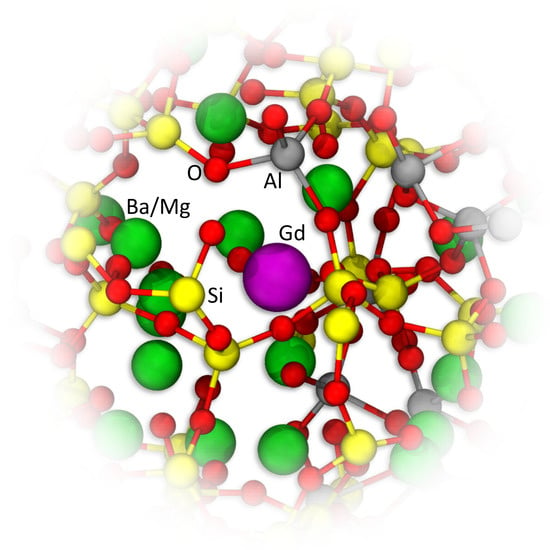
3.1. Comparison of Simulated Annealing (ANN) and Inherent Structure Sampling (ISS)
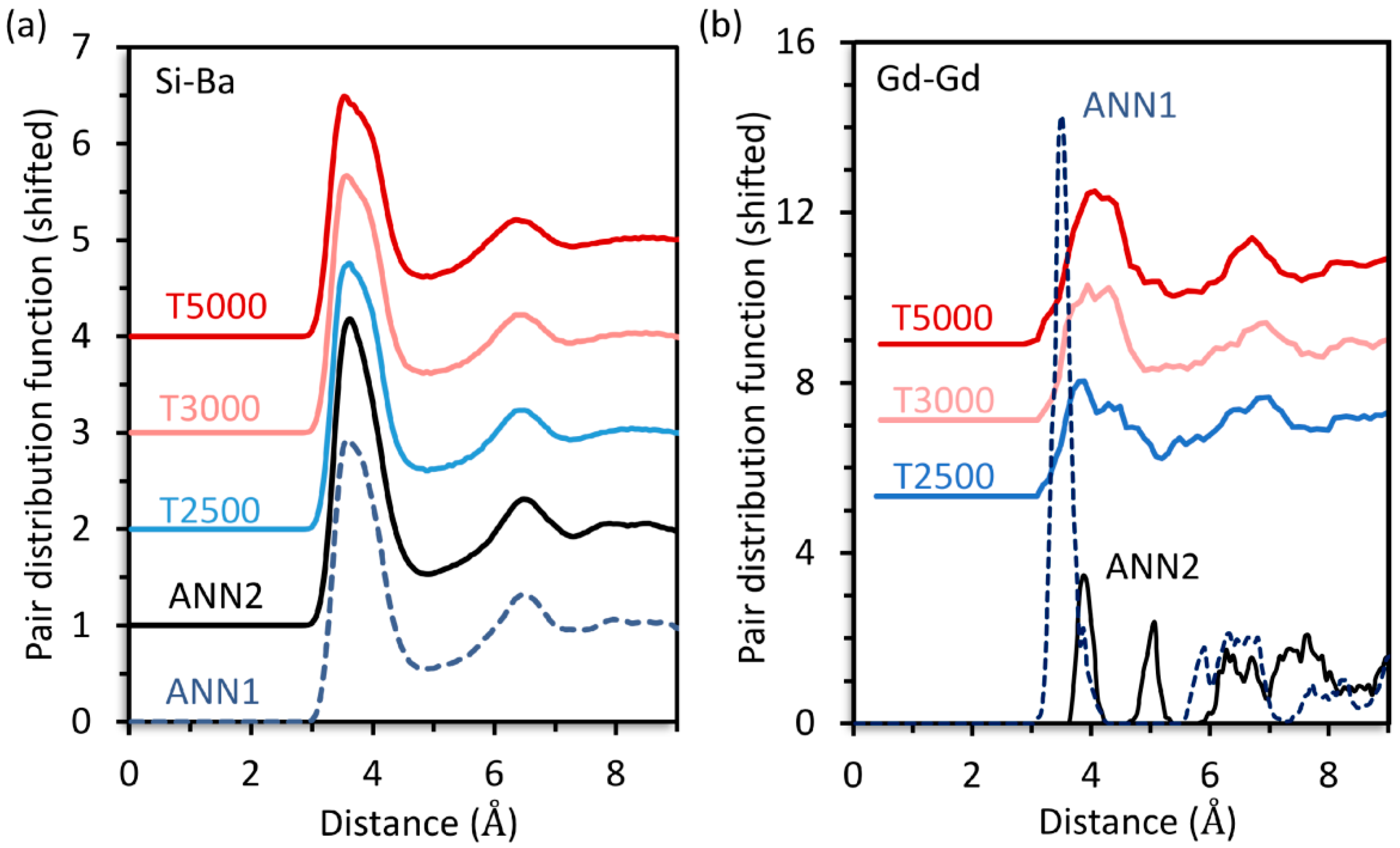
The Si-Ba,
Figure 1a, and Gd-Gd,
Figure 1b, pair distribution functions (PDF) for
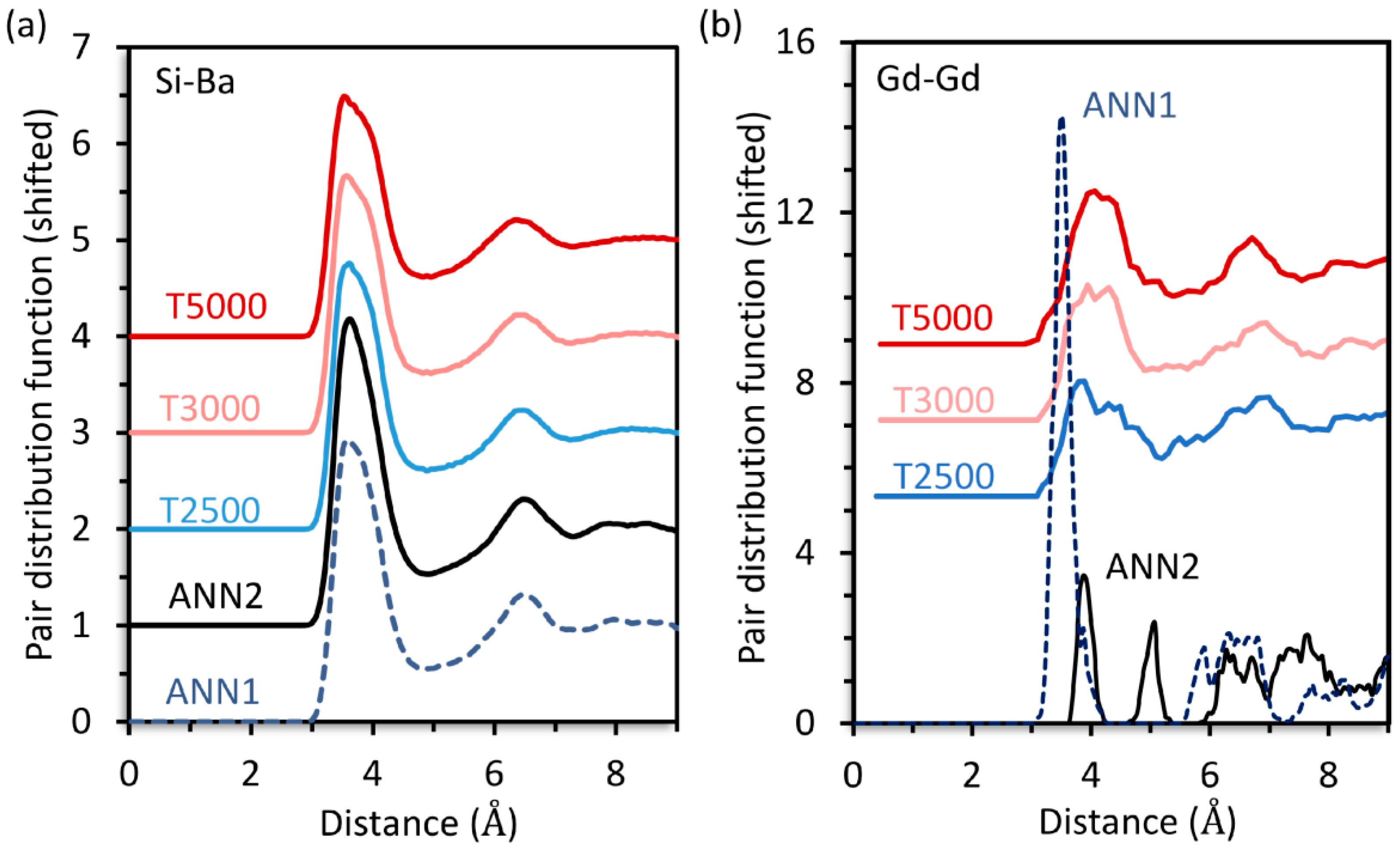
30Ba were calculated by simulated annealing (ANN), yielding the structure models ANN1 and ANN2, as well as inherent structure sampling (ISS) at different temperatures (T2500, T3000, T5000). For all predicted structures, the Si-Ba PDF are virtually identical, see
Figure 1a. This also applies to every other PDF of the network former and modifier ions (not shown). However, the Gd-Gd PDF are very different for ANN1 and ANN2, see
Figure 1b. By contrast, the ISS yields structures with similar Gd-Gd PDF that are nearly independent of the employed sampling temperature, see
Figure 1b.
Table 2 summarizes the mass densities and relative potential energies Δ
Ep with respect to the lowest energy structure (ANN1) for
30Ba predicted by ANN and ISS. The calculated mass densities are 3.5 g/cm
3 for ANN1 and ANN2, which is in good agreement with the experimentally observed value (3.54 g/cm
3 [
12]). In addition, the potential energies of both structures are almost the same. By contrast, the average mass densities and relative energies predicted by ISS increase with increasing sampling temperature ranging from 65 (T2500) to 119 meV/atom (T5000). Similarly, the mass densities are up to 0.2 g/cm
