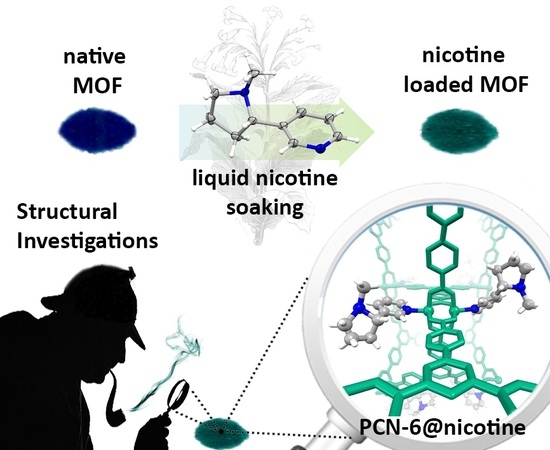
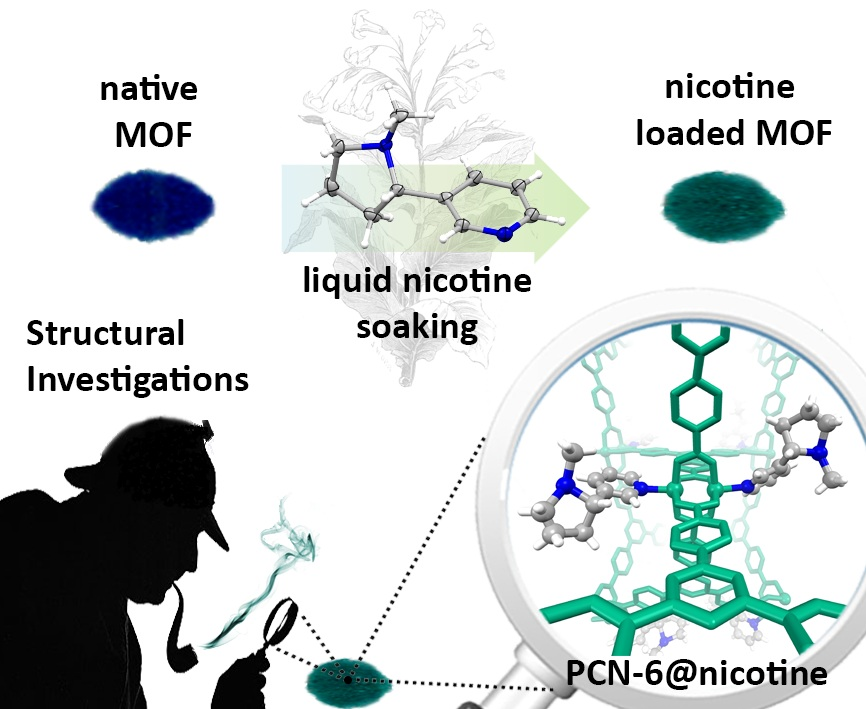
