
Zirconia/Hydroxyapatite Composites Synthesized Via Sol-Gel: Influence of Hydroxyapatite Content and Heating on Their Biological Properties


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
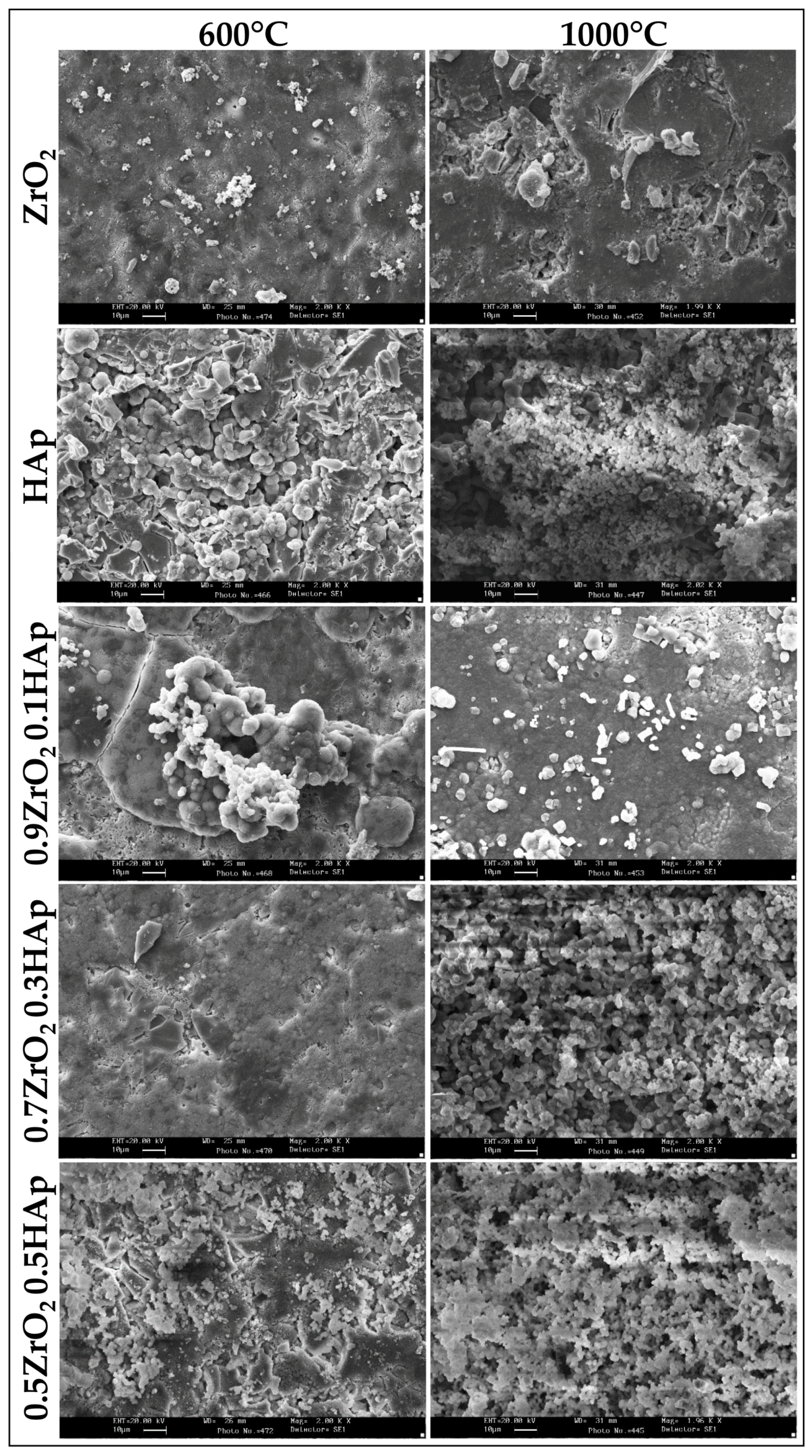
