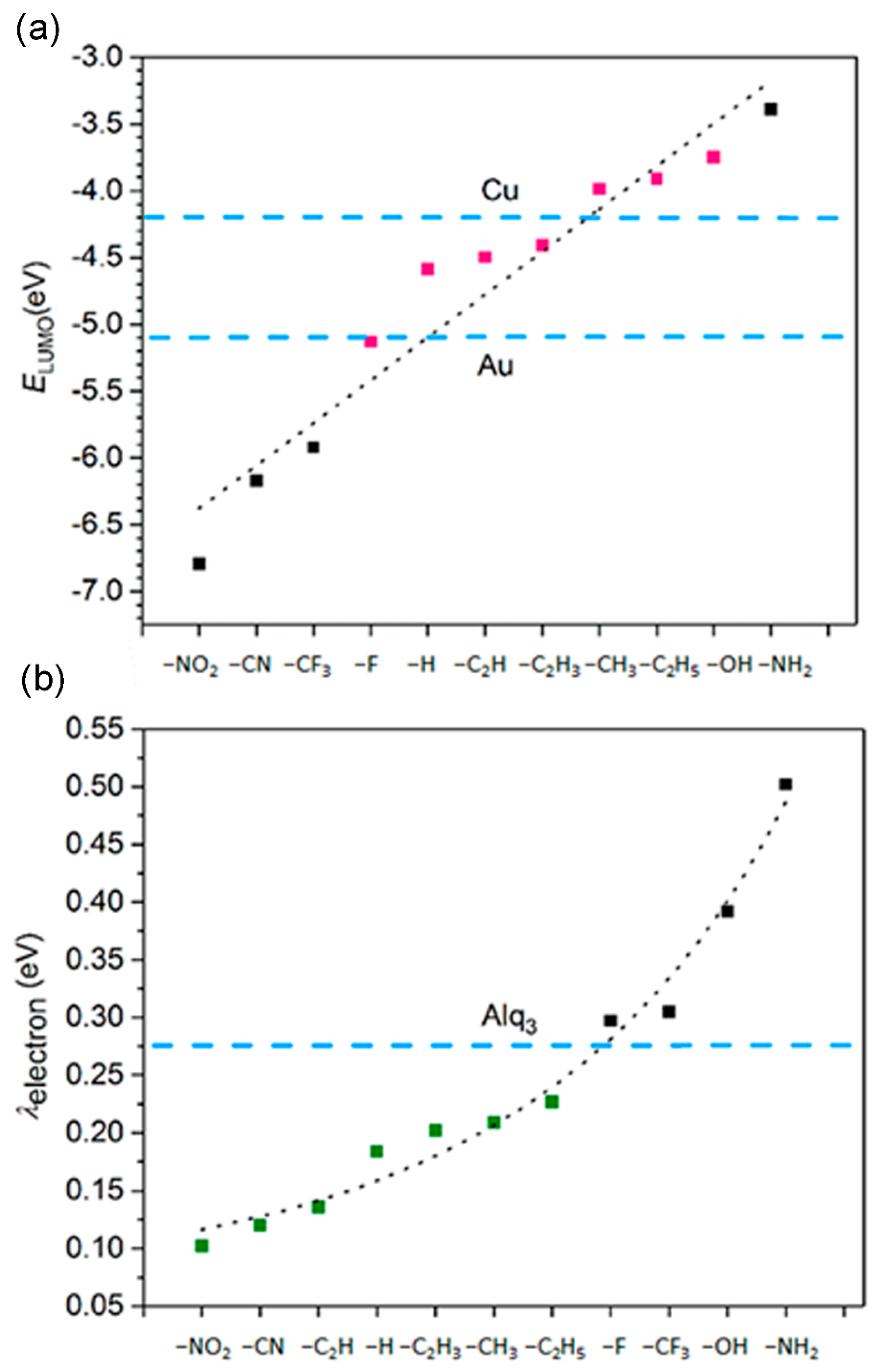
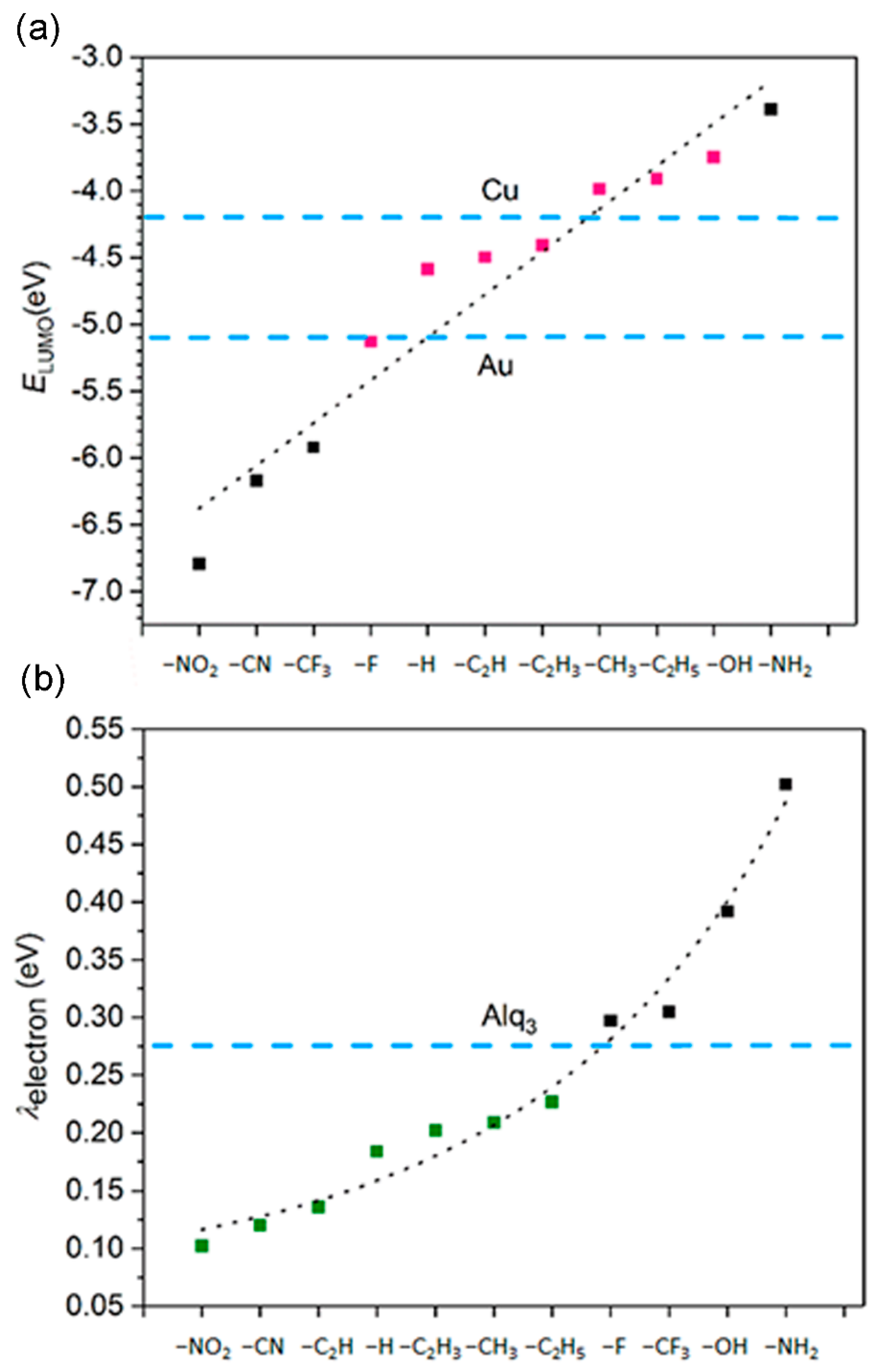
3.3. Ionization Potentials, Electron Affinities, and Reorganization Energy
3.4. Electron Transition from the Ground State to the First Low-Lying Excited State
3.5. The Electron Injection and Transportation Controlled by the Substituents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pilia, L.; Espa, D.; Barsella, A.; Fort, A.; Makedonas, C.; Marchio, L.; Mercuri, M.L.; Serpe, A.; Mitsopoulou, C.A.; Deplano, P. Combined experimental and theoretical study on redox-active d8 metal dithione-dithiolato complexes showing molecular second-order nonlinear optical activity. Inorg. Chem. 2011, 50, 10015–10027. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Nguyen, M.T. Silole-based nickel bisdithiolene complexes: A theoretical design for optoelectronic applications. J. Phys. Chem. C 2016, 120, 16418–16426. [Google Scholar] [CrossRef]
- Charrier, D.S.H.; de Vries, T.; Mathijssen, S.G.J.; Geluk, E.J.; Smits, E.C.P.; Kemerink, M.; Janssen, R.A.J. Bimolecular recombination in ambipolar organic field effect transistors. Org. Electron. 2009, 10, 994–997. [Google Scholar] [CrossRef]
- Anthopoulos, T.D.; Anyfantis, G.C.; Papavassiliou, G.C.; de Leeuw, D.M. Air-stable ambipolar organic transistors. Appl. Phys. Lett. 2007, 90, 122105. [Google Scholar] [CrossRef] [Green Version]
- Wada, H.; Taguchi, T.; Noda, B.; Kambayashi, T.; Mori, T.; Ishikawa, K.; Takezoe, H. Air stability of n-channel organic transistors based on nickel coordination compounds. Org. Electron. 2007, 8, 759–766. [Google Scholar] [CrossRef]
- Espa, D.; Pilia, L.; Makedonas, C.; Marchio, L.; Mercuri, M.L.; Serpe, A.; Barsella, A.; Fort, A.; Mitsopoulou, C.A.; Deplano, P. Role of the acceptor in tuning the properties of metal [m(ii) = ni, pd, pt] dithiolato/dithione (donor/acceptor) second-order nonlinear chromophores: Combined experimental and theoretical studies. lnorg. Chem. 2014, 53, 1170–1183. [Google Scholar] [CrossRef] [PubMed]