Coordination Driven Capture of Nicotine Inside a Mesoporous MOF


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
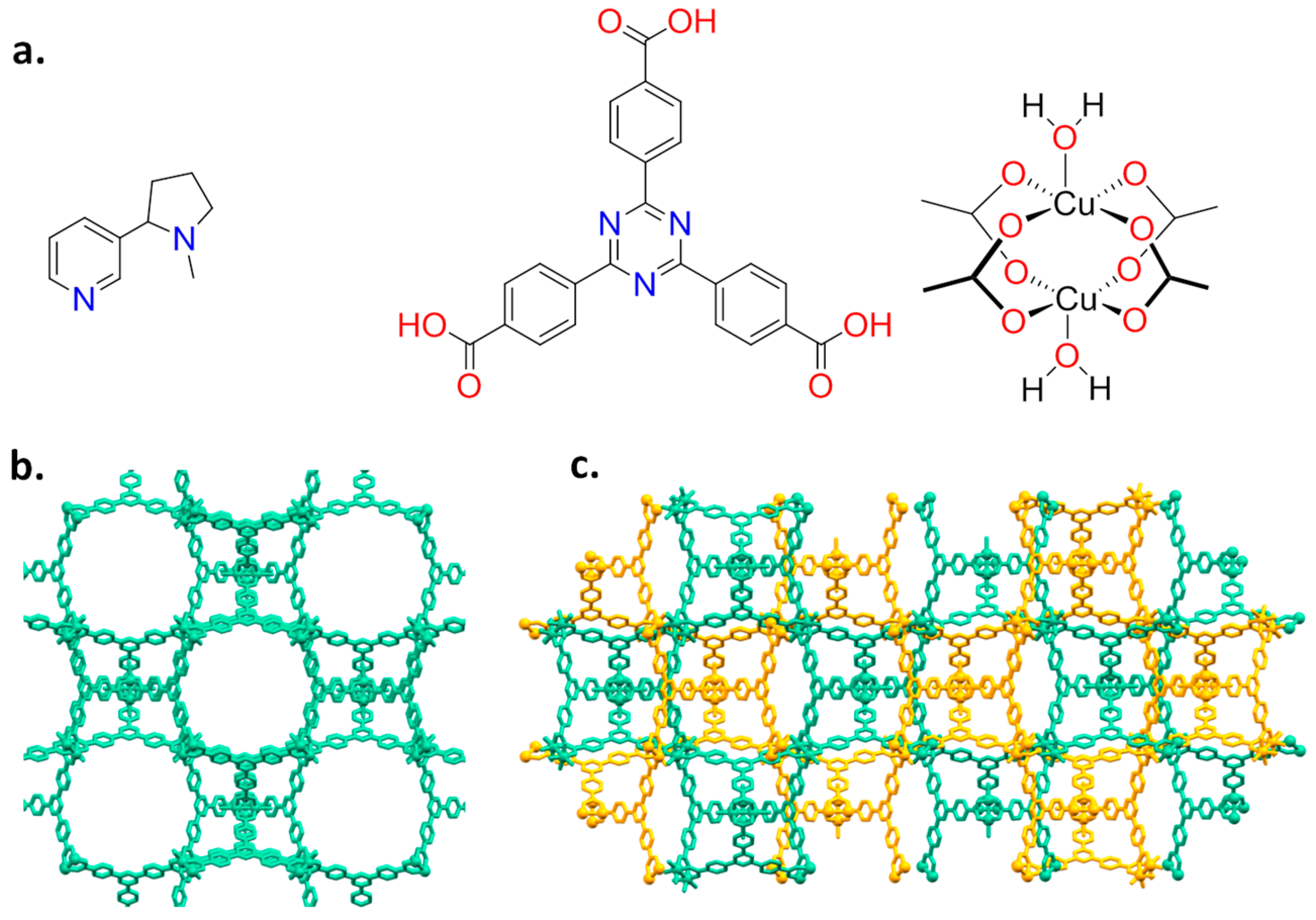