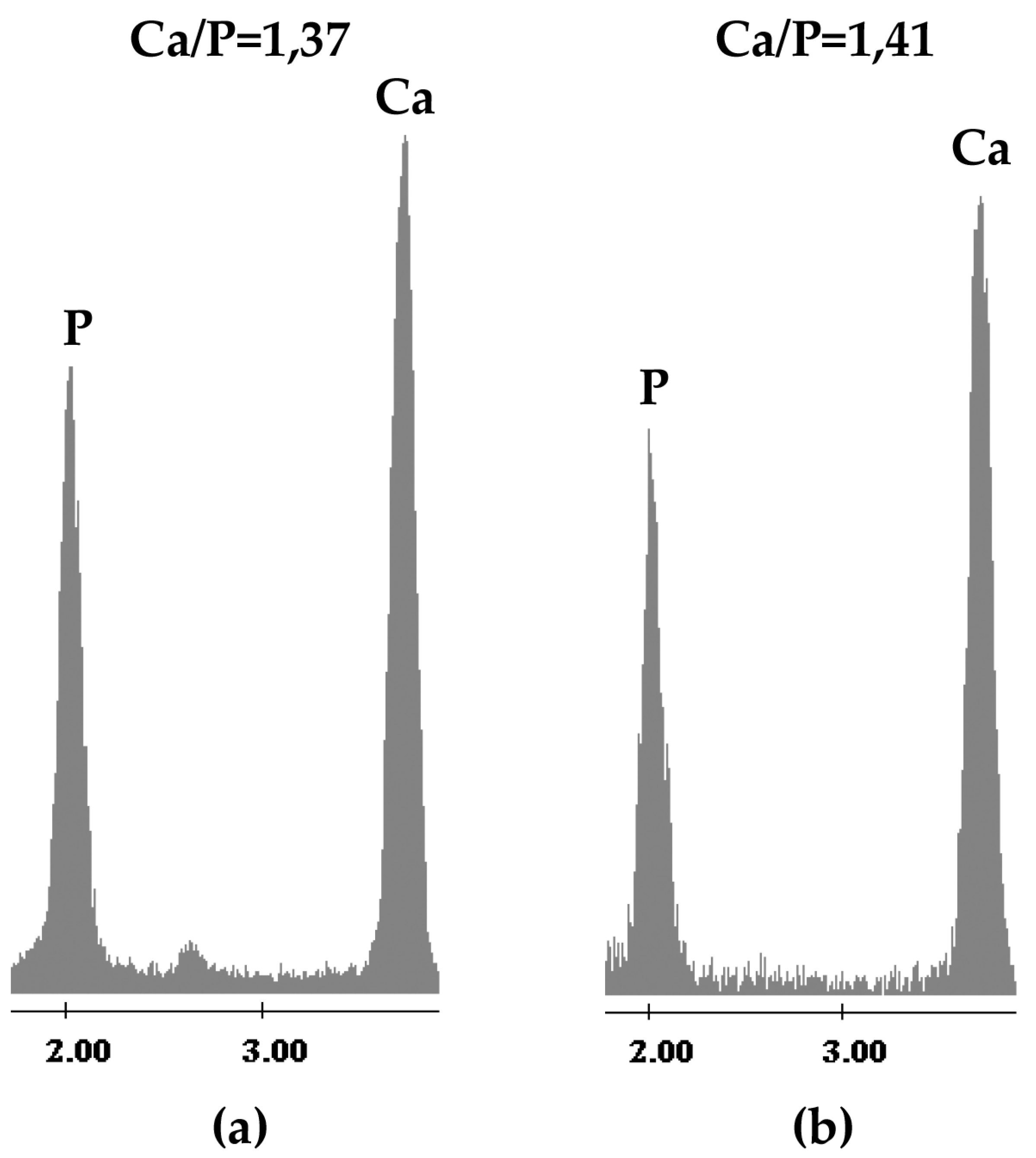
2. Results and Discussion
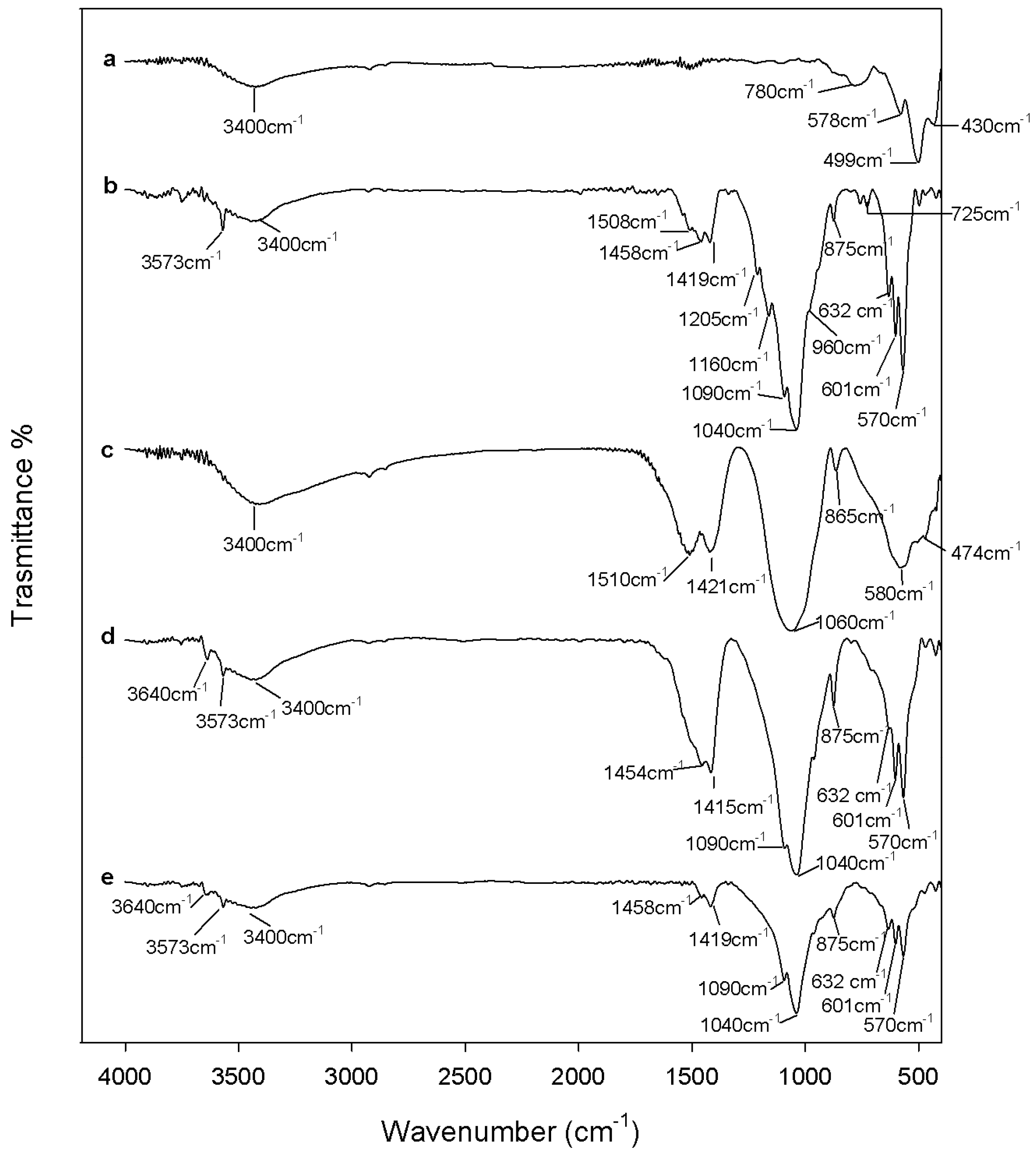
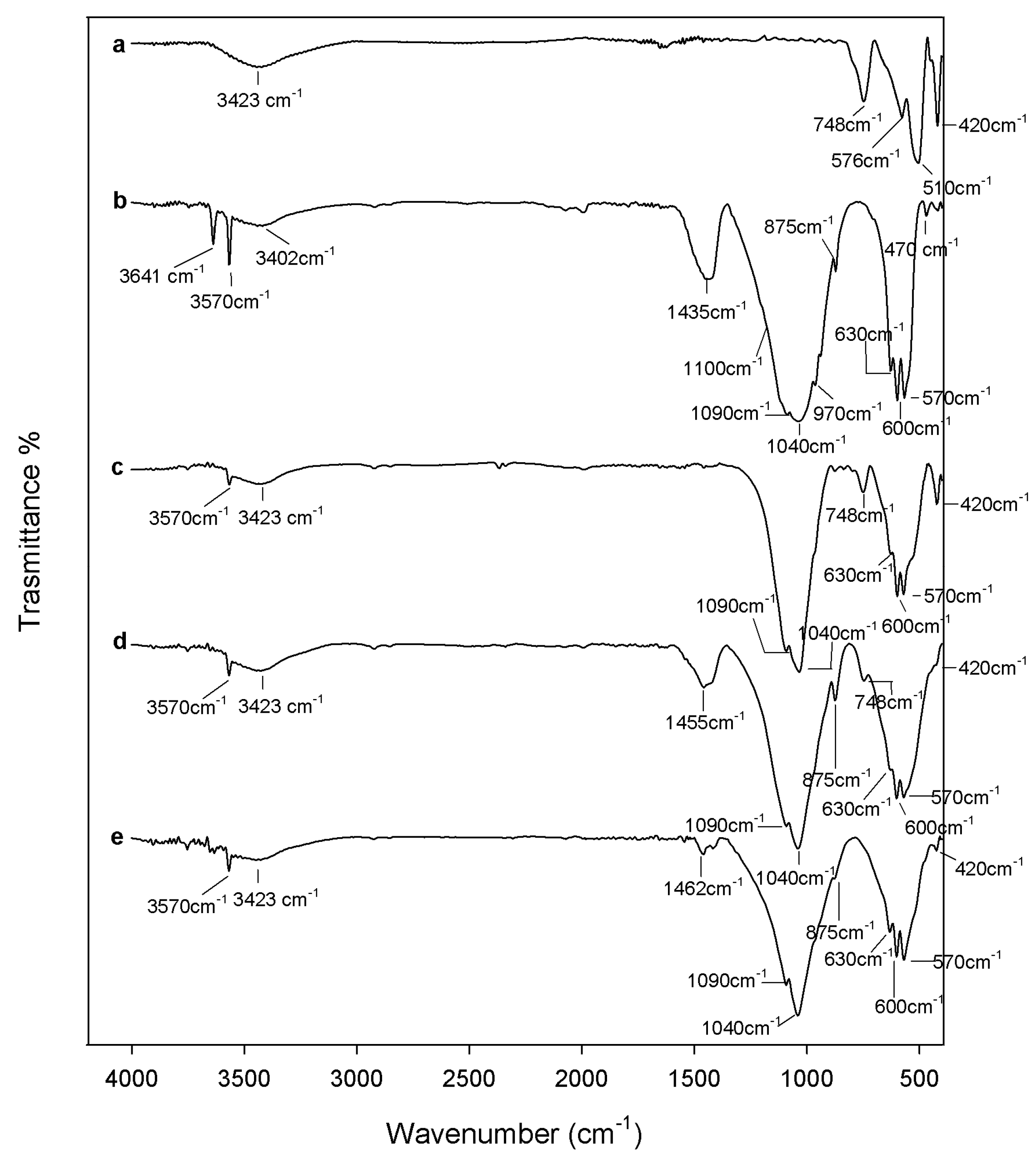
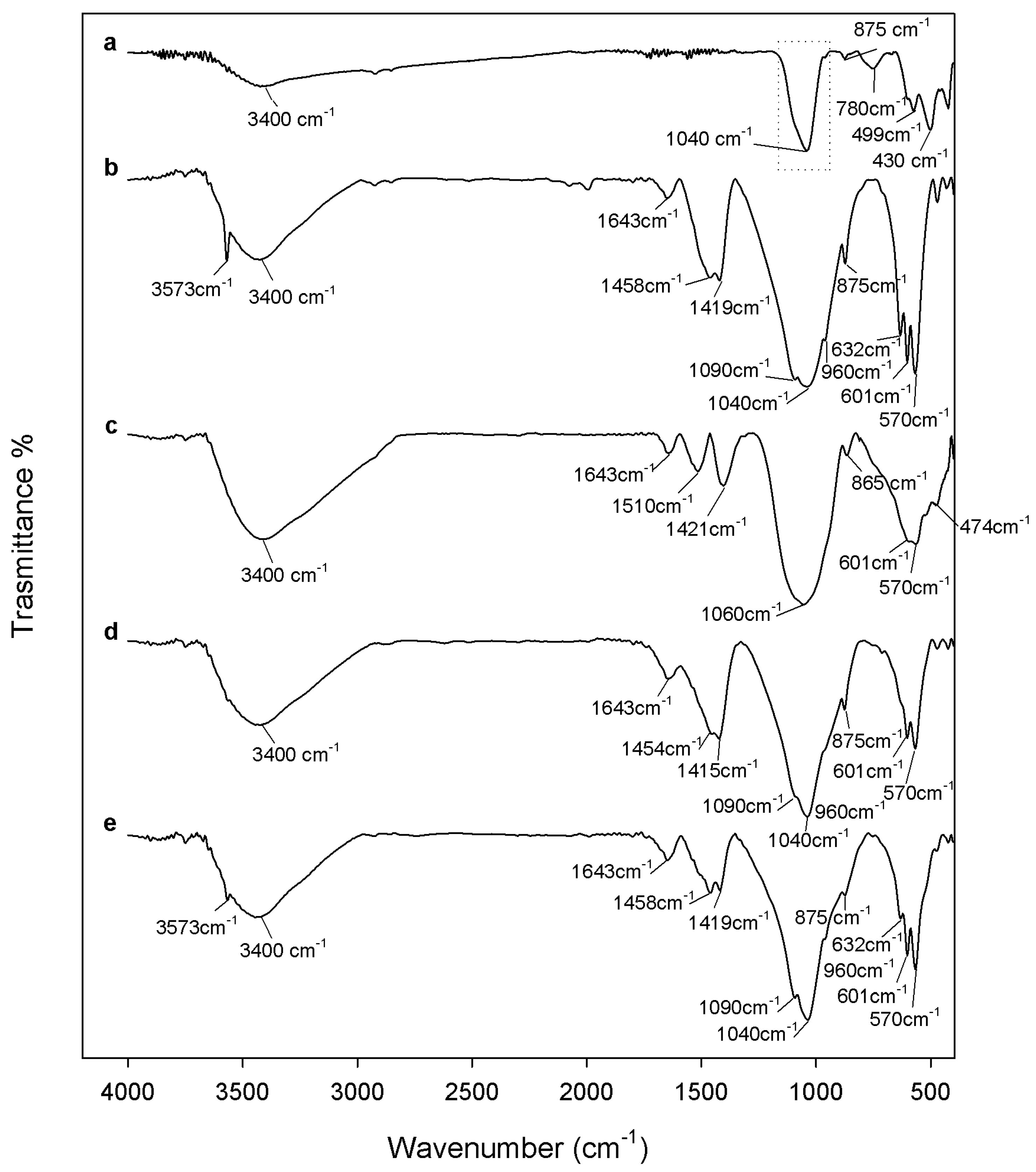
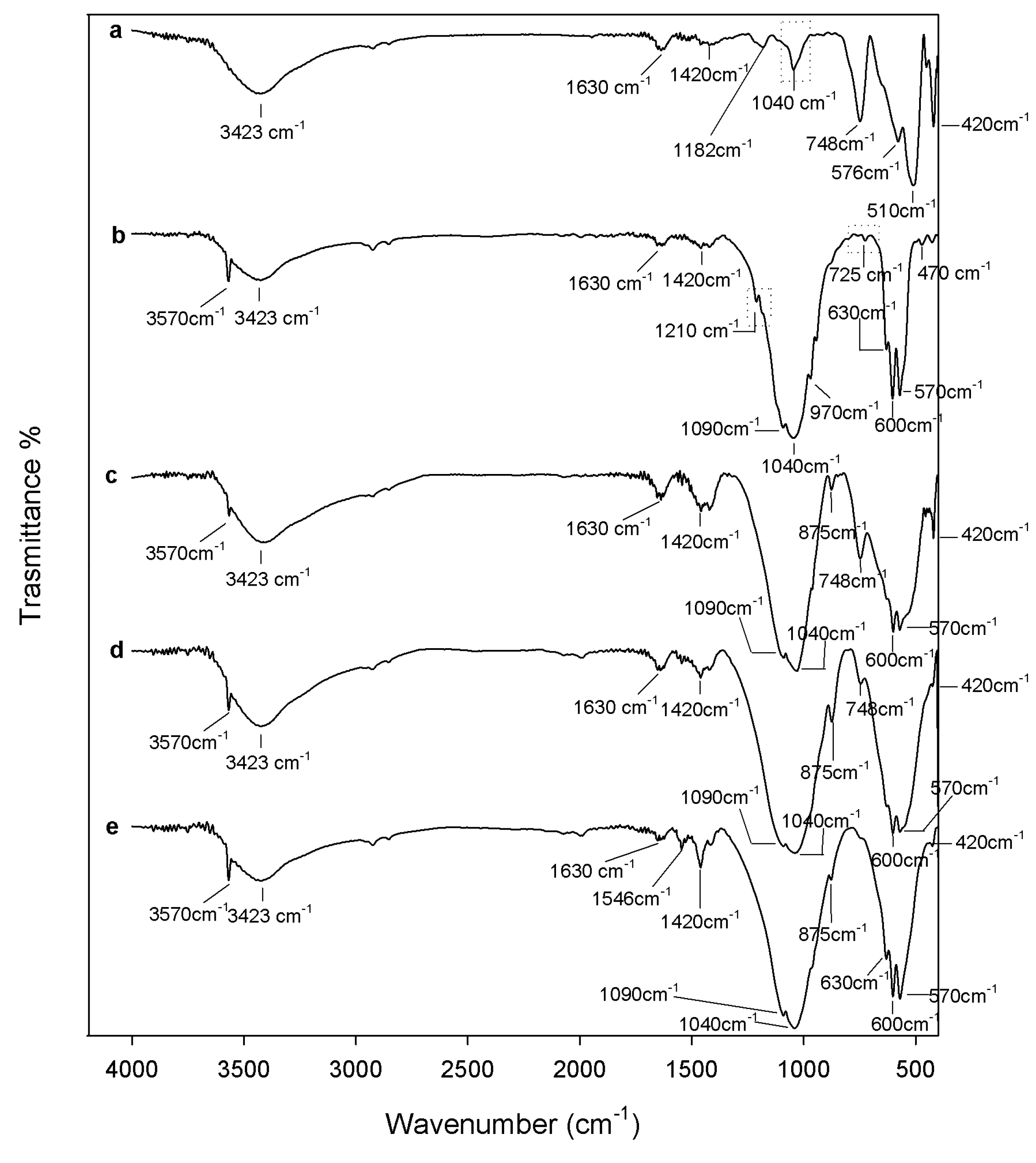
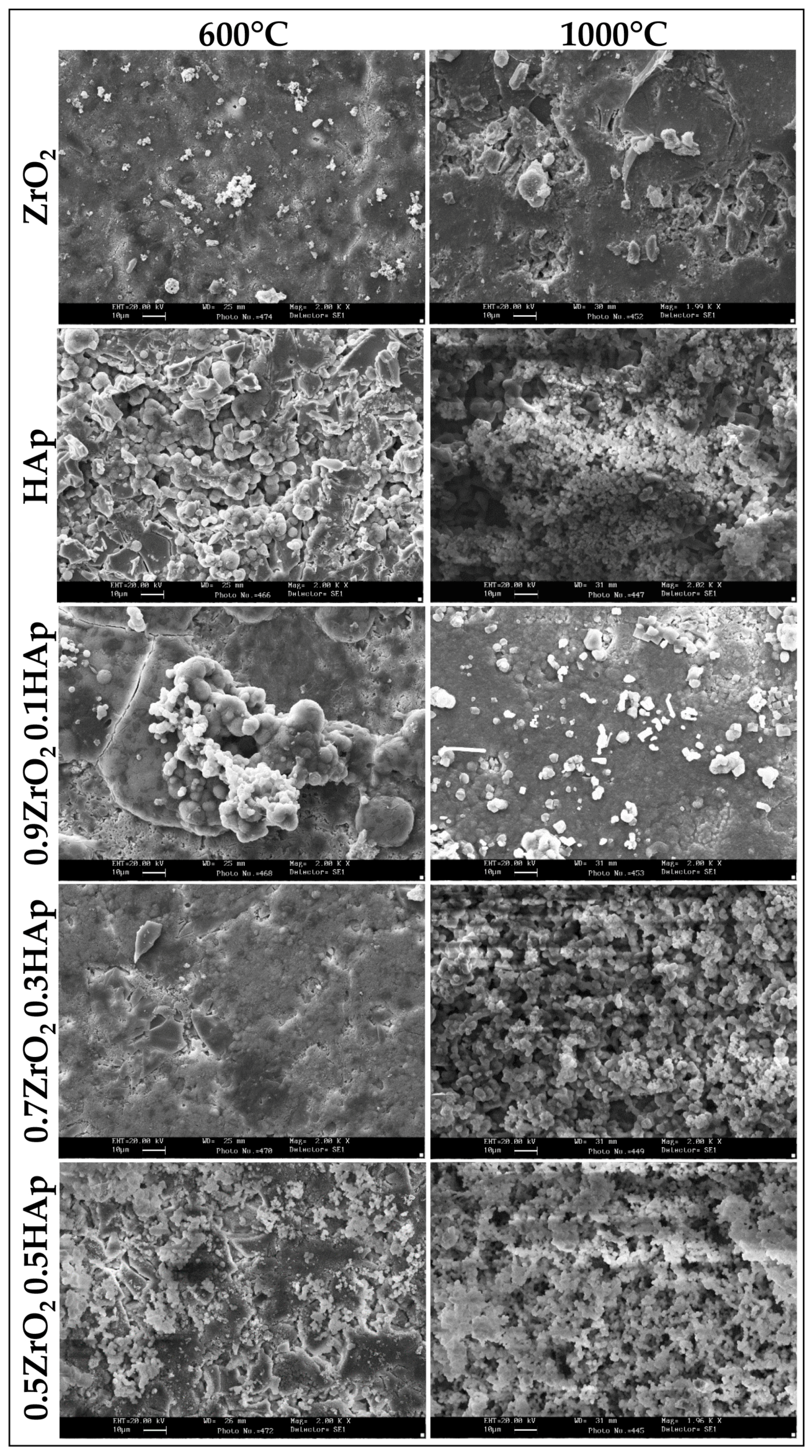