3 (T5000) higher compared to ANN1 and ANN2.
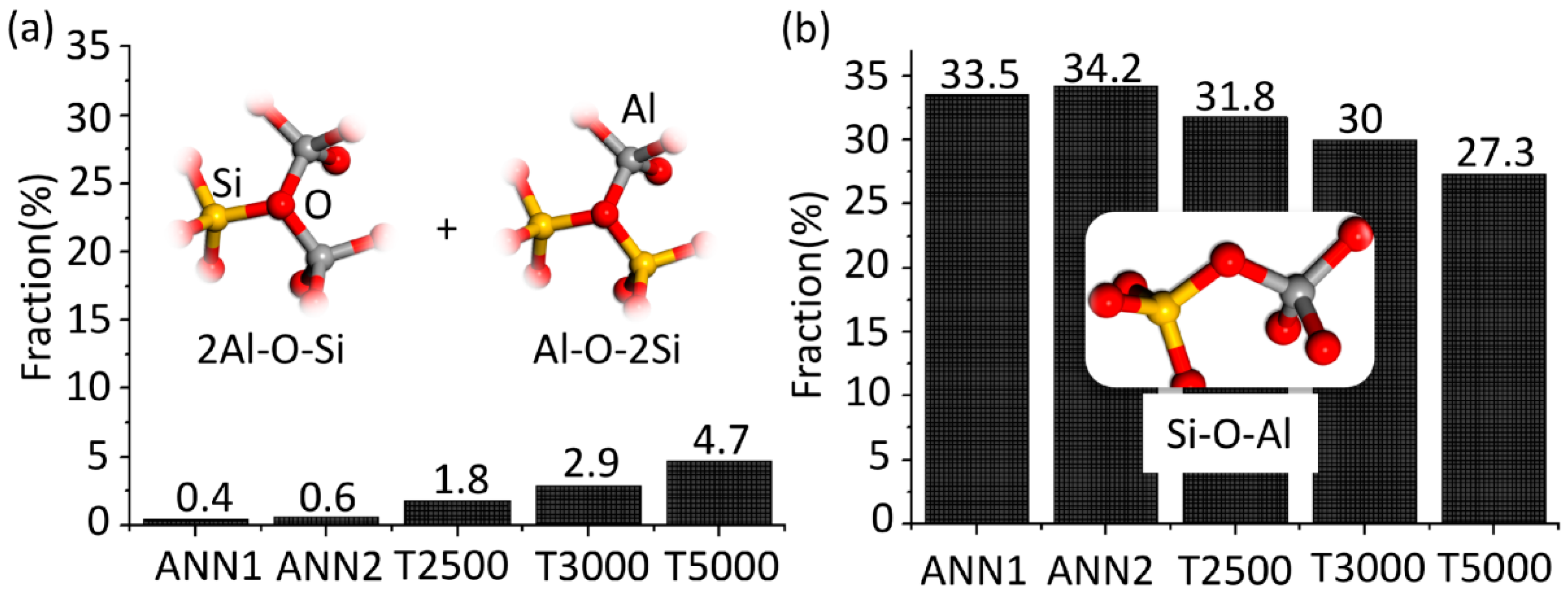
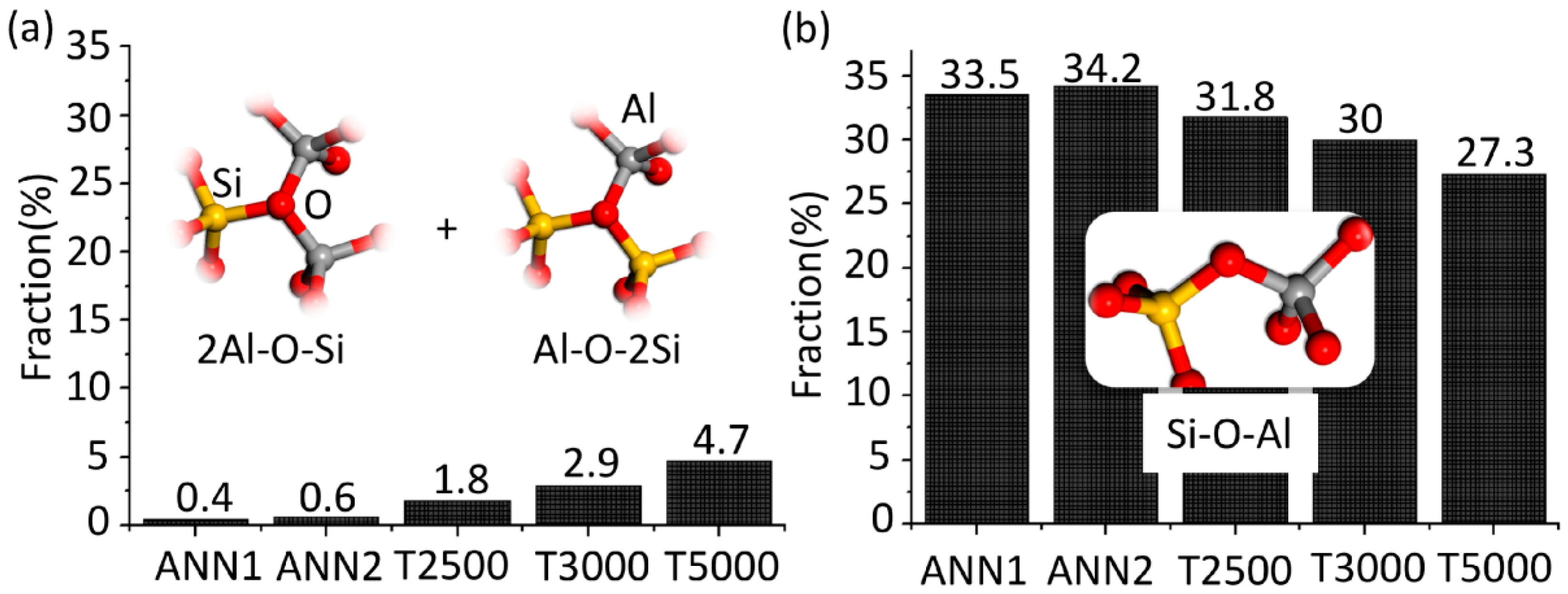
The most significant differences between the atomic structures obtained by ISS and ANN, which lead to higher densities and relative energies of the ISS predicted structures, are the fractions of triclusters (2Al-O-Si and Al-O-2Si) and bridging oxygen (Al-O-Si) ions bonded to [AlO
x] or [SiO
4], depicted in
Figure 2. While almost none of such tricluster oxygen ions are present in ANN1 and ANN2, their fraction increases from 1.8 to 4.7% with increasing sampling temperature in the case of the ISS and is accompanied by a decrease of the Al-O-Si bridging oxygen fraction by approximately the same amount.
Both glass structure models predicted using the simulated annealing procedure (ANN1, ANN2) show virtually identical potential energies as well as short- and medium-range structures of network former and modifier ions. These findings demonstrate that the employed unit cell containing 6460 ions provides a sufficient sampling of atomic configurations to describe the glass network. However, including only 32 Gd
3+ ions results in an inadequate sampling of the Gd
3+ medium-range order, such as the Gd-Gd PDF, making precise modeling of the atomic structure impossible. Indeed, it was shown that structural models containing at least 80 rare earth ions are required [
16] for reliable structure predictions of rare earth doped glasses using ANN. This means that in case of
30Ba, a unit cell containing at least 16600 atoms is necessary to obtain a sufficient sampling of atomic configurations, which would result in computationally demanding simulations.
In contrast, the ISS uses unit cells containing only 2 Gd
3+ and about 400 ions in total along with comparably short durations of MD simulations; hence considerably less computational effort is required. However, the sampling of the configuration space is not achieved by atomic structures within one single unit cell but by a large set of inherent structures (IS). Since the distribution
P(
eIS,
T) of the IS energies
eIS corresponds to the probability that a configuration in the equilibrated liquid state is related to an IS with energy
eIS [
21,
22], it is possible to determine an average structure, see Equation (1), representing the liquid state at temperature
T. The resulting medium-range structures of
30Ba, i.e., the atomic structure starting from the second coordination shell of the ions such as the Si-Ba distribution, shows a very good agreement with the structures obtained by ANN. In addition, independent ISS at different temperatures yield very similar Gd-Gd PDF, showing that the sampling of the medium-range order of Gd
3+ is sufficient to provide reproducible results.