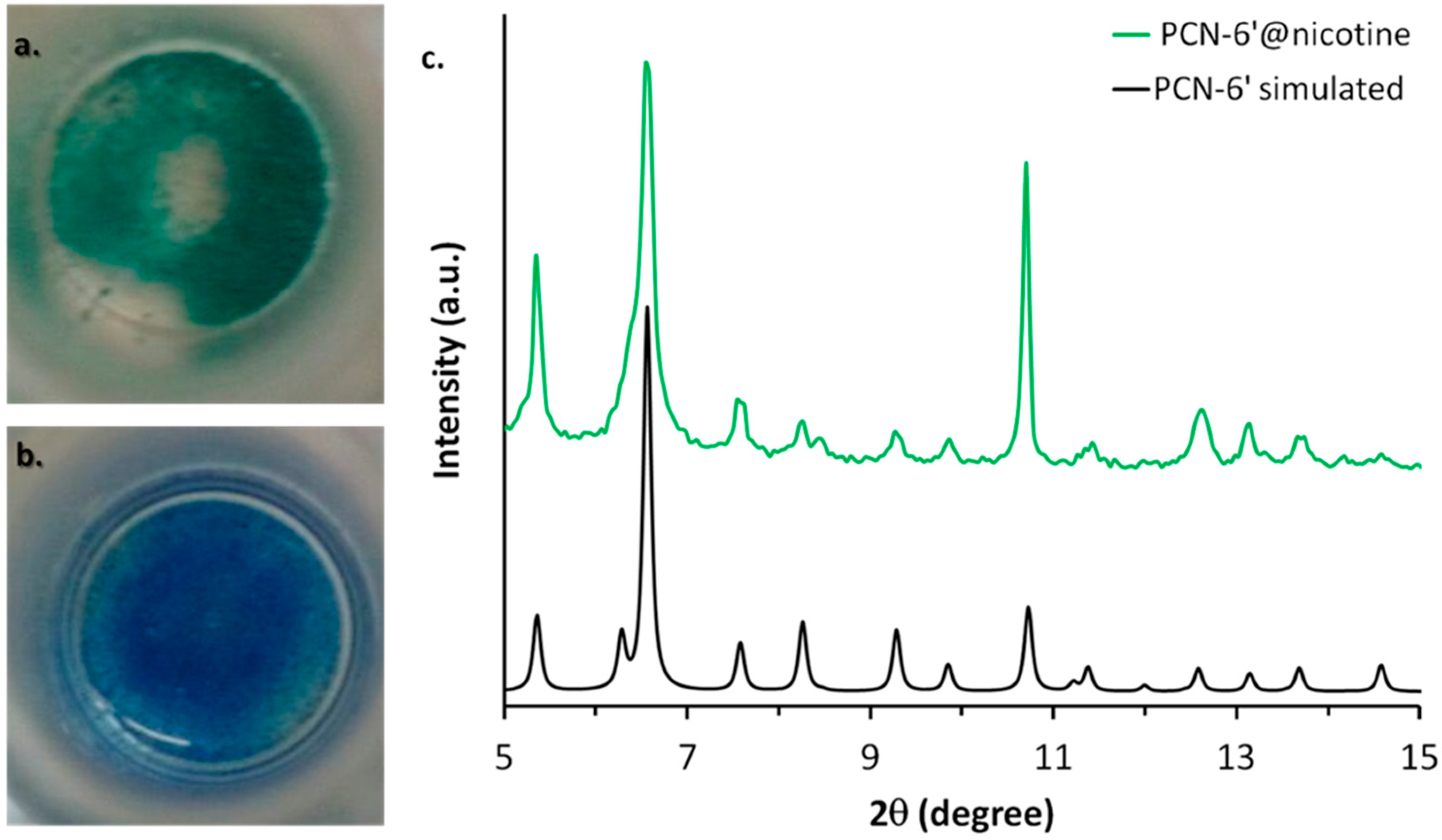