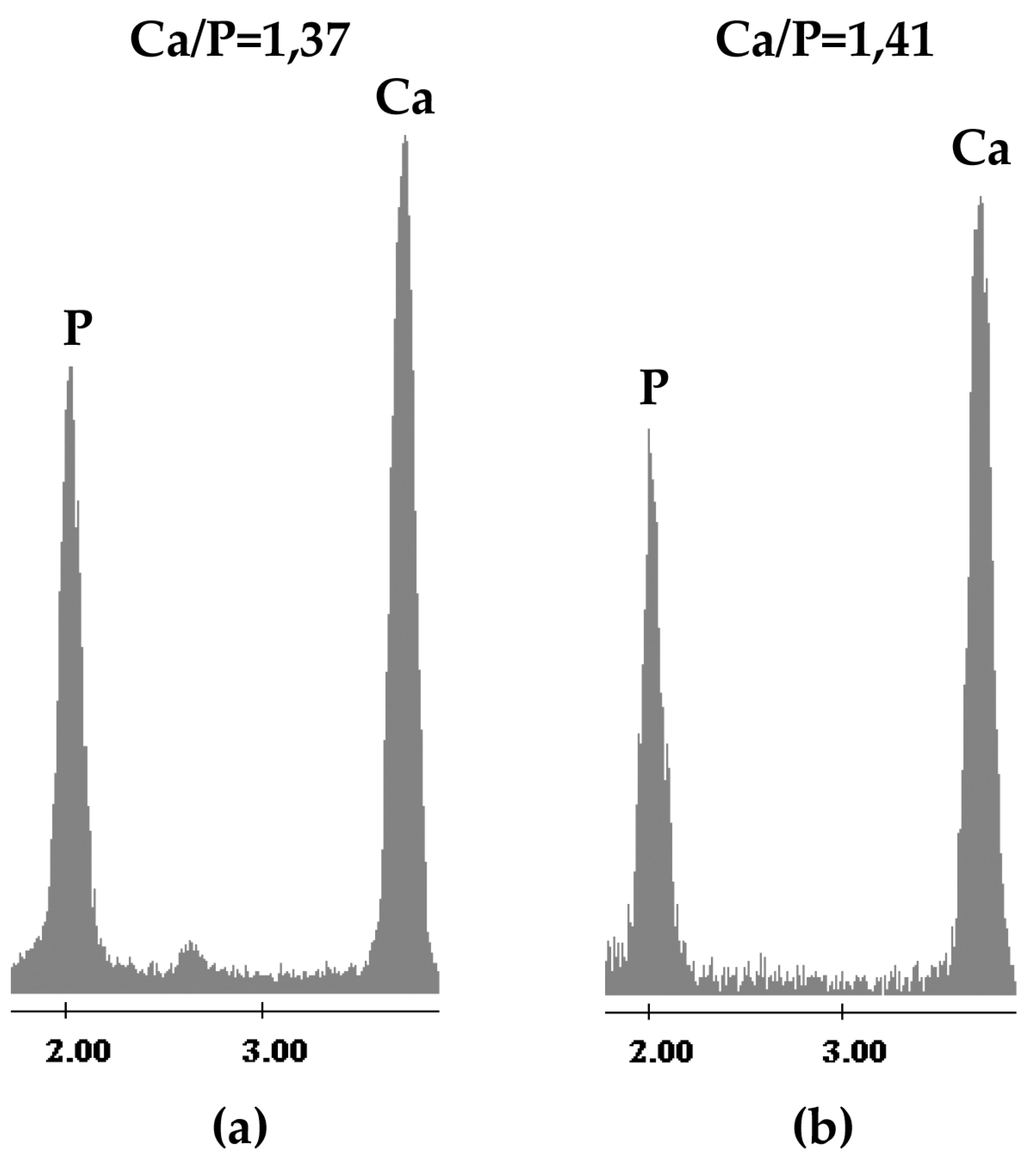
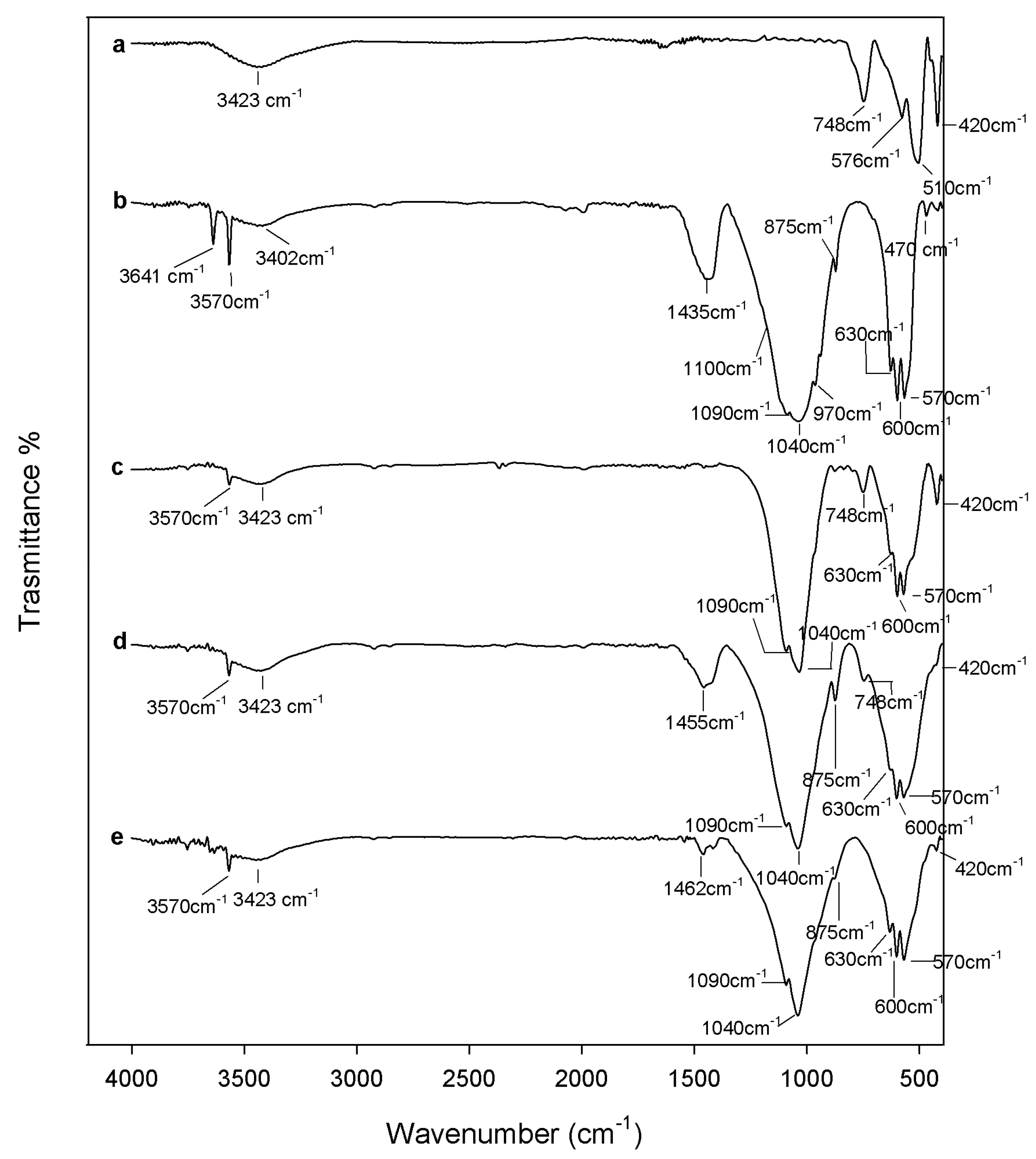
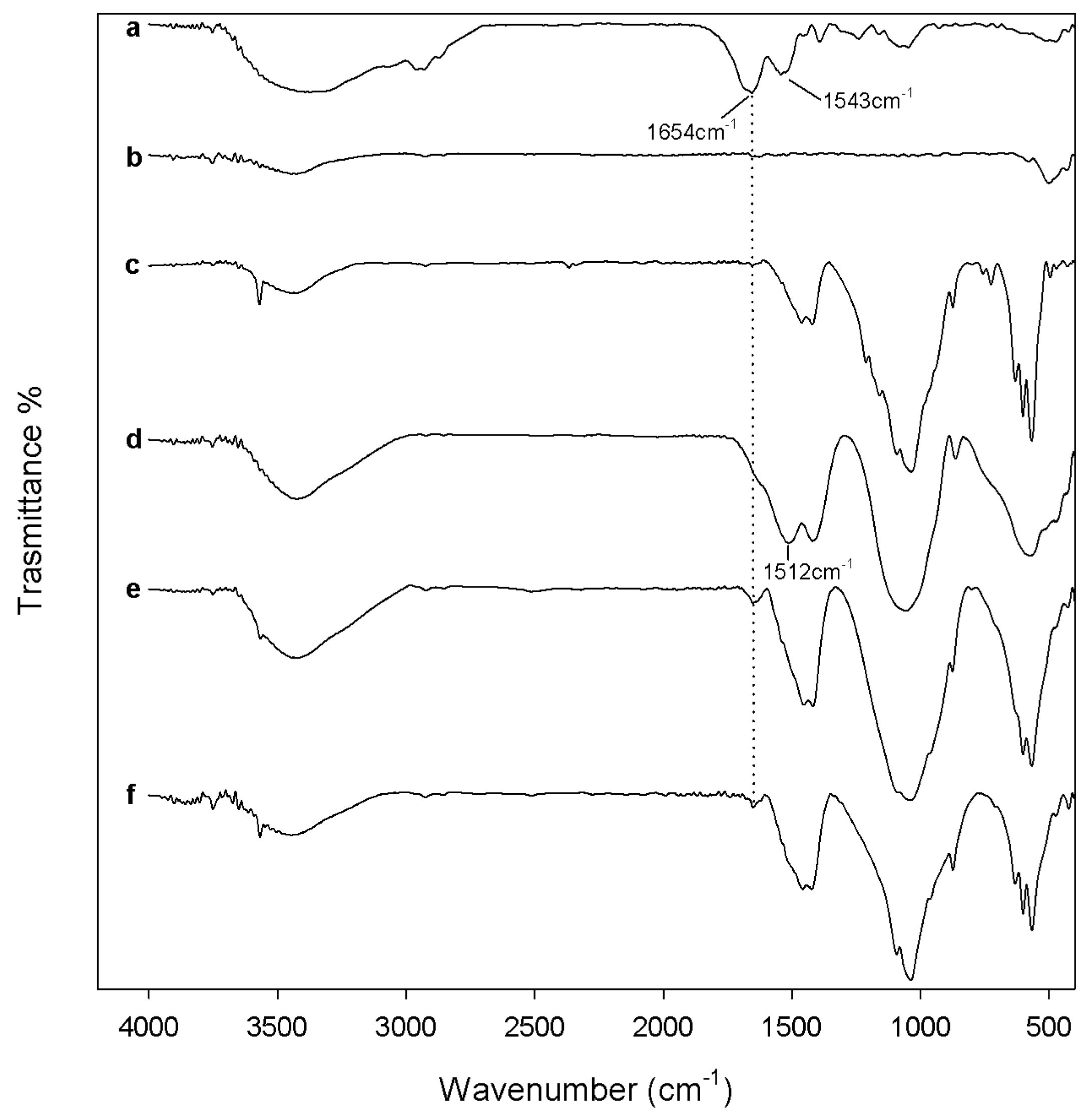
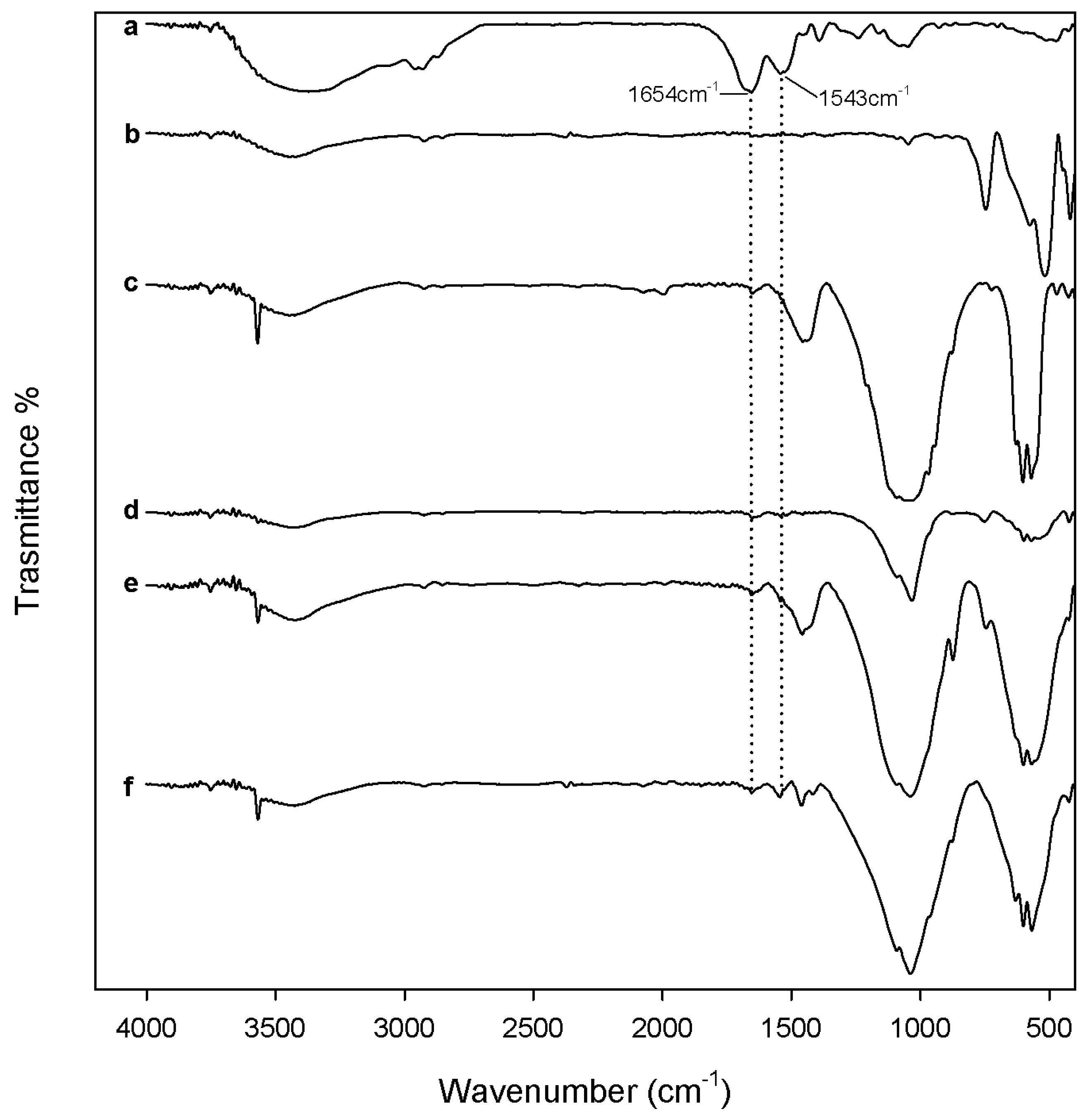
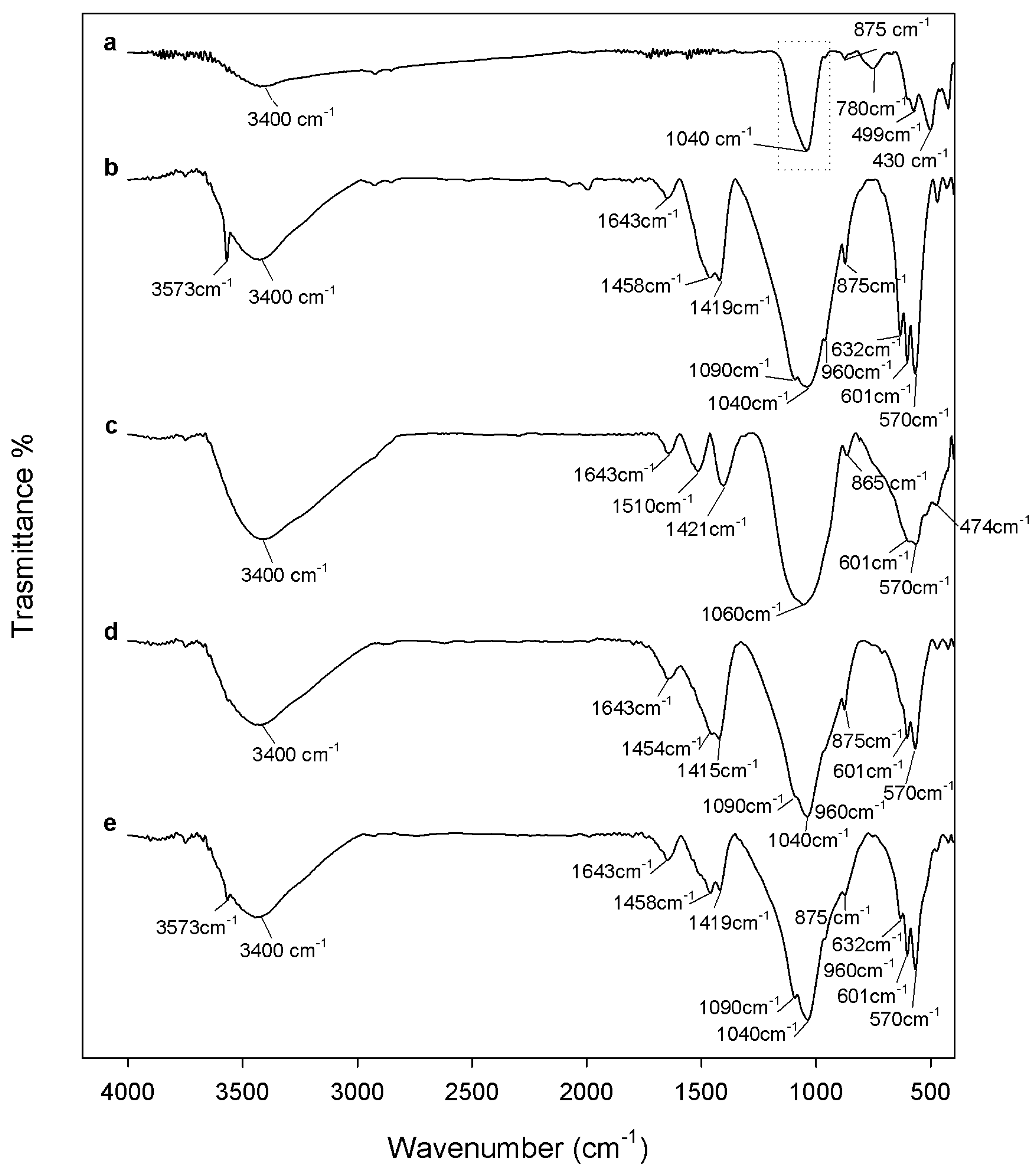
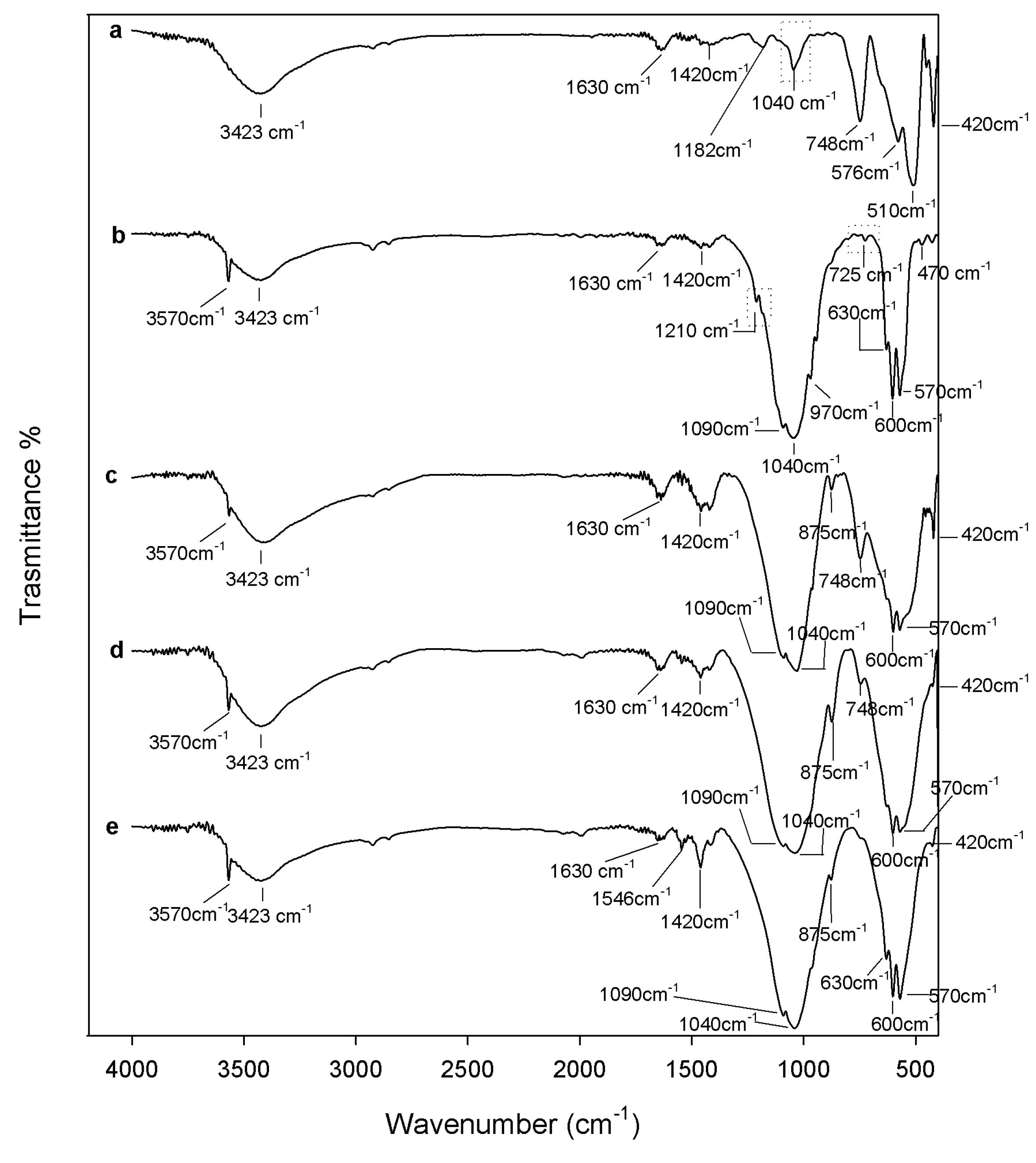