- McNamara, W.R.; Han, Z.J.; Yin, C.J.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. Cobalt-dithiolene complexes for the photocatalytic and electrocatalytic reduction of protons in aqueous solutions. Proc. Natl. Acad. Sci. USA 2012, 109, 15594–15599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, B.H.; Hammes-Schiffer, S. Computational study of anomalous reduction potentials for hydrogen evolution catalyzed by cobalt dithiolene complexes. J. Am. Chem. Soc. 2012, 134, 15253–15256. [Google Scholar] [CrossRef] [PubMed]
- Zarkadoulas, A.; Koutsouri, E.; Mitsopoulou, C.A. A perspective on solar energy conversion and water photosplitting by dithiolene complexes. Coord. Chem. Rev. 2012, 256, 2424–2434. [Google Scholar] [CrossRef]
- Miao, Q.Q.; Gao, J.X.; Wang, Z.Q.; Yu, H.; Luo, Y.; Ma, T.L. Syntheses and characterization of several nickel bis(dithiolene) complexes with strong and broad near-ir absorption. Inorg. Chim. Acta 2011, 376, 619–627. [Google Scholar] [CrossRef]
- Das, S.K.; Biswas, D.; Maiti, R.; Sarkar, S. Modeling the tungsten sites of inactive and active forms of hyperthermophilic pyrococcus furiosus aldehyde ferredoxin oxidoreductase. J. Am. Chem. Soc. 1996, 118, 1387–1397. [Google Scholar] [CrossRef]
- Barriere, F.; Camire, N.; Geiger, W.E.; Mueller-Westerhoff, U.T.; Sanders, R. Use of medium effects to tune the δe1/2 values of bimetallic and oligometallic compounds. J. Am. Chem. Soc. 2002, 124, 7262–7263. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.S.; Fomitchev, D.V.; Holm, R.H. Nickel dithiolenes revisited: Structures and electron distribution from density functional theory for the three-member electron-transfer series [ni(s2c2me2)2]0, 1−, 2−. lnorg. Chem. 2001, 40, 4257–4262. [Google Scholar] [CrossRef]
- Amb, C.M.; Heth, C.L.; Evenson, S.J.; Pokhodnya, K.I.; Rasmussen, S.C. Thiophene-fused nickel dithiolenes: A synthetic scaffold for highly delocalized π-electron systems. lnorg. Chem. 2016, 55, 10978–10989. [Google Scholar] [CrossRef] [PubMed]
- Mas-Torrent, M.; Rovira, C. Novel small molecules for organic field-effect transistors: Towards processability and high performance. Chem. Soc. Rev. 2008, 37, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Huong, V.T.T.; Tai, T.B.; Nguyen, M.T. A theoretical study on charge transport of dithiolene nickel complexes. Phys. Chem. Chem. Phys. 2016, 18, 6259–6267. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.; Moore, A.J.; Gibson, J.E.; Bryce, M.R.; Petty, M.C. A field effect transistor based on langmuir-blodgett films of an ni(dmit)2 charge transfer complex. Thin Solid Films 1994, 244, 932–935. [Google Scholar] [CrossRef]