However, structures predicted by ISS are slightly higher in energy and mass density due to the overestimation of the concentration of tricluster oxygen ions compared to ANN1 and ANN2. This is connected with the sampling of IS in the liquid state above
Tg. At higher sampling temperatures, i.e., higher kinetic energies of the ions, a larger number of IS of the PES are available lowering the probability of finding low energy structures. This leads to a shift of
P(
eIS,
T) to higher
eIS with increasing sampling temperature [
34] and, consequently, the predicted average IS show structural features such as tricluster oxygen ions that are higher in energy. In contrast, during the ANN such structural features are able to turn into lower energy structures (mainly bridging oxygen ions) until
Tg is reached and the glass structure gets “frozen”. An additional equilibration performed for a geometrically optimized random structure (
30Ba) for 1 ns at 2000 K showed no changes of the short- and medium-range structure. This means that
Tg, predicted by MD simulations along with the employed interatomic potential functions, is located between 2000 and 2500 K. An accurate calculation of thermodynamic quantities including
Tg requires several ISS in this temperature range for evaluation of the temperature dependence of
P(
eIS,
T) [
21,
34]. In contrast to the ISS, the ANN allows short-ranged relaxations between 2000 and 2500 K explaining the lower number of tricluster oxygen ions compared to the ISS. However, the slight deviations of tricluster oxygen fractions of less than 3% for T2500 and T3000 are expected to be smaller than the accuracy of structure predictions in general if empirically parameterized interatomic potential functions are applied.
One drawback of the ISS is that in the case of clustering of rare earth ions, the actual cluster structure comprising several Gd
3+ ions cannot be predicted when only two Gd
3+ ions are included in the structural model as in the present study. However, the employed ISS procedure allows estimation of the driving force for the formation of Gd-O-Gd contacts and, therefore, the determination of whether the tendency for Gd
3+ clustering is preferred for the investigated glass compositions or not, see
Section 3.3. Nevertheless, one advantage of the ISS, is the generation of a large set of structures which represent the liquid state close to
Tg, and show a close resemblance to the atomic configurations predicted by ANN but contain a considerably lower number of ions in the unit cells. This facilitates a simple yet efficient parallelization of computationally more demanding, quantum mechanical simulations. For instance, simulations at the density functional theory (DFT) level for unit cells containing several thousand atoms are tremendously challenging. In contrast, using at least a subset of atomic configurations obtained by ISS for structural optimizations at the DFT level would allow a refinement of the IS as well as
P(
eIS,
T). Such a simulation approach could provide a deeper understanding of the relationship between the atomic structure and optical properties of rare earth doped aluminosilicate glasses.
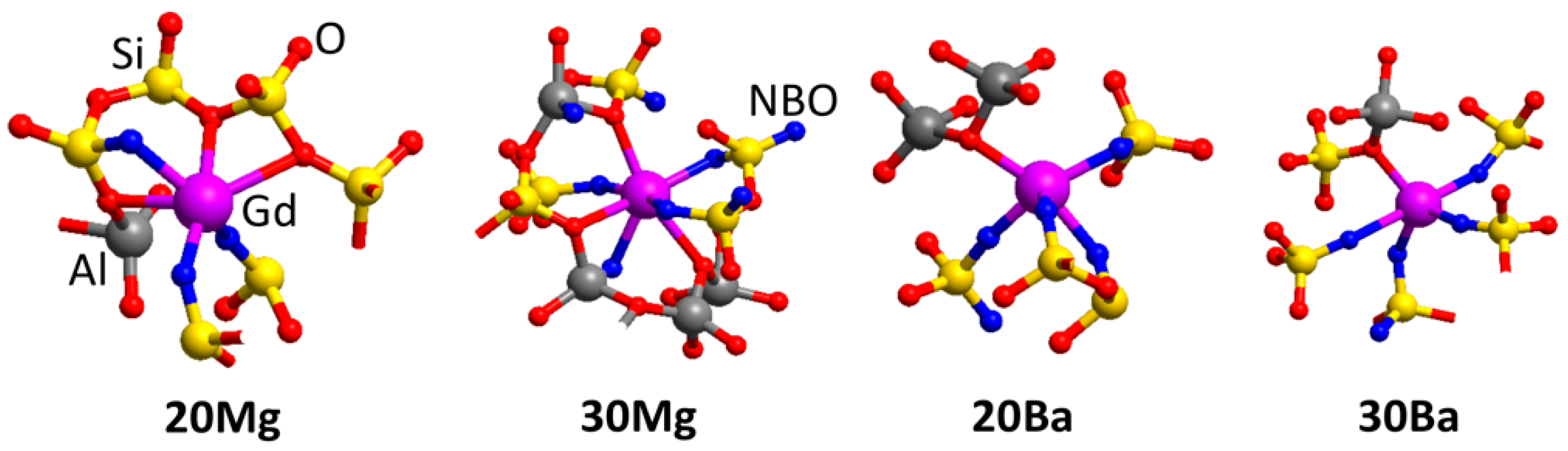
3.2. Atomic Structure of Glass Network Formers and Modifiers Ions
The short-range order of the network builder ions is characterized by the first peak of the Si-O and Al-O pair distribution functions as well as the corresponding coordination numbers (not shown). The Si-O pair distribution functions of all glass compositions are as expected: The Si-O bond lengths show a narrow distribution and the mean bond length is 1.59 Å (1.60 Å in crystalline SiO
2 [
35], 1.61 Å in Mg
2Al
3[AlSi
5O
18] [
36]). The Si-O coordination number is four. The Al-O bonds show a somewhat larger bond length of 1.71 Å in all cases (1.75 Å in Mg
2Al
3[AlSi
5O
18] [
36]). The mean Al-O coordination number is 4.3 and 4.2 for
20Mg and
20Ba, respectively. Hence, the coordination number is a little bit higher than expected for an overall tetrahedral coordination. That means that small quantities of aluminum occur with a higher coordination number, i.e., with 5-fold (22.5% in
20Mg and 16.7% in
20Ba) or 6-fold (2.0% in
20Mg and 1.2% in
20Ba) coordination. Consequently, a corresponding small concentration of non-bridging oxygen should also occur. The Al-O coordination numbers for
30Mg and
30Ba are 4.2 and 4.1, respectively. They are somewhat smaller than in the metaluminous glasses due to the lower amount of 5- and 6-fold coordinated Al
3+. The percentages are: 5-fold: 20.0% in
30Mg and 9.4% in
30Ba and 6-fold: 1.5% in
30Mg and 0.4% in
30Ba.
Experimental studies were carried out using high-resolution magic angle spinning (HRMAS) NMR investigations on calcium aluminosilicate glasses with the same molar compositions of the undoped glass models (corresponding to
20Ca and
30Ca, cf.