2.1. Chemical Characterization
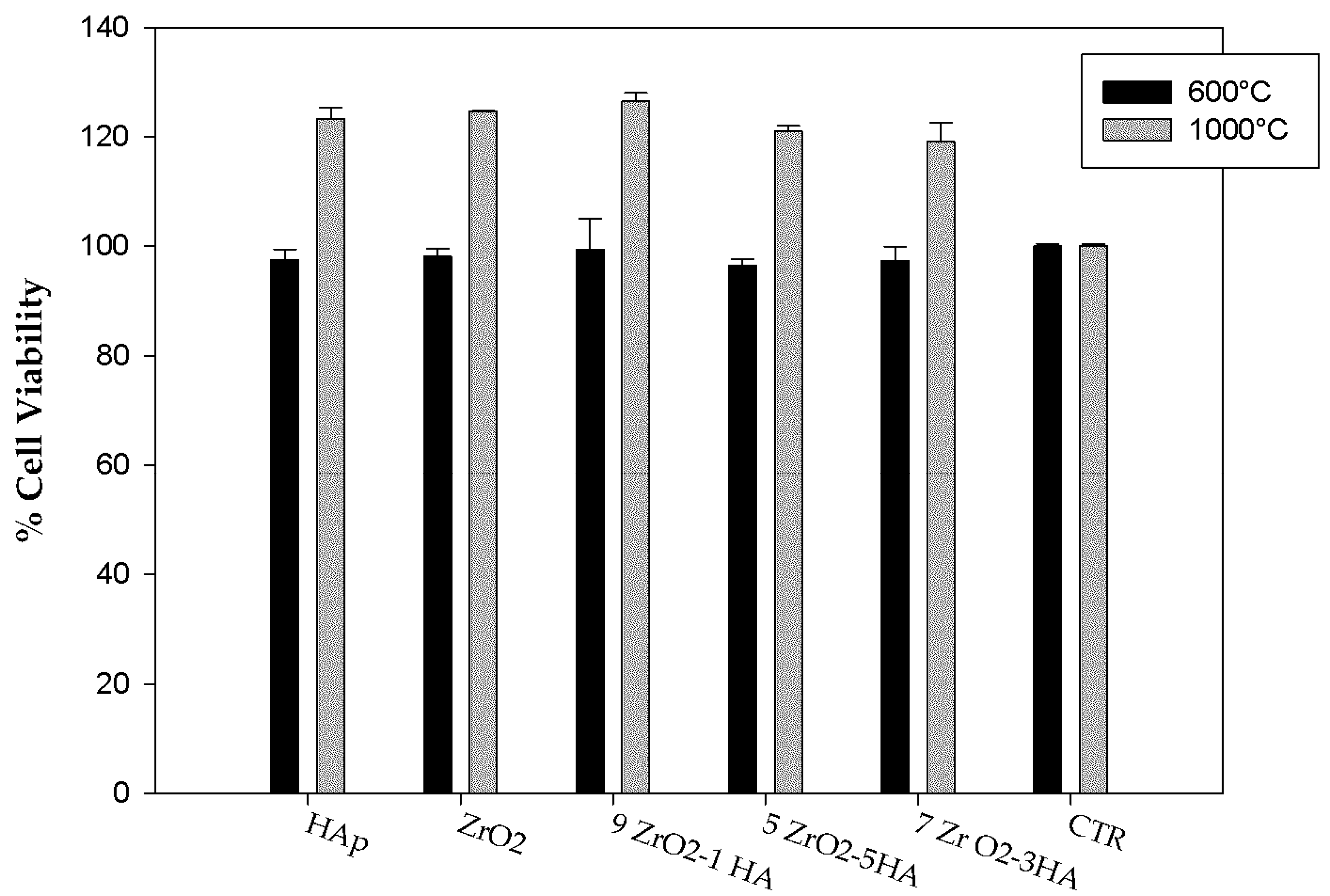
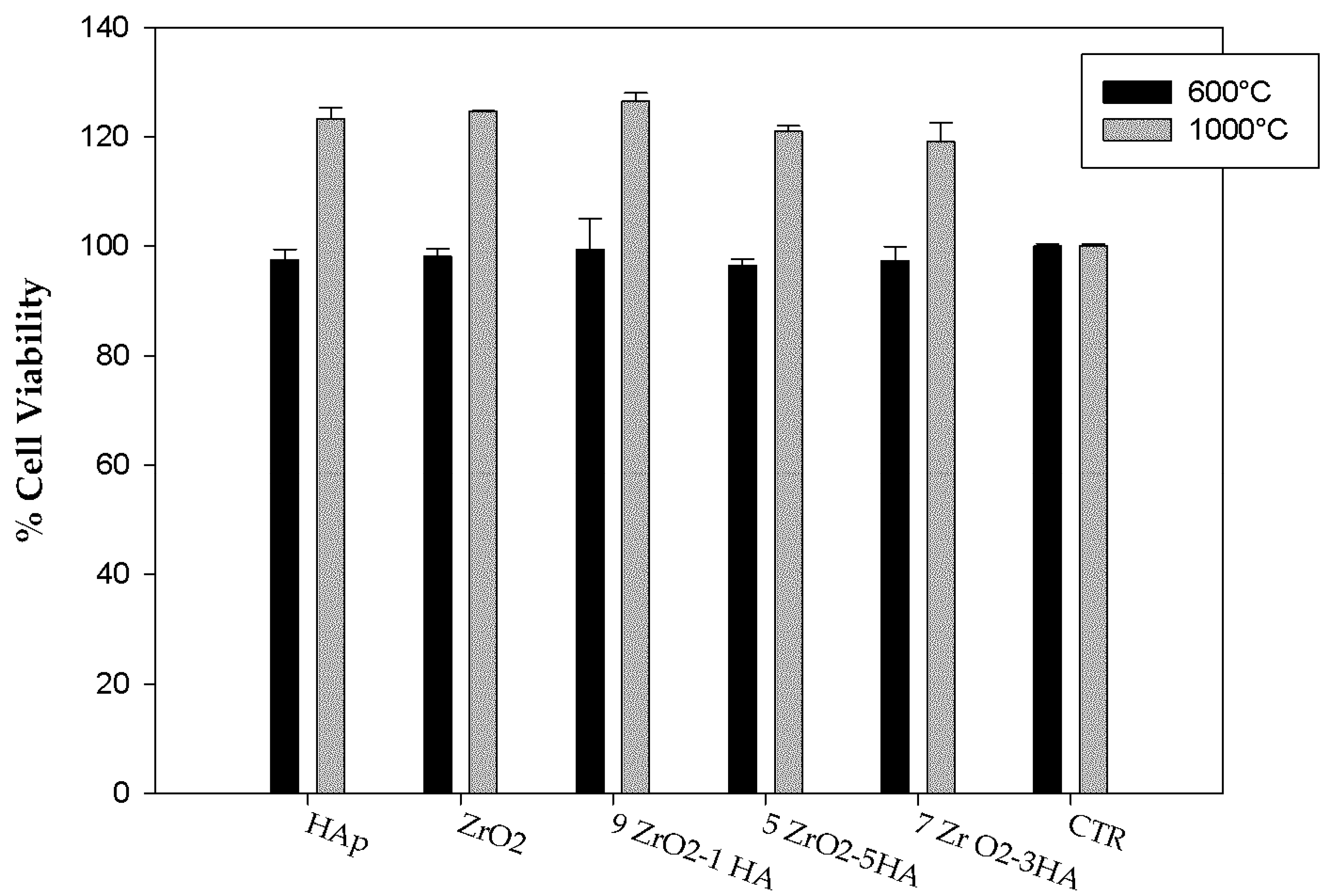
2.2. Evaluation of Biological Properties
3. Materials and Methods
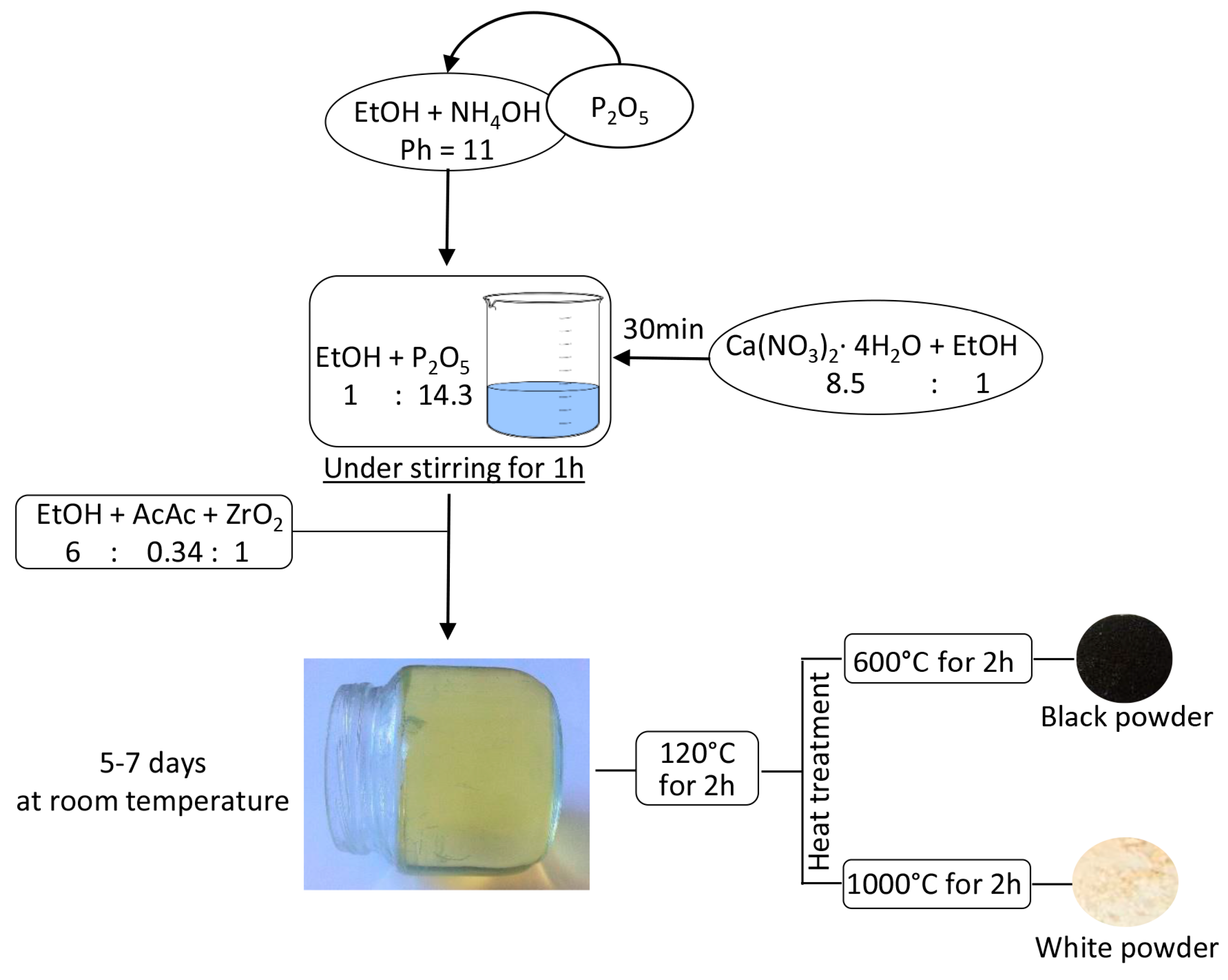
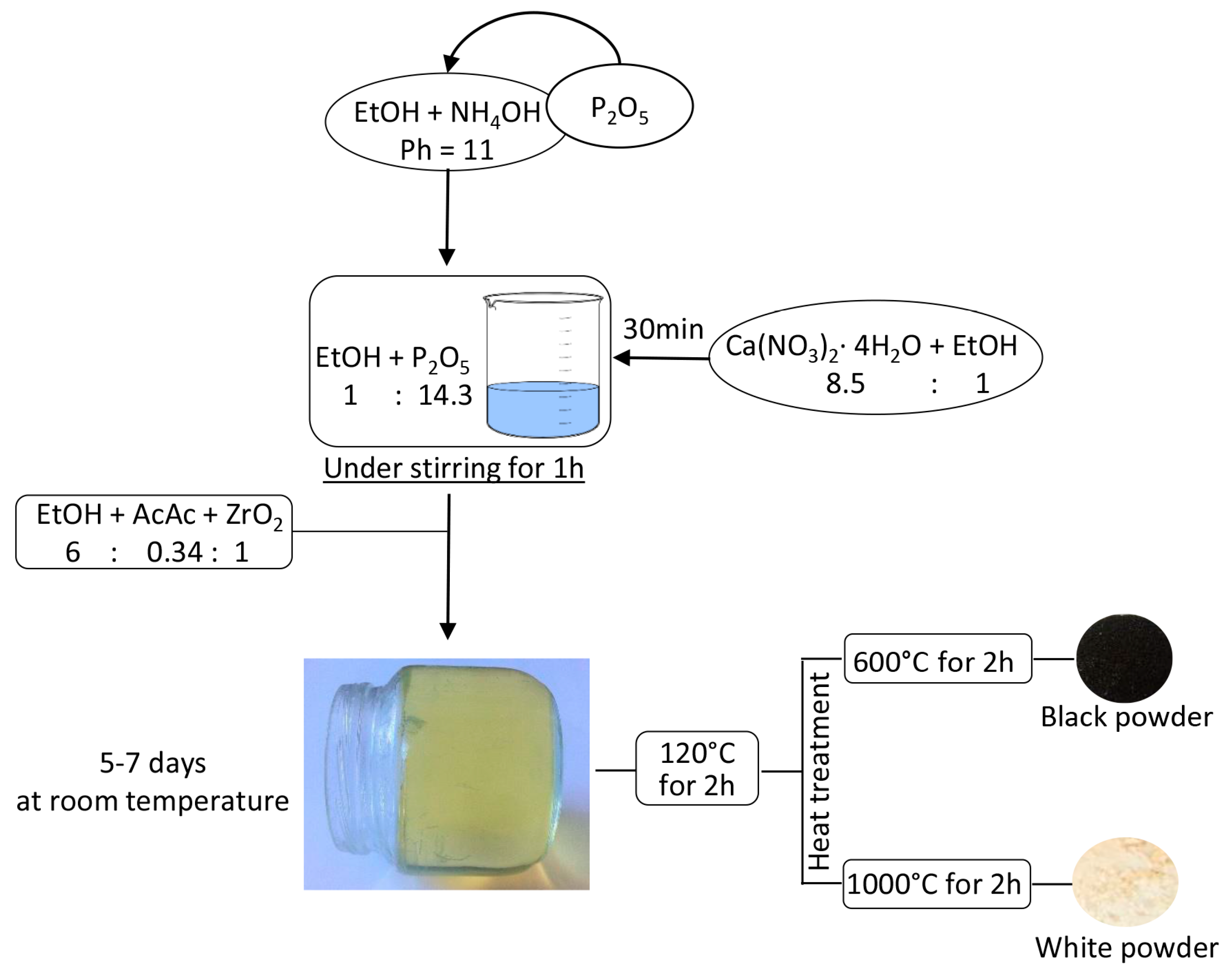
3.1. Sol-Gel Synthesis of the Composites
- Calcium nitrate tetrahydrate (Ca(NO3)2·4H2O) was dissolved in ethanol 99.8% under stirring;
- Phosphorus pentoxide was added to a solution of NH4OH in Ethanol with pH = 11 under stirring.