- Bozic-Weber, B.; Constable, E.C.; Housecroft, C.E. Light harvesting with earth abundant d-block metals: Development of sensitizers in dye-sensitized solar cells (dscs). Coord. Chem. Rev. 2013, 257, 3089–3106. [Google Scholar] [CrossRef]
- Linfoot, C.L.; Richardson, P.; McCall, K.L.; Durrant, J.R.; Morandeira, A.; Robertson, N. A nickel-complex sensitiser for dye-sensitised solar cells. Sol. Energy 2011, 85, 1195–1203. [Google Scholar] [CrossRef]
- Robertson, N. Cui versus ruii: Dye-sensitized solar cells and beyond. Chemsuschem 2008, 1, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.B.; Hall, M.B. How electron flow controls the thermochemistry of the addition of olefins to nickel dithiolenes: Predictions by density functional theory. J. Am. Chem. Soc. 2002, 124, 12076–12077. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, C.; Fabian, J. Density functional derived structures and molecular properties of nickel dithiolenes and related complexes. Eur. J. Inorg. Chem. 1999, 1995–2004. [Google Scholar] [CrossRef]
- Gaussian, version 09; Gaussian, Inc.: Wallingford, UK, 2009.
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for k to au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Couty, M.; Hall, M.B. Basis sets for transition metals: Optimized outer p functions. J. Comput. Chem. 1996, 17, 1359–1370. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Bohme, M.; Dapprich, S.; Gobbi, A.; Hollwarth, A.; Jonas, V.; Kohler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of f-polarization functions for pseudo-potential basis sets of the transition metals sc cu, y ag and la au. Chem. Phys. Lett 1993, 208, 111–114. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Separation between fast and slow polarizations in continuum solvation models. J. Phys. Chem. A 2000, 104, 10614–10622. [Google Scholar] [CrossRef]