Table 1). It was observed that the concentrations of 5-fold coordinated Al
3+ are about 3% (
20Ca) [
37] and 5% (
30Ca), respectively [
38]. In addition, it was found that alkaline earth ions with higher field strengths (Mg
2+ > Ca
2+ > Ba
2+) favor the formation of 5-fold and 6-fold coordinated Al
3+, while the concentration of Al
3+ in 6-fold coordination is considerably lower than that in 5-fold coordination [
39]. These observations are qualitatively in agreement with the simulation results, yet the absolute value of 5-fold coordinated Al
3+ is clearly overestimated. This is possibly connected with the inaccuracy of the empirical parameterization employed for the Al-O interatomic potential functions. However, the employed simulation procedure facilitates the refinement of the obtained structure models using higher-level simulations, e.g., at the DFT level, see
Section 3.1.
Table 3 shows the distances and coordination numbers (CN) of the first and second coordination sphere of the network-modifier (NM) ions. In the case of
20Mg and
20Ba, the calculated Mg-O and Ba-O bond lengths are 2.01 and 2.79 Å and the CN 4.6 and 9.1, respectively. Of course, the larger ionic radius of the Ba
2+ ions should result in longer bonds and a higher CN than for the smaller Mg
2+. Since the [AlO
4]
− tetrahedra need a cation for charge compensation, the distances Mg-Al and Ba-Al are also of interest. They are 3.27 and 3.57 Å, while the calculated CN are 2.9 and 4.3 for Al-Mg and Al-Ba, respectively. The bond lengths Mg–O and Ba–O for
30Mg and
30Ba are 2.01 Å and 2.79 Å and hence identical with those obtained for
20Mg and
20Ba. The calculated CN are 4.7 and 8.3, respectively. Hence, the NM–O CN for
30Mg is significantly smaller than for
30Ba.
The CN of NM ions shown in
Table 3 for the second coordination sphere, i.e., their coordination with other cations, is substantially different for the peralkaline glass structure models (
30Mg,
30Ba) compared to the metaluminous compositions (
20Mg,
20Ba). As expected from the chemical compositions, the coordination of NM ions with [AlO
x] polyhedra is more likely in the metaluminous glasses than in peralkaline compositions, while in the peralkaline compositions the coordination with other NM ions is more likely. This also shows that the so-called depolymerized regions in the glass structure must be larger in the peralkaline glasses. However, the most numerous coordination partners of the NM ions are [SiO
4] tetrahedra. Interestingly, the number of coordinating [SiO
4] tetrahedra is nearly constant for each of the network-modifying atoms (Mg
2+, Ba
2+) in peralkaline and metaluminous glasses. That means their coordination with [SiO
4] tetrahedra, similarly to their coordination with oxygen, depends only on the radius of the network-modifying ion.
In