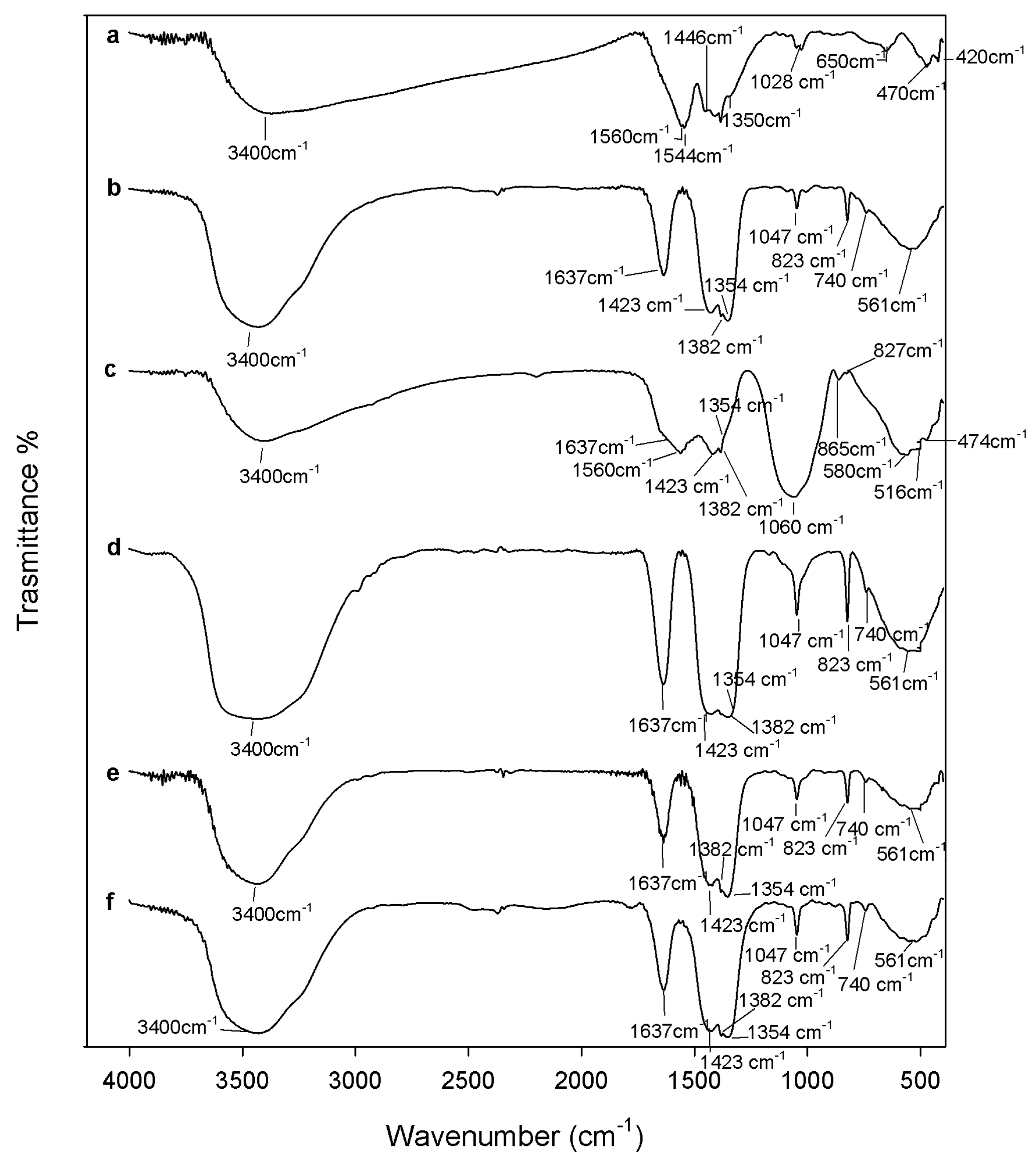
3.2. Composites Chemical Structure
3.3. Biological Properties
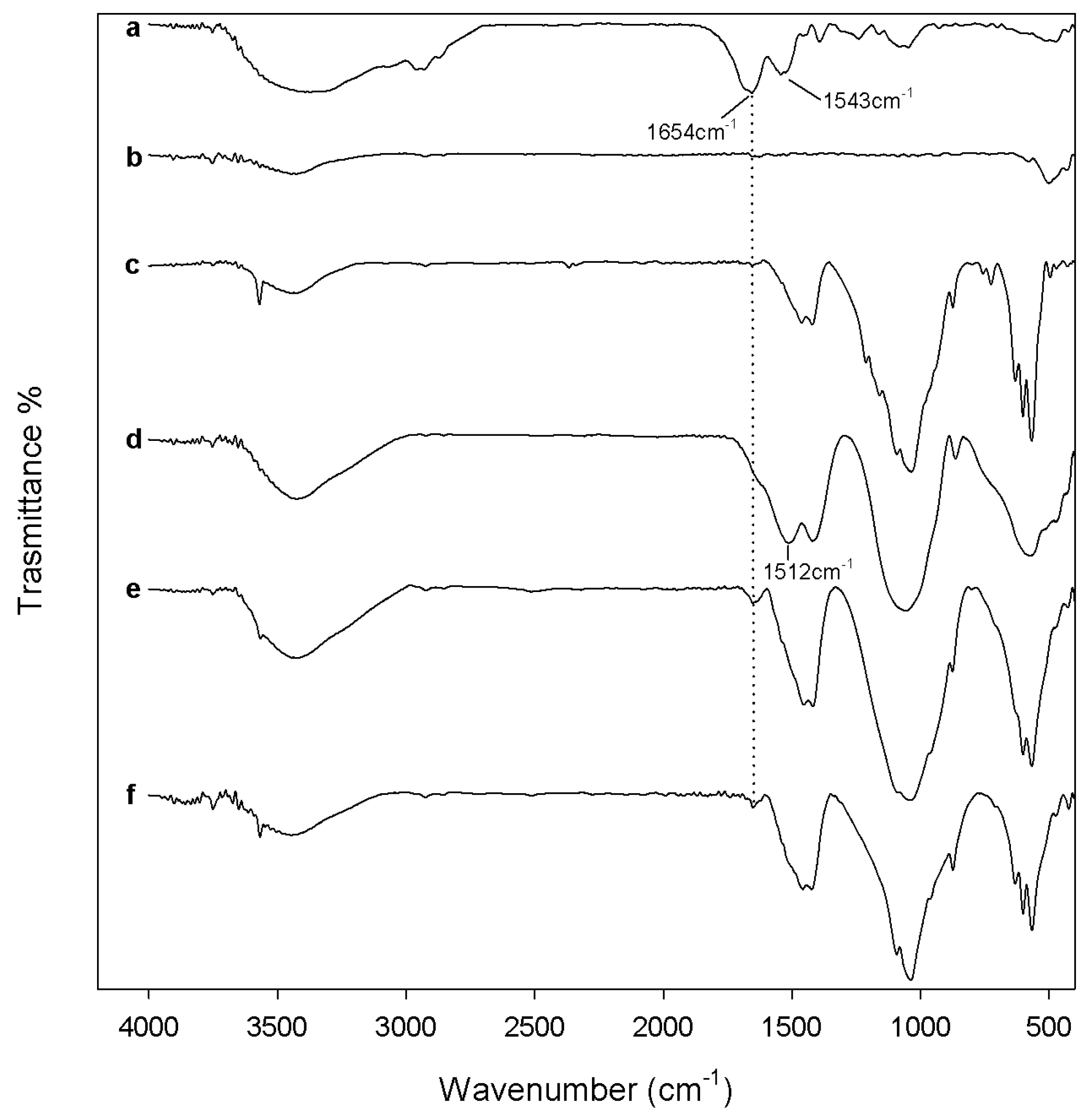
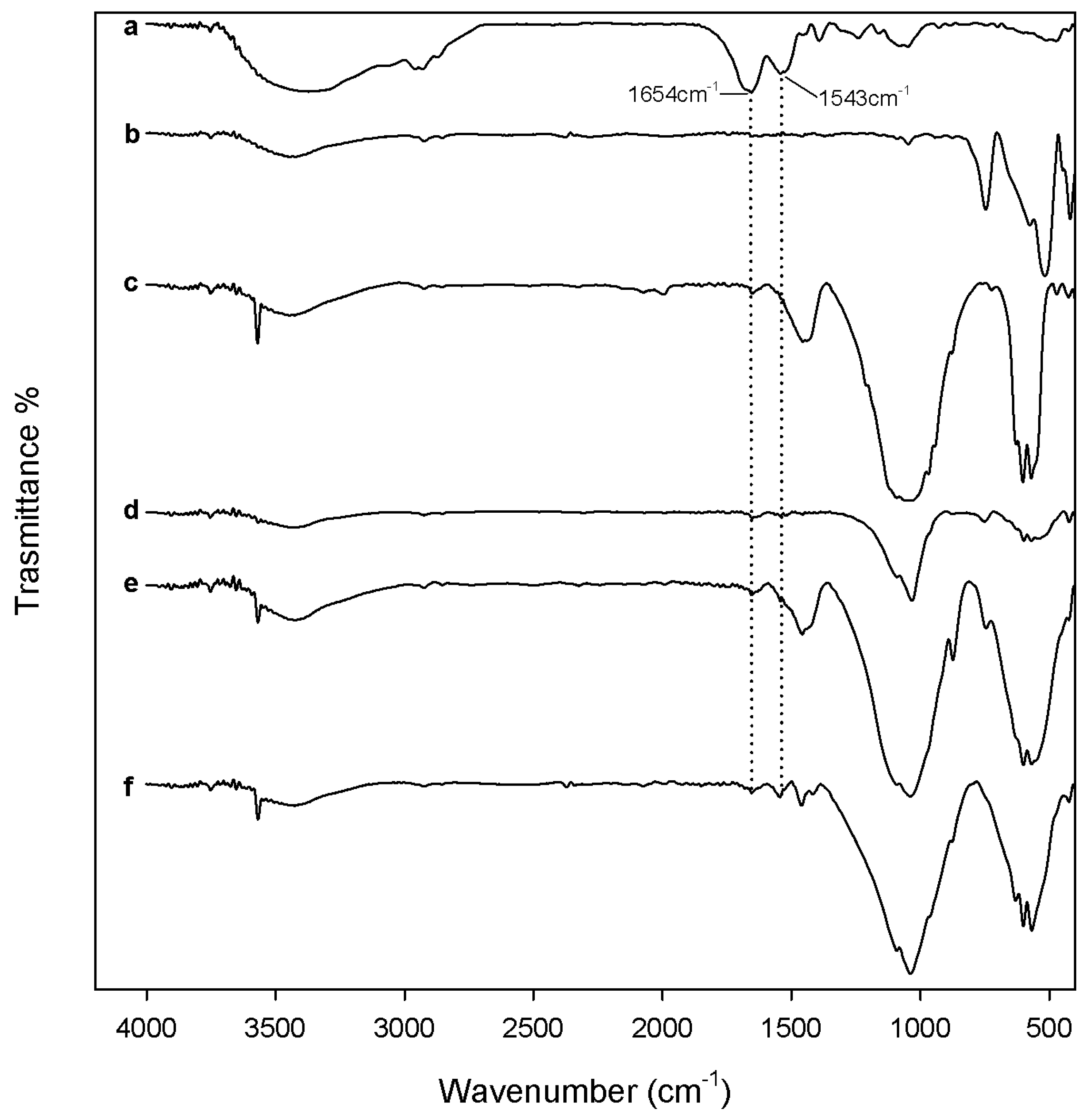
3.3.1. Protein Adsorption Evaluation
3.3.2. WST-8 Assay
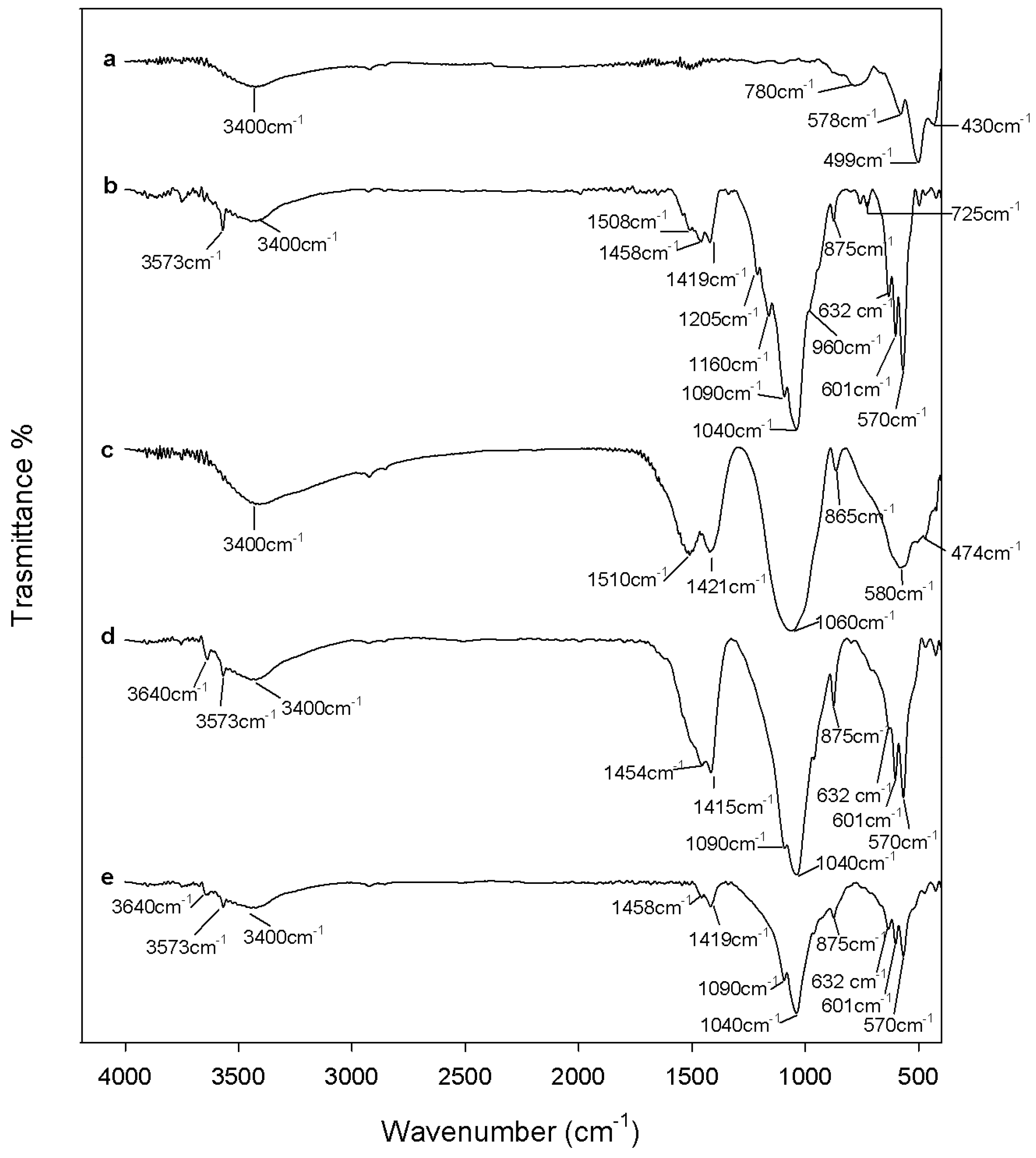
3.3.3. Apatite-Forming Ability Test
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Abd El-Ghany, O.S.; Sherief, A.H. Zirconia based ceramics, some clinical and biological aspects: Review. Future Dent. J. 2016, 2, 55–64. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F.; Pacifico, S.; Galasso, S.; Ferrara, C.; Mustarelli, P. Synthesis of zirconia/polyethylene glycol hybrid materials by sol-gel processing and connections between structure and release kinetic of indomethacin. Drug Deliv. 2014, 21, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Harianawala, H.; Kheur, M.; Kheur, S.; Sethi, T.; Bal, A.; Burhanpurwala, M.; Sayed, F. Biocompatibility of Zirconia. J. Adv. Med. Dent. Sci. Res. 2016, 4, 35–39. [Google Scholar]
- Catauro, M.; Papale, F.; Bollino, F.; Gallicchio, M.; Pacifico, S. Biological evaluation of zirconia/PEG hybrid materials synthesized via sol-gel technique. Mater. Sci. Eng. C 2014, 40, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, T.R.; Gangaiah, M.; Harish, P.V.; Krishnakumar, U.; Nandakishore, B. Zirconia Ceramics as a Dental Biomaterial—An Over view. Trends Biomater. Artif. Organs 2012, 26, 154–160. [Google Scholar]
- Hannink, R.H.J.; Kelly, P.M.; Muddle, B.C. Transformation toughening in zirconia-containing ceramics. J. Am. Ceram. Soc. 2000, 83, 461–487. [Google Scholar] [CrossRef]
- Burger, W.; Richter, H.G.; Piconi, C.; Vatteroni, R.; Cittadini, A.; Boccalari, M. New Y-TZP powders for medical grade zirconia. J. Mater. Sci. Mater. Med. 1997, 8, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Readey, M.J. Effect of Heat Treatment on Grain Size, Phase Assemblage, and Mechanical Properties of 3 mol% Y-TZP. J. Am. Ceram. Soc. 1996, 79, 2331–2340. [Google Scholar] [CrossRef]
- Gupta, T.K.; Lange, F.F.; Bechtold, J.H. Effect of stress-induced phase transformation on the properties of polycrystalline zirconia containing metastable tetragonal phase. J. Mater. Sci. 1978, 13, 1464–1470. [Google Scholar] [CrossRef]
- Piconi, C.; Maccauro, G. Zirconia as a ceramic biomaterial. Biomaterials 1999, 20, 1–25. [Google Scholar] [CrossRef]
- Evans, A.G.; Heuer, A.H. Review—Transformation Toughening in Ceramics: Martensitic Transformations in Crack-Tip Stress Fields. J. Am. Ceram. Soc. 1980, 63, 241–248. [Google Scholar] [CrossRef]
- Coli, P.; Karlsson, S. Fit of a New Pressure-Sintered Zirconium Dioxide Coping. Int. J. Prosthodont. 2004, 17, 59–64. [Google Scholar] [PubMed]
- Salehi, S.; Fathi, M.H. Fabrication and characterization of sol–gel derived hydroxyapatite/zirconia composite nanopowders with various yttria contents. Ceram. Int. 2010, 36, 1659–1667. [Google Scholar] [CrossRef]
- Qiu, D.; Wang, A.; Yin, Y. Characterization and corrosion behavior of hydroxyapatite/zirconia composite coating on NiTi fabricated by electrochemical deposition. Appl. Surf. Sci. 2010, 257, 1774–1778. [Google Scholar] [CrossRef]
- Say, Y.; Aksakal, B. Effects of hydroxyapatite/Zr and bioglass/Zr coatings on morphology and corrosion behaviour of Rex-734 alloy. J. Mater. Sci. Mater. Med. 2016, 27, 105. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-J.; Long, D.; Wu, Y.-Z.; Zhou, C.-Z. Mechanical properties of hydroxyapatite-zirconia coatings prepared by magnetron sputtering. Trans. Nonferrous Met. Soc. China 2012, 22, 104–110. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics. J. Am. Ceram. Soc. 1998, 81, 1705–1728. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F.; Pacifico, S. Modulation of indomethacin release from ZrO2/PCL hybrid multilayers synthesized via sol-gel dip coating. J. Drug Deliv. Sci. Technol. 2015, 26, 10–16. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F. Biocompatibility improvement of titanium implants by coating with hybrid materials synthesized by sol-gel technique. J. Biomed. Mater. Res. Part A 2014, 102, 4473–4479. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Bollino, F.; Papale, F. Preparation, characterization, and biological properties of organic-inorganic nanocomposite coatings on titanium substrates prepared by sol-gel. J. Biomed. Mater. Res. Part A 2014, 102, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Bollino, F.; Papale, F.; Giovanardi, R.; Veronesi, P. Corrosion behavior and mechanical properties of bioactive sol-gel coatings on titanium implants. Mater. Sci. Eng. C 2014, 43, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Bollino, F.; Papale, F.; Mozetic, P.; Rainer, A.; Trombetta, M. Biological response of human mesenchymal stromal cells to titanium grade 4 implants coated with PCL/ZrO2 hybrid materials synthesized by sol-gel route: In vitro evaluation. Mater. Sci. Eng. C 2014, 45, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Faure, J.; Drevet, R.; Lemelle, A.; Ben Jaber, N.; Tara, A.; El Btaouri, H.; Benhayoune, H. A new sol–gel synthesis of 45S5 bioactive glass using an organic acid as catalyst. Mater. Sci. Eng. C 2015, 47, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Laudisio, G.; Costantini, A.; Fresa, R.; Branda, F. Low Temperature Synthesis, Structure and Bioactivity of 2CaO·3SiO2 Glass. J. Sol-Gel Sci. Technol. 1997, 10, 231–237. [Google Scholar] [CrossRef]