- Katoh, R.; Furube, A.; Yoshihara, T.; Hara, K.; Fujihashi, G.; Takano, S.; Murata, S.; Arakawa, H.; Tachiya, M. Efficiencies of electron injection from excited n3 dye into nanocrystalline semiconductor (ZrO2, TiO2, ZnO, Nb2O5, SnO2, In2O3) films. J. Phys. Chem. B 2004, 108, 4818–4822. [Google Scholar] [CrossRef]
- Bundgaard, E.; Krebs, F.C. Low band gap polymers for organic photovoltaics. Sol. Energy Mater. Sol. Cells 2007, 91, 954–985. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.K.; Hussain, S.Q.; Lee, Y.J.; Lee, Y.; Ahn, S.; Yi, J. Study on the ITO work function and hole injection barrier at the interface of ito/a-si: H(p) in amorphous/crystalline silicon heterojunction solar cells. Mater. Res. Bull. 2012, 47, 3032–3035. [Google Scholar] [CrossRef]
- Wang, K. Dithiolene Chemistry: Synthesis, Properties, and Applications; Progress in Inorganic Chemistry; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Costa, J.C.S.; Lima, C.F.R.A.C.; Santos, L.M.N.B.F. Electron transport materials for organic light-emitting diodes: Understanding the crystal and molecular stability of the tris(8-hydroxyquinolines) of Al, Ga, and In. J. Phys. Chem. C 2014, 118, 21762–21769. [Google Scholar] [CrossRef]
- Lin, B.C.; Cheng, C.P.; You, Z.Q.; Hsu, C.P. Charge transport properties of tris(8-hydroxyquinolinato)aluminum(iii): Why it is an electron transporter. J. Am. Chem. Soc. 2005, 127, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Bourouina, A.; Rekhis, M.; Trari, M. Dft/td-dft study of ruthenium bipyridyl-based dyes with a chalcogen donor (x = s, se, te), for application as dye-sensitized solar cells. Polyhedron 2017, 127, 217–224. [Google Scholar] [CrossRef]
- Hansch, C.; LEO, A.; TAFT, R.W. A survey of hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituent | EHOMO/eV | ELUMO/eV | ΔEH−L/eV |
|---|---|---|---|
| –NO2 | −8.307 | −6.793 | 1.514 |
| –CN | −7.796 | −6.173 | 1.623 |
| –CF3 | −7.717 | −5.922 | 1.795 |
| –F | −6.761 | −5.130 | 1.631 |
| –H | −6.158 | −4.410 | 1.748 |
| –CH3 | −5.581 | −3.910 | 1.671 |
| –C2H5 | −5.442 | −3.749 | 1.693 |
| –CH=CH2 | −5.933 | −4.498 | 1.435 |
| –C≡CH | −6.108 | −4.590 | 1.518 |
| –OH | −5.263 | −3.988 | 1.275 |
| –NH2 | −4.593 | −3.389 | 1.204 |
| Substituent | IP/eV | IP/eV | EA/eV | EA/eV | λhole/eV | λelectron/eV |