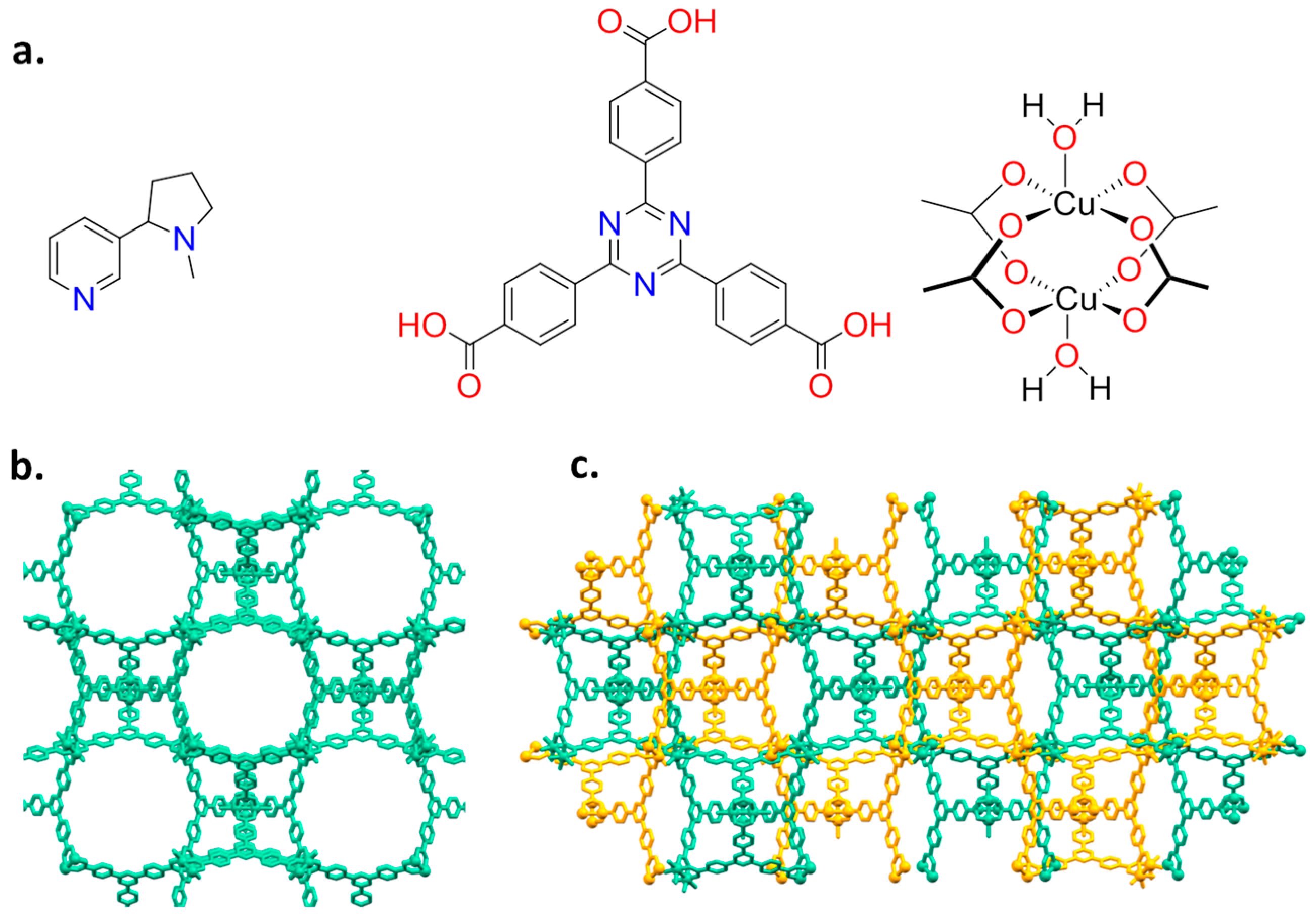
2.1. Synthesis and Loading
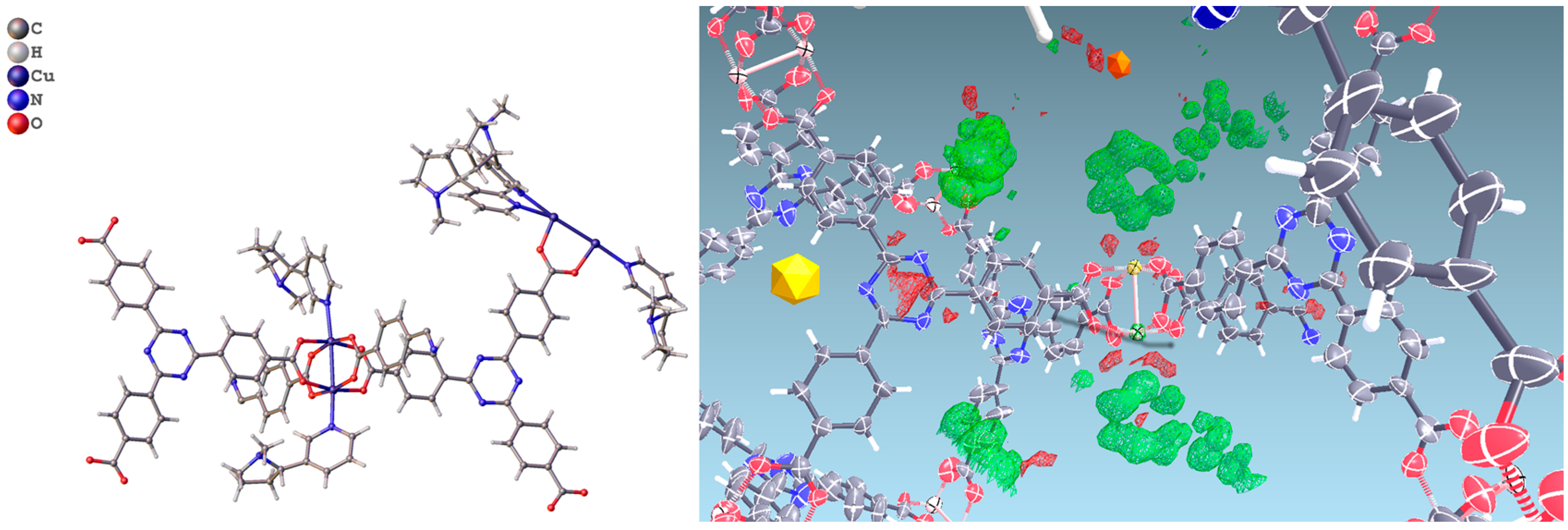