Table 4 the oxygen coordination numbers of the NM ions are separated into coordination with bridging oxygen (BO), i.e., ≡(Si, Al)−O−(Si, Al)≡, and non-bridging oxygen (NBO), i.e., ≡Si−O, as well as oxygen ions in triclusters (Tri). As can be seen from the data, the coordination with NBO is generally higher for the peralkaline compositions
30Mg and
30Ba. As explained in the introduction section, this is due to the higher NMO/Al
2O
3 ratio in these glasses. Interestingly, the data shows that a notably higher number of oxygen ions are of the non-bridging species in the metaluminous magnesium containing
20Mg (31.1%) compared to its barium containing counterpart
20Ba (16.5%). Due to the smaller size of the Mg
2+ ion and the repulsion between two [AlO
4]
− tetrahedra the charge compensation of two [AlO
4]
− tetrahedra by only one NM
2+ is less likely in metaluminous magnesium aluminosilicate glasses than in barium aluminosilicate glasses and NBO sites are formed instead [
40,
41]. In a macroscopic scale, this effect results in a lower glass transition temperature,
Tg, and lower melt viscosity of the
20Mg composition in comparison to the
20Ba composition, while exactly the opposite effect occurs for the peralkaline compositions. Here,
30Ba has a lower glass transition temperature and lower viscosity than
30Mg [
14].
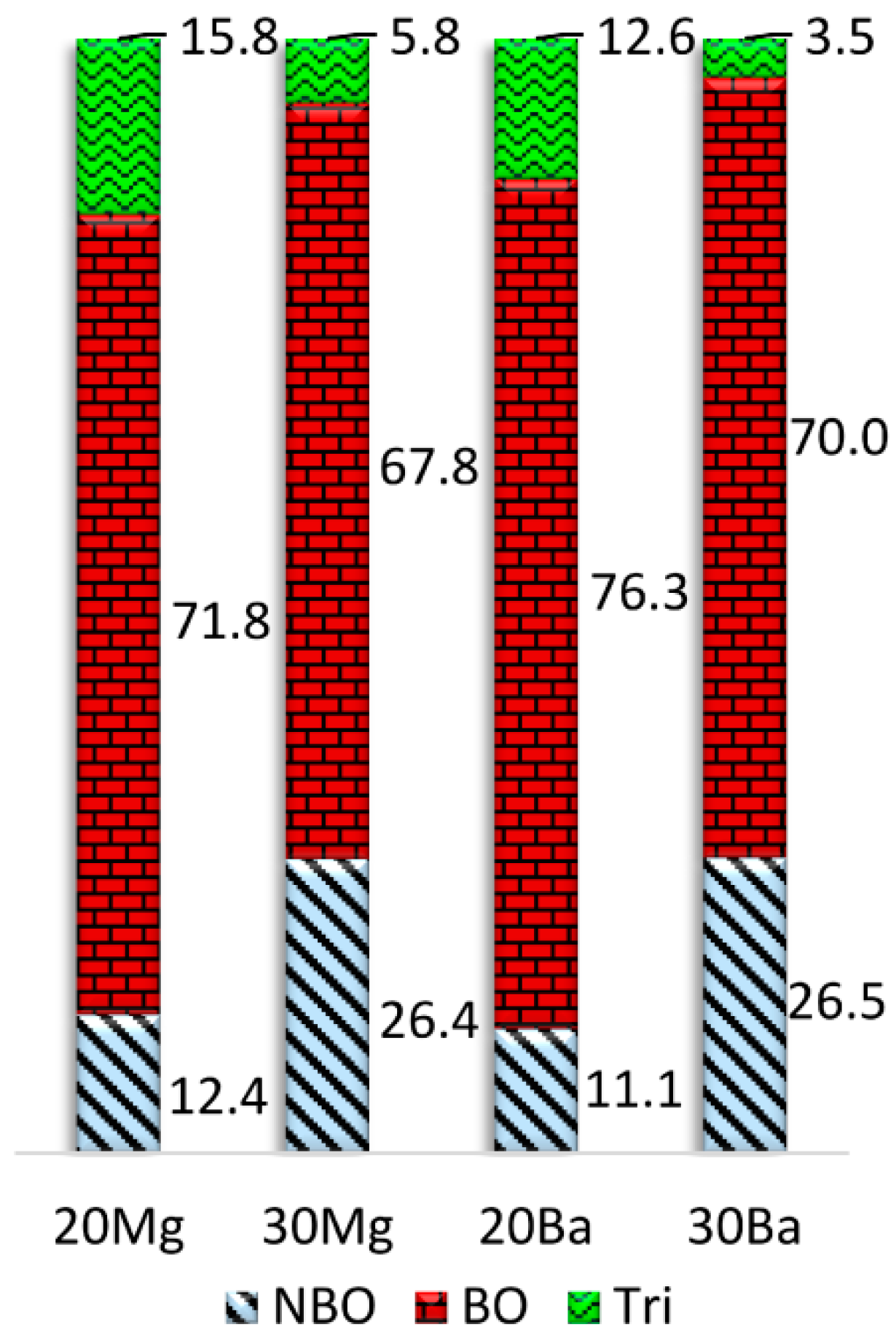
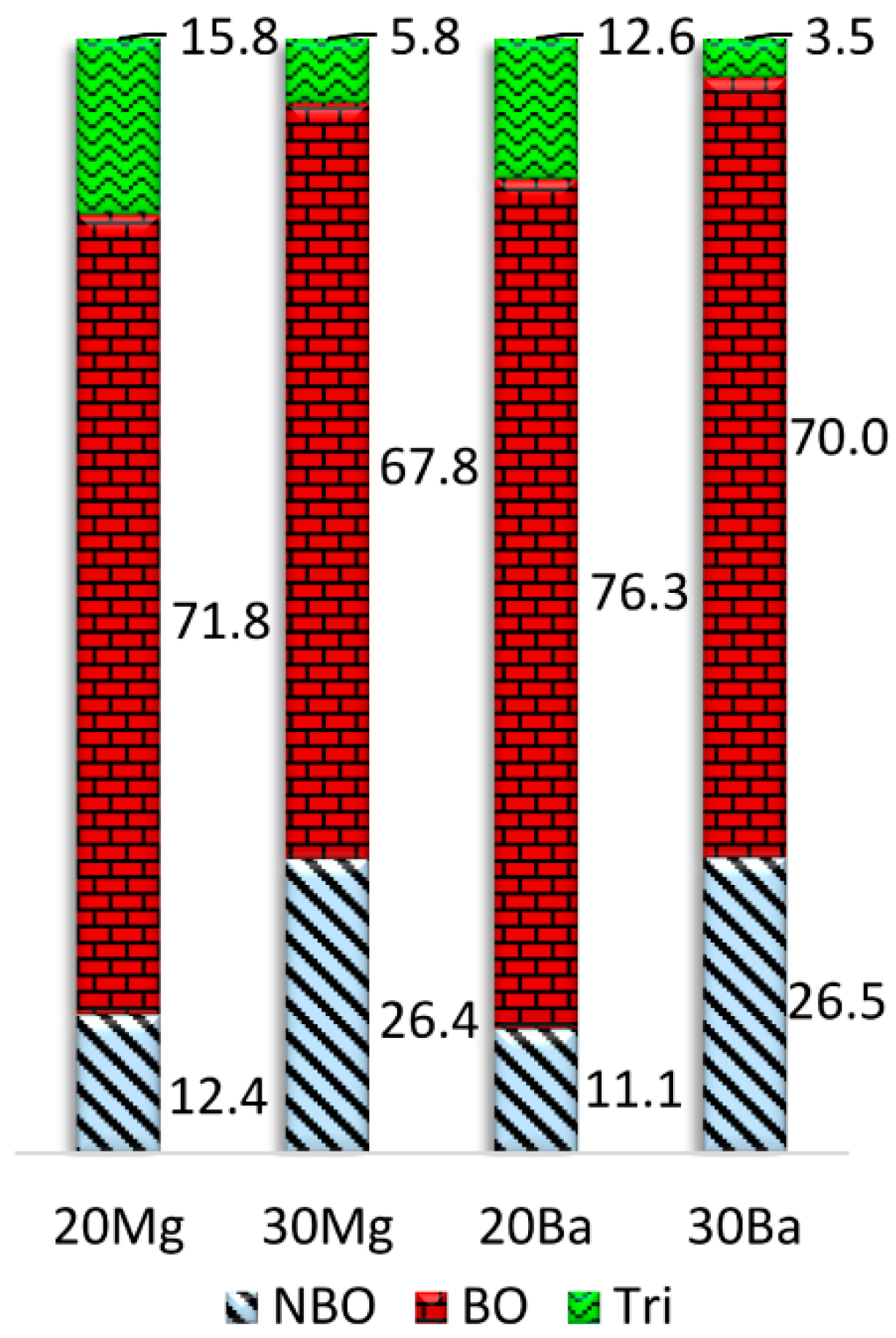
A comparison of the average bridging oxygen (BO), NBO, and oxygen tricluster fractions calculated for all oxygen ions of the glass structure models is depicted in
Figure 3. While the number of bridging oxygen slightly increases, a considerable number of NBO turn into oxygen triclusters when the concentration of NM ions is increased. However, it must be noted that the number of oxygen triclusters is slightly overestimated by the employed structure prediction procedure. The higher number of NBO sites in
20Mg compared to
20Ba additionally results in a higher number of oxygen triclusters in this composition, since the triclusters ensure charge compensation of the remaining [AlO
4]
− tetrahedra. In addition, the number of triclusters is drastically reduced in the peralkaline compositions
30Mg and
30Ba compared to
20Mg and
20Ba due to their higher NMO/Al
2O
3 ratio. Accordingly, the coordination of the NM ions with oxygen triclusters is notably smaller for the peralkaline glasses
30Mg and
30Ba, see
Table 4. In the case of calcium aluminosilicate glasses (
20Ca,
30Ca), HRMAS NMR investigations found concentrations of about 4% NBO for
20Ca [
37] and 23% NBO for
30Ca [
38]. Furthermore, it was observed that the NBO content is virtually independent of the used alkaline earth oxide [
39]. Except for the overestimation of the NBO fraction for the metaluminous glass composition (
20Mg,
20Ba), the simulation results are in good agreement with these experimental observations. As mentioned above, a further refinement of the structural models can be achieved by employing higher-level simulations, see
Section 3.1.