|---|---|---|---|---|---|---|
| –NO2 | 9.44 | 9.27 | 4.87 | 4.97 | 0.181 | 0.102 |
| –CN | 9.21 | 9.11 | 4.75 | 4.81 | 0.211 | 0.120 |
| –CF3 | 9.08 | 8.87 | 4.17 | 4.33 | 0.418 | 0.305 |
| –F | 8.23 | 8.03 | 3.37 | 3.52 | 0.391 | 0.297 |
| –H | 7.83 | 7.71 | 2.76 | 2.85 | 0.245 | 0.184 |
| –CH3 | 7.06 | 6.93 | 2.41 | 2.51 | 0.248 | 0.202 |
| –C2H5 | 6.82 | 6.69 | 2.31 | 2.43 | 0.255 | 0.227 |
| –CH=CH2 | 6.92 | 6.83 | 2.89 | 3.01 | 0.179 | 0.209 |
| –C≡CH | 7.37 | 7.28 | 3.28 | 3.35 | 0.177 | 0.136 |
| –OH | 6.81 | 6.60 | 2.42 | 2.61 | 0.430 | 0.392 |
| –NH2 | 6.05 | 5.77 | 1.92 | 2.13 | 0.574 | 0.502 |
| Substituent | λ/nm | Excitation Energy/eV | Oscillator Strength | LHE |
|---|---|---|---|---|
| –NO2 | 751.23 | 1.6504 | 0.1891 | 0.3530 |
| –CN | 774.91 | 1.6000 | 0.2231 | 0.4017 |
| –CF3 | 688.66 | 1.8004 | 0.1587 | 0.3061 |
| –F | 740.17 | 1.6751 | 0.1447 | 0.2834 |
| –H | 670.76 | 1.8484 | 0.1372 | 0.2709 |
| –CH3 | 707.58 | 1.7522 | 0.2006 | 0.3699 |
| –C2H5 | 726.68 | 1.7062 | 0.2151 | 0.3906 |
| –CH=CH2 | 859.00 | 1.4434 | 0.2644 | 0.4560 |
| –C≡CH | 823.66 | 1.5053 | 0.2927 | 0.4903 |
| –OH | 822.66 | 1.5071 | 0.1868 | 0.3496 |
| –NH2 | 1022.86 | 1.2121 | 0.0706 | 0.1500 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Shu, S.; Zhou, Y.; Hou, S.; Liu, Y.; Ke, Z. Regulating the Optoelectronic Properties of Nickel Dithiolene by the Substituents: A Theoretical Study. Materials 2018, 11, 2192. https://doi.org/10.3390/ma11112192
Sun L, Shu S, Zhou Y, Hou S, Liu Y, Ke Z. Regulating the Optoelectronic Properties of Nickel Dithiolene by the Substituents: A Theoretical Study. Materials. 2018; 11(11):2192. https://doi.org/10.3390/ma11112192
Chicago/Turabian StyleSun, Lili, Siwei Shu, Yi Zhou, Sen Hou, Yan Liu, and Zhuofeng Ke. 2018. "Regulating the Optoelectronic Properties of Nickel Dithiolene by the Substituents: A Theoretical Study" Materials 11, no. 11: 2192. https://doi.org/10.3390/ma11112192
APA StyleSun, L., Shu, S., Zhou, Y., Hou, S., Liu, Y., & Ke, Z. (2018). Regulating the Optoelectronic Properties of Nickel Dithiolene by the Substituents: A Theoretical Study. Materials, 11(11), 2192. https://doi.org/10.3390/ma11112192