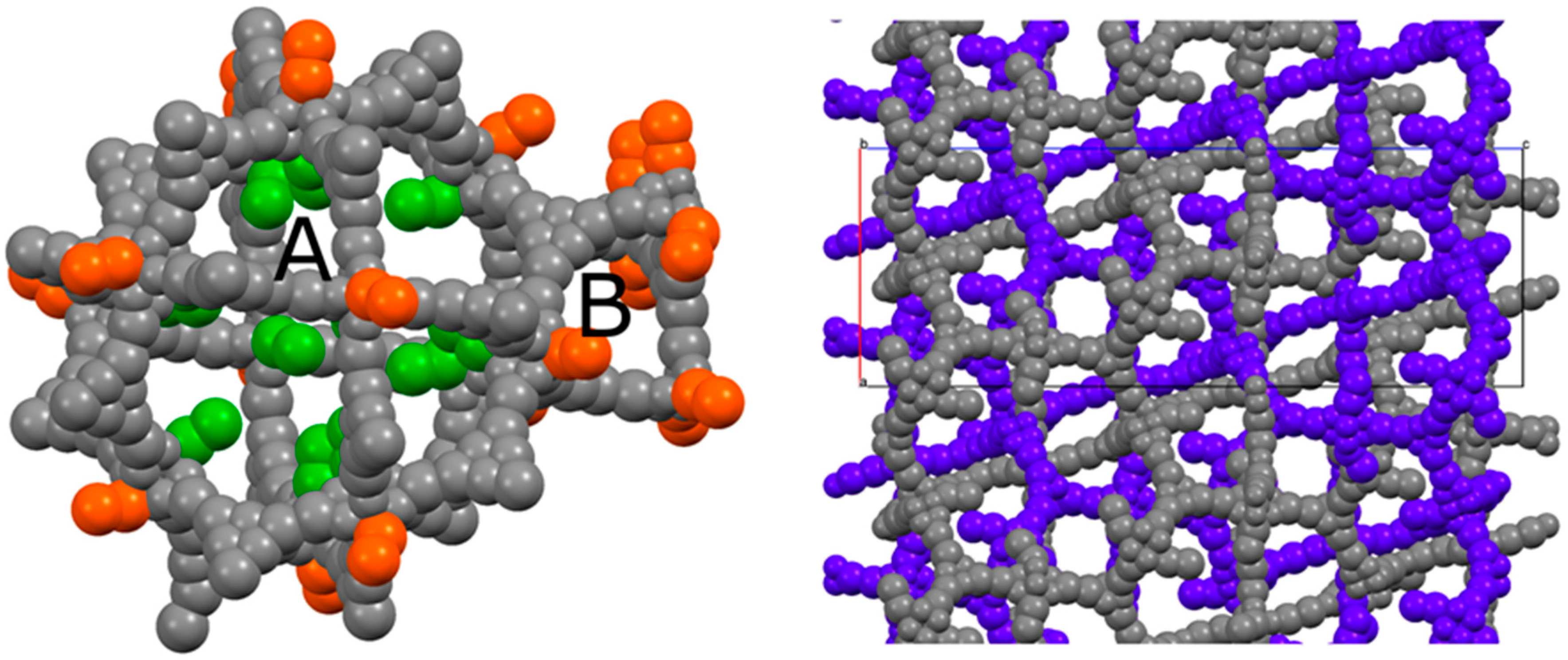