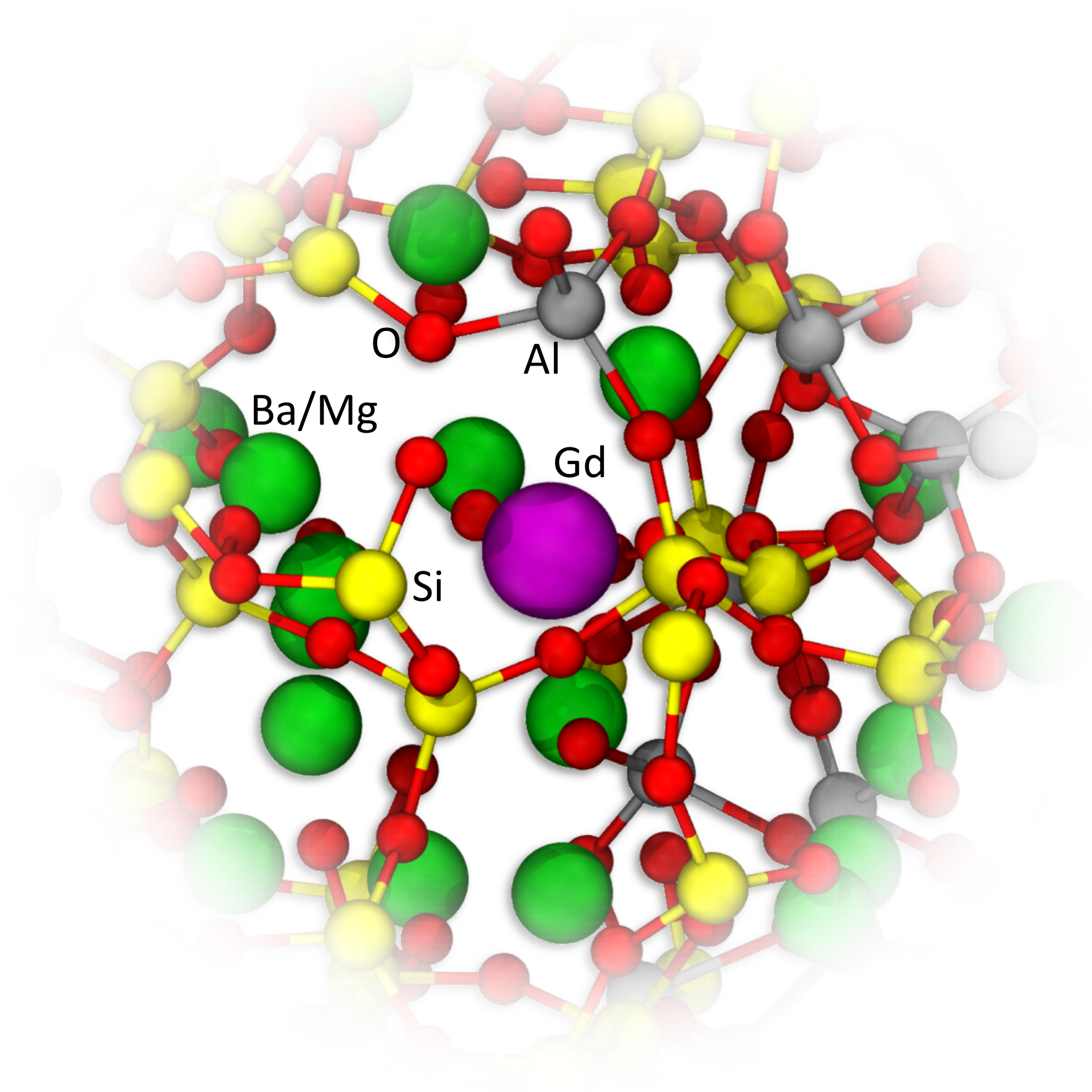
3.3. Medium-Range Structure of Gd3+
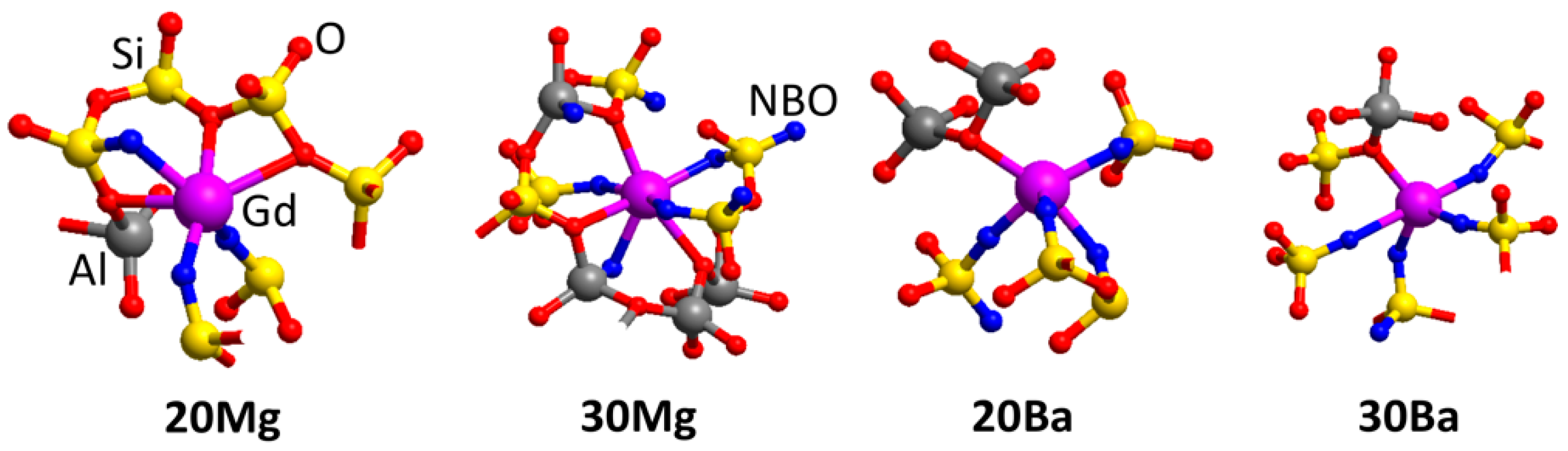
Table 5 summarizes the coordination numbers (CN) of Gd
3+ ions for all glass structural models. Examples of the atomic structure for the first and second coordination shell of Gd
3+ of the glass models are depicted in
Figure 4. The coordination of Gd
3+ with [AlO
x] and [SiO
4] shows virtually the same dependence on the chemical composition as the NM ions. While the number of [SiO
4] units in the second coordination shell is approximately constant, the coordination with [AlO
x] polyhedra increases with increasing Al
2O
3 concentration.