- Pirayesh, H.; Nychka, J.A. Sol–Gel Synthesis of Bioactive Glass-Ceramic 45S5 and its in vitro Dissolution and Mineralization Behavior. J. Am. Ceram. Soc. 2013, 96, 1643–1650. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F. Anti-inflammatory entrapment in polycaprolactone/silica hybrid material prepared by sol-gel route, characterization, bioactivity and in vitro release behavior. J. Appl. Biomater. Funct. Mater. 2013, 11, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, A.; Sockalingum, G.; Michel, J.; Fauré, J.; Banchet, V.; Wortham, L.; Bouthors, S.; Laurent-Maquin, D.; Balossier, G. Synthesis and characterisation of sol gel derived bioactive glass for biomedical applications. Mater. Lett. 2006, 60, 3752–3757. [Google Scholar] [CrossRef]
- Brinker, C.; Scherer, G. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing; Academic Press: San Diego, CA, USA, 1989. [Google Scholar]
- Gupta, R.; Kumar, A. Bioactive materials for biomedical applications using sol-gel technology. Biomed. Mater. 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.A.; Yue, S.; Hanna, J.V.; Lee, P.D.; Newport, R.J.; Smith, M.E.; Jones, J.R. Characterizing the hierarchical structures of bioactive sol-gel silicate glass and hybrid scaffolds for bone regeneration. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2012, 370, 1422–1443. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, P.; Jones, J.R.; Hench, L.L. Characterization of melt-derived 45S5 and sol-gel–derived 58S bioactive glasses. J. Biomed. Mater. Res. 2001, 58, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Picquart, M.; López, T.; Gómez, R.; Torres, E.; Moreno, A.; Garcia, J. Dehydration and crystallization process in sol-gel zirconia. J. Therm. Anal. Calorim. 2004, 76, 755–761. [Google Scholar] [CrossRef]
- Agrawal, K.; Singh, G.; Puri, D.; Prakash, S. Synthesis and Characterization of Hydroxyapatite Powder by Sol-Gel Method for Biomedical Application. J. Miner. Mater. Charact. Eng. 2011, 10, 727–734. [Google Scholar] [CrossRef]
- Mutlu, H.I.; Hascicek, Y.S. Insulation for Wind and React High Temperature Superconducting Coils. In Advances in Cryogenic Engineering Materials; Balachandran, U.B., Gubser, D.G., Hartwig, K.T., Reed, R., Warnes, W.H., Bardos, V.A., Eds.; Springer Science + Business Media, LLC: New York, NY, USA, 1998. [Google Scholar]
- Wachsman, E.D.; Henn, F.E.G.; Jiang, N.; Leezenberg, P.B.; Buchanan, R.M.; Frank, C.W.; Stevenson, D.A.; Wenckus, J.F. Luminescence of Anion Vacancies and Dopant-Vacancy Associated in Stabilized Zirconia. In Science and Technology of Zirconia V; Badwal, S.P.S., Bannister, M.J., Hannink, R.H.J., Eds.; Technomic Publishing Company: Lancaster, PA, USA, 1993; pp. 584–592. [Google Scholar]
- Catauro, M.; Bollino, F.; Papale, F.; Mozzati, M.C.; Ferrara, C.; Mustarelli, P. ZrO2/PEG hybrid nanocomposites synthesized via sol-gel: Characterization and evaluation of the magnetic properties. J. Non-Cryst. Solids 2015, 413, 1–7. [Google Scholar] [CrossRef]
- Santos, V.; Zeni, M.; Bergmann, C.; Hohemberger, J. Correlation between thermal treatment and tetragonal/monoclinic nanostructured zirconia powder obtained by sol–gel process. Rev. Adv. Mater. Sci. 2008, 17, 62–70. [Google Scholar]
- Petkova, N.; Dlugocz, S.; Gutzov, S. Preparation and optical properties of transparent zirconia sol–gel materials. J. Non-Cryst. Solids 2011, 357, 1547–1551. [Google Scholar] [CrossRef]
- Georgieva, I.; Danchova, N.; Gutzov, S.; Trendafilova, N. DFT modeling, UV-Vis and IR spectroscopic study of acetylacetone-modified zirconia sol-gel materials. J. Mol. Model 2012, 18, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Elvira, M.R.; Mazo, M.A.; Tamayo, A.; Rubio, F.; Rubio, J.; Oteo, J.L. Study and characterization of organically modified silica-zirconia anti-Graffiti coatings obtained by sol-gel. J. Chem. Chem. Eng. 2013, 7, 120–131. [Google Scholar]
- Hao, Y.; Li, J.; Yang, X.; Wang, X.; Lu, L. Preparation of ZrO2–Al2O3 composite membranes by sol–gel process and their characterization. Mater. Sci. Eng. A 2004, 367, 243–247. [Google Scholar] [CrossRef]
- Irish, D.E.; Walrafen, G.E. Raman and infrared spectral studies of aqueous calcium nitrate solutions. J. Chem. Phys. 1967, 46, 378–384. [Google Scholar] [CrossRef]
- Brauer, D.S. Phosphate glass. In Bio-Glasses: An Introduction; Jones, J.R., Clare, A.G., Eds.; John Wiley & Sons, Ltd.: Oxford, UK, 2012; pp. 46–49. [Google Scholar]
- De Oliveira, A.A.; de Souza, D.A.; Dias, L.L.; de Carvalho, S.M.; Mansur, H.S.; de Magalhães Pereira, M. Synthesis, characterization and cytocompatibility of spherical bioactive glass nanoparticles for potential hard tissue engineering applications. Biomed. Mater. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Bollino, F.; Renella, R.A.; Papale, F. Sol-gel synthesis of SiO2-CaO-P2O5 glasses: Influence of the heat treatment on their bioactivity and biocompatibility. Ceram. Int. 2015, 41, 12578–12588. [Google Scholar] [CrossRef]
- Brangule, A.; Gross, K.A. Importance of FTIR Spectra Deconvolution for the Analysis of Amorphous Calcium Phosphates; IOP Conference Series: Materials Science and Engineering; IOP Publishing Ltd.: Bristol, UK, 2015. [Google Scholar]
- Kurajica, S.; Lozić, I.; Pantaler, M. Thermal decomposition of calcium(II) bis(acetylacetonate) n-hydrate. Polimeri 2015, 35, 4–9. [Google Scholar]
- Li, P.; Ohtsuki, C.; Kokubo, T.; Nakanishi, K.; Soga, N.; Nakamura, T.; Yamamuro, T. Process of formation of bone-like apatite layer on silica gel. J. Mater. Sci. Mater. Med. 1993, 4, 127–131. [Google Scholar] [CrossRef]
- Ohtsuki, C.; Kokubo, T.; Yamamuro, T. Mechanism of apatite formation on CaOSiO2P2O5 glasses in a simulated body fluid. J. Non-Cryst. Solids 1992, 143, 84–92. [Google Scholar] [CrossRef]
- Beganskienė, A.; Dudko, O.; Sirutkaitis, R.; Giraitis, R. Water Based Sol-Gel Synthesis of Hydroxyapatite. Mater. Sci. 2003, 9, 383–386. [Google Scholar]