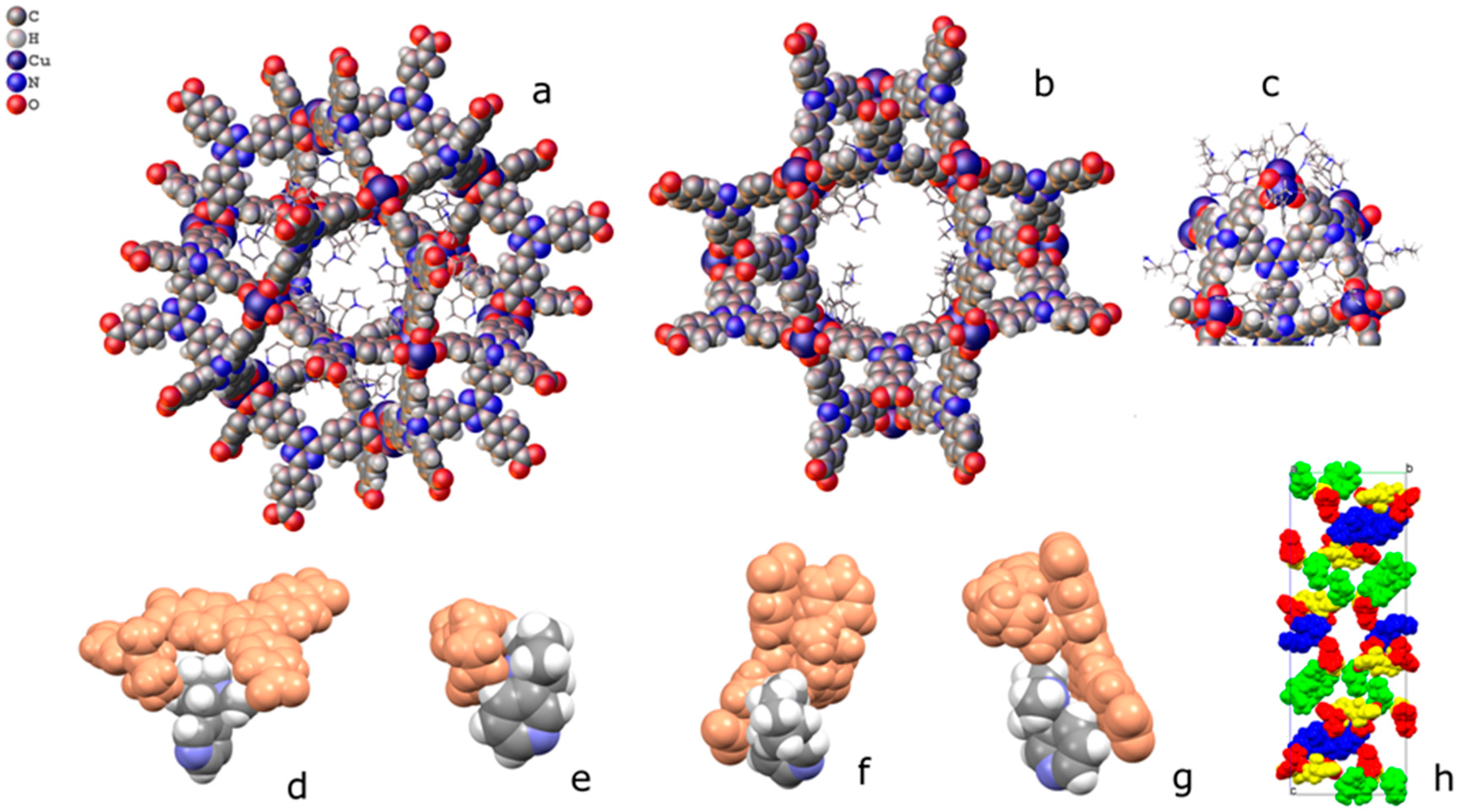
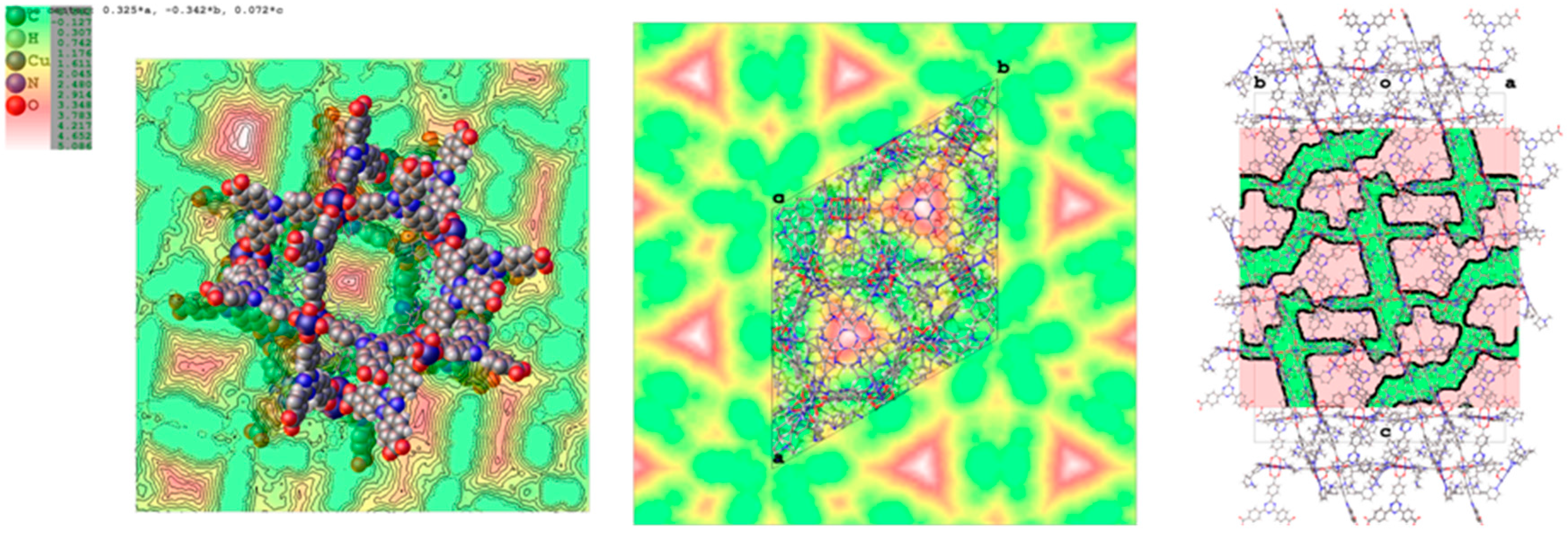
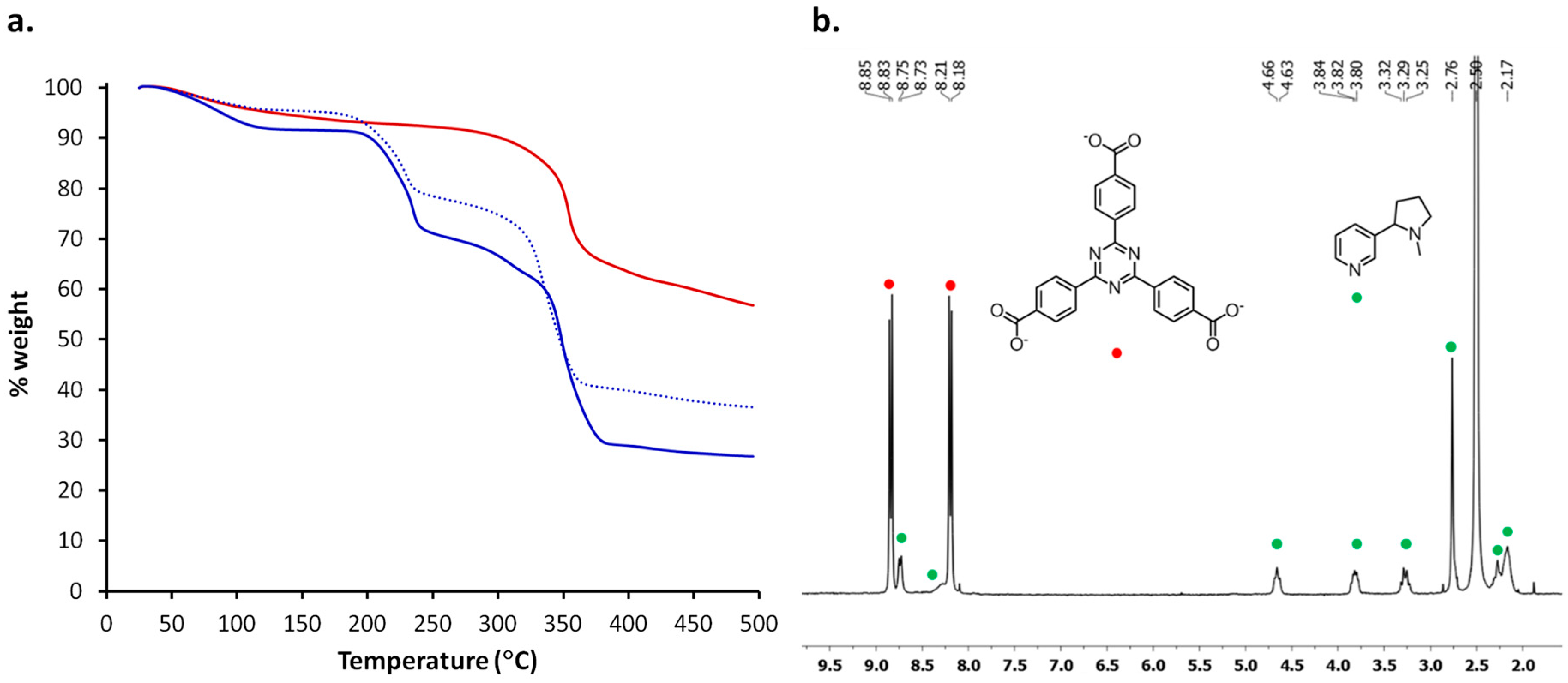
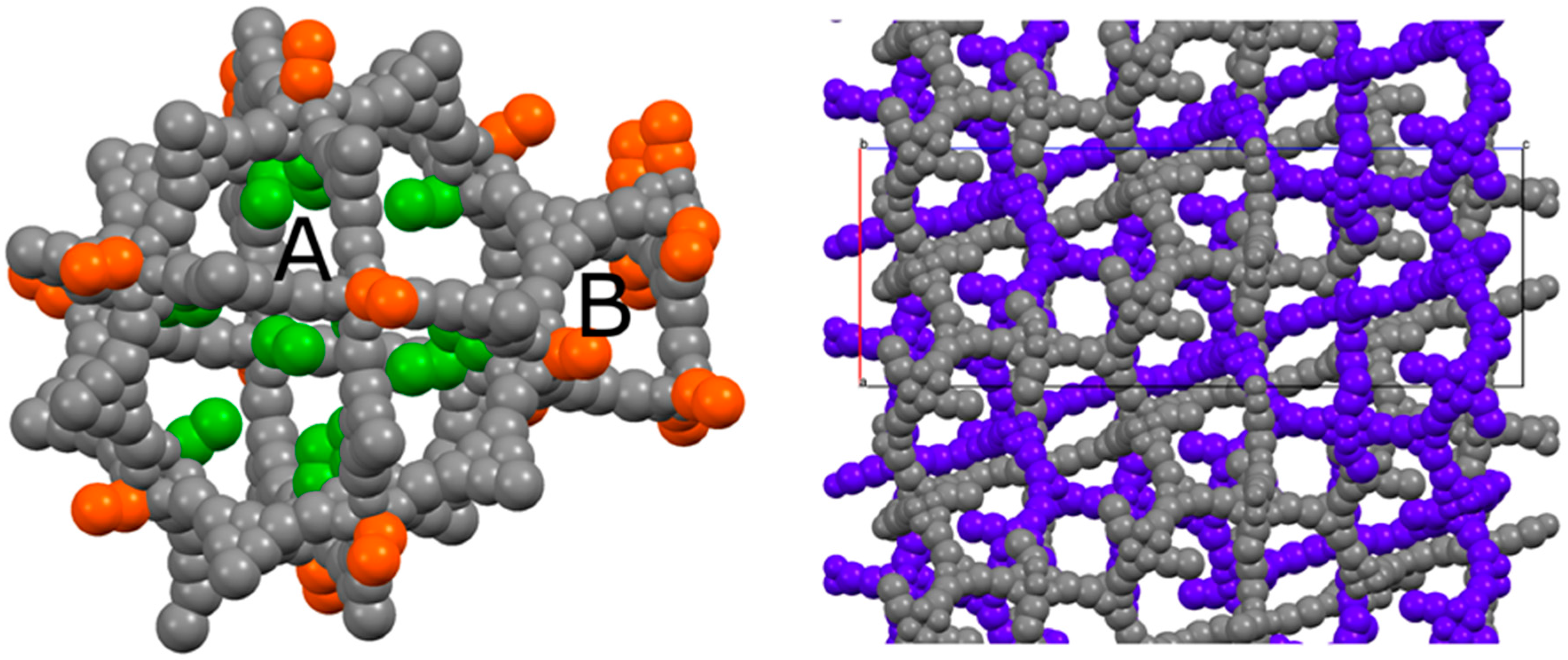