In contrast, significant changes are found for the coordination of Gd
3+ ions to the NM ions, which is steadily increasing in the order
20Mg <
20Ba <
30Mg <
30Ba. For the peralkaline composition
30Ba, Gd
3+ ions have by far the highest coordination number to other network modifier (NM) ions (Ba in this case). In fact, this composition is the only the case where the Gd
3+ coordination is notably different from all other glass structural models. The same applies to the Ba
2+ ions showing a considerably higher NM-NM coordination compared to the other glass structure models, see
Table 3. This clearly shows that Gd
3+ is incorporated into the glass structure in the same way as a network-modifying ion. However, despite its increasing coordination to NM ions and increasing Gd-NM distances depending on the glass composition, the distance to directly neighboring oxygen atoms is constant at 2.25 Å for all four glass structure models. Also, the number of neighboring oxygen ions is mostly independent of any change in the glass composition. It varies only between 5.4 and 5.8 in all four predicted glass structures.
The Gd-Gd CNs shown in
Table 5 are lower than 0.1 for all structural models showing that, on average, less than 10% of the Gd
3+ ions of the IS form Gd-O-Gd contacts. This indicates a low driving force for clustering of Gd
3+ ions in the investigated aluminosilicate glasses. Therefore, the formation of larger Gd
xO
y clusters appears to be unlikely. In addition, the Gd-Gd CN is almost independent of the chemical composition since such small changes are expected to be lower than the accuracy of present structural predictions using empirically parameterized interatomic potential functions.
Table 6 summarizes the coordination numbers of Gd
3+ with bridging (BO), non-bridging (NBO), and tricluster (Tri) oxygen ions. The coordination of Gd
3+ with NBO is somewhat higher in the barium-containing glass compositions than in the respective magnesium-containing glass structure models. Obviously, ions of higher electric field strength, i.e., ions with a smaller ionic radius or higher charge, such as Mg
2+ and Gd
3+ prefer coordination with NBO sites. In addition, in analogy to the NM ions, the coordination of Gd
3+ with oxygen ions in triclusters is lower in the peralkaline compositions (
30Mg,
30Ba) compared to the metaluminous compositions (
20Mg,
20Ba), which correlates with the total concentration of tricluster oxygen ions, see
Figure 3.
Table 7 shows the coordination numbers of Gd
3+ ions with BO and NBO separated according to the network former ions present in the second coordination sphere. It can be differentiated between Si-bound (
−O−Si≡) and Al-bound (
−O−Al≡) NBO and BO, i.e., ≡(Si, Al)−O−(Si, Al)≡, or BO in triclusters. The overall coordination of Gd
3+ with NBO increases with the increasing radius and concentration of the network-modifying ions, see
Table 6. However, as shown in
Table 7, this increase is only caused by additional Si-bound oxygen. The percentage of Al-bound non-bridging oxygen is virtually constant for all glasses. The increase in the numbers of Gd-coordinated NBO is realized at the expense of the numbers of BO. But here the number of ≡Si−O−Si≡ species remains almost unchanged, while the number of ≡Al−O−Al≡ species is the most diminished. In summary, it can be stated that the results show a clear tendency for an increased number of Si-bound NBO in the coordination sphere of the doped Gd
3+ ions with increasing size and concentration of the network-modifying ions, while the absolute number of coordinating Si
4+ in the second coordination sphere remains approximately constant, as stated earlier, see
Table 5. Additionally, the Al
3+ ions in the second coordination sphere with their high field strength are replaced by the relatively low field strength network-modifier ions Mg
2+ and Ba
2+. This replacement is more apparent in the order
20Mg <
20Ba <
30Mg <
30Ba. A calculation of the summed-up field strengths at the Gd
3+ positions resulting from the coordinating cations Si
4+, Al
3+, Mg
2+, and Ba
2+, according to the data in
Table 5, gives an order
20Mg ≈
30Mg >
20Ba >
30Ba. This very simplified model can be applied since the number and distance of the directly neighboring oxygen atoms is mostly independent of the glass composition, see
Table 5. Despite different network-modifier CN and distances, the field strengths for
20Mg and
30Mg are almost the same. This result correlates perfectly with the peak splitting in Tb
3+ doped glasses [
14]: Higher concentrations of low field strength ions in the glass composition increase the peak splitting and therefore induce an increased electric field at the rare earth positions; most likely due to their lower polarization power on the neighboring oxygen atoms. On the other hand, these results do not correlate with the experimentally derived luminescence lifetime data: The lifetimes for all tested rare earth ions Sm
3+, Eu
3+, and Tb
3+ increase in the order
20Ba <
20Mg =
30Mg <
30Ba [
11,
12,
14]. The respective lifetimes for the samples
20Mg and
30Mg are almost the same. However, it must be stated that in the cases of Sm
3+ and Tb
3+, lifetimes from
30Ba compositions were not reported. Instead, the results for the very similar composition with 35 mol% BaO, 10 mol% Al
2O
3, and 55 mol% SiO
2 have been used for these comparisons. In summary, the structural models of the glasses presented here are not sufficient to explain all the observed luminescence effects. However, a deeper understanding of the relation between the local structure of rare earth ions in aluminosilicate glasses and the optical properties can possibly be achieved using higher level quantum mechanical simulations employing the provided structural models.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}