- Vecchio Ciprioti, S.; Bollino, F.; Tranquillo, E.; Catauro, M. Synthesis, thermal behavior and physicochemical characterization of ZrO/PEG inorganic/organic hybrid materials via sol–gel technique. J. Therm. Anal. Calorim. 2017, 1–6. [Google Scholar] [CrossRef]
- Jayakumar, S.; Ananthapadmanabhan, P.V.; Perumal, K.; Thiyagarajan, T.K.; Mishra, S.C.; Su, L.T.; Tok, A.I.Y.; Guo, J. Characterization of nano-crystalline ZrO2 synthesized via reactive plasma processing. Mater. Sci. Eng. B 2011, 176, 894–899. [Google Scholar] [CrossRef]
- Chen, S.; Yin, Y.; Wang, D.; Liu, Y.; Wang, X. Structures, growth modes and spectroscopic properties of small zirconia clusters. J. Cryst. Growth 2005, 282, 498–505. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of Calcium Phosphates Using Fourier Transform Infrared Spectroscopy. In Infrared Spectroscopy—Materials Science, Engineering and Technology; Theophile, T., Ed.; InTech: Rijeka, Croatia, 2012; pp. 123–148. [Google Scholar]
- Gozalian, A.; Behnamghader, A.; Daliri, M.; Moshkforoush, A. Synthesis and thermal behavior of Mg-doped calcium phosphate nanopowders via the sol gel method. Sci. Iran. 2011, 18, 1614–1622. [Google Scholar] [CrossRef]
- Meejoo, S.; Maneeprakorn, W.; Winotai, P. Phase and thermal stability of nanocrystalline hydroxyapatite prepared via microwave heating. Thermochim. Acta 2006, 447, 115–120. [Google Scholar] [CrossRef]
- Fleet, M.E.; Liu, X.; King, P.L. Accommodation of the carbonate ion in apatite: An FTIR and X-ray structure study of crystals synthesized at 2–4 GPa. Am. Mineral. 2004, 89, 1422–1432. [Google Scholar] [CrossRef]
- Eichert, D.; Drouet, C.; Sfihi, H.; Rey, C.; Combes, C. Nanocrystalline apatite-based biomaterials: Synthesis, processing and characterization. In Biomaterials Research Advances; Kendall, J.B., Ed.; Nova Science Publishers: New York, NY, USA, 2007; pp. 93–143. [Google Scholar]
- Barralet, J.; Knowles, J.C.; Best, S.; Bonfield, W. Thermal decomposition of synthesised carbonate hydroxyapatite. J. Mater. Sci. Mater. Med. 2002, 13, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Latifi, S.M.; Fathi, M.; Varshosaz, J.; Ghochaghi, N. Mechanisms controlling ca ion release from sol-gel derived in situ apatite-silica nanocomposite powder. Ceram. Silik. 2015, 59, 64–69. [Google Scholar]
- Mansour, S.F.; El-dek, S.I.; Ahmed, M.K. Physico-mechanical and morphological features of zirconia substituted hydroxyapatite nano crystals. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Lemaitre, J.; Rouxhet, P.G. Temperature-programmed characterization of synthetic calcium-deficient phosphate apatites. Thermochim. Acta 1989, 143, 265–282. [Google Scholar] [CrossRef]
- Kalinkin, A.M.; Kalinkina, E.V.; Zalkind, O.A.; Makarova, T.I. Chemical interaction of calcium oxide and calcium hydroxide with CO2 during mechanical activation. Inorg. Mater. 2005, 41, 1073–1079. [Google Scholar] [CrossRef]
- Popescu, R.A.; Magyari, K.; Vulpoi, A.; Trandafir, D.L.; Licarete, E.; Todea, M.; Stefan, R.; Voica, C.; Vodnar, D.C.; Simon, S.; et al. Bioactive and biocompatible copper containing glass-ceramics with remarkable antibacterial properties and high cell viability designed for future in vivo trials. Biomater. Sci. 2016, 4, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, E.; Costa, A.M.; Costa, L.T.; Achete, C.A.; Mello, A.; Granjeiro, J.M.; Rossi, A.M. Adsorption and bioactivity studies of albumin onto hydroxyapatite surface. Coll. Surf. B Biointerfaces 2011, 83, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Münstedt, H. Polyamide/silver antimicrobials: Effect of crystallinity on the silver ion release. Polym. Int. 2005, 54, 1180–1186. [Google Scholar] [CrossRef]
- Ogose, A.; Hotta, T.; Kawashima, H.; Kondo, N.; Gu, W.; Kamura, T.; Endo, N. Comparison of hydroxyapatite and beta tricalcium phosphate as bone substitutes after excision of bone tumors. J. Biomed. Mater. Res. Part B Appl. Biomater. 2005, 72B, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Jalota, S.; Bhaduri, S.B.; Tas, A.C. In vitro testing of calcium phosphate (HA, TCP, and biphasic HA-TCP) whiskers. J. Biomed. Mater. Res. Part A 2006, 78A, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Papale, F.; Sapio, L.; Naviglio, S. Biological influence of Ca/P ratio on calcium phosphate coatings by sol-gel processing. Mater. Sci. Eng. C 2016, 65, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Radev, L. Influence of thermal treatment on the structure and in vitro bioactivity of sol-gel prepared CaO-SiO2-P2O5 glass-ceramics. Process. Appl. Ceram. 2014, 8, 155–166. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bollino, F.; Armenia, E.; Tranquillo, E. Zirconia/Hydroxyapatite Composites Synthesized Via Sol-Gel: Influence of Hydroxyapatite Content and Heating on Their Biological Properties. Materials 2017, 10, 757. https://doi.org/10.3390/ma10070757
Bollino F, Armenia E, Tranquillo E. Zirconia/Hydroxyapatite Composites Synthesized Via Sol-Gel: Influence of Hydroxyapatite Content and Heating on Their Biological Properties. Materials. 2017; 10(7):757. https://doi.org/10.3390/ma10070757
Chicago/Turabian StyleBollino, Flavia, Emilia Armenia, and Elisabetta Tranquillo. 2017. "Zirconia/Hydroxyapatite Composites Synthesized Via Sol-Gel: Influence of Hydroxyapatite Content and Heating on Their Biological Properties" Materials 10, no. 7: 757. https://doi.org/10.3390/ma10070757
APA StyleBollino, F., Armenia, E., & Tranquillo, E. (2017). Zirconia/Hydroxyapatite Composites Synthesized Via Sol-Gel: Influence of Hydroxyapatite Content and Heating on Their Biological Properties. Materials, 10(7), 757. https://doi.org/10.3390/ma10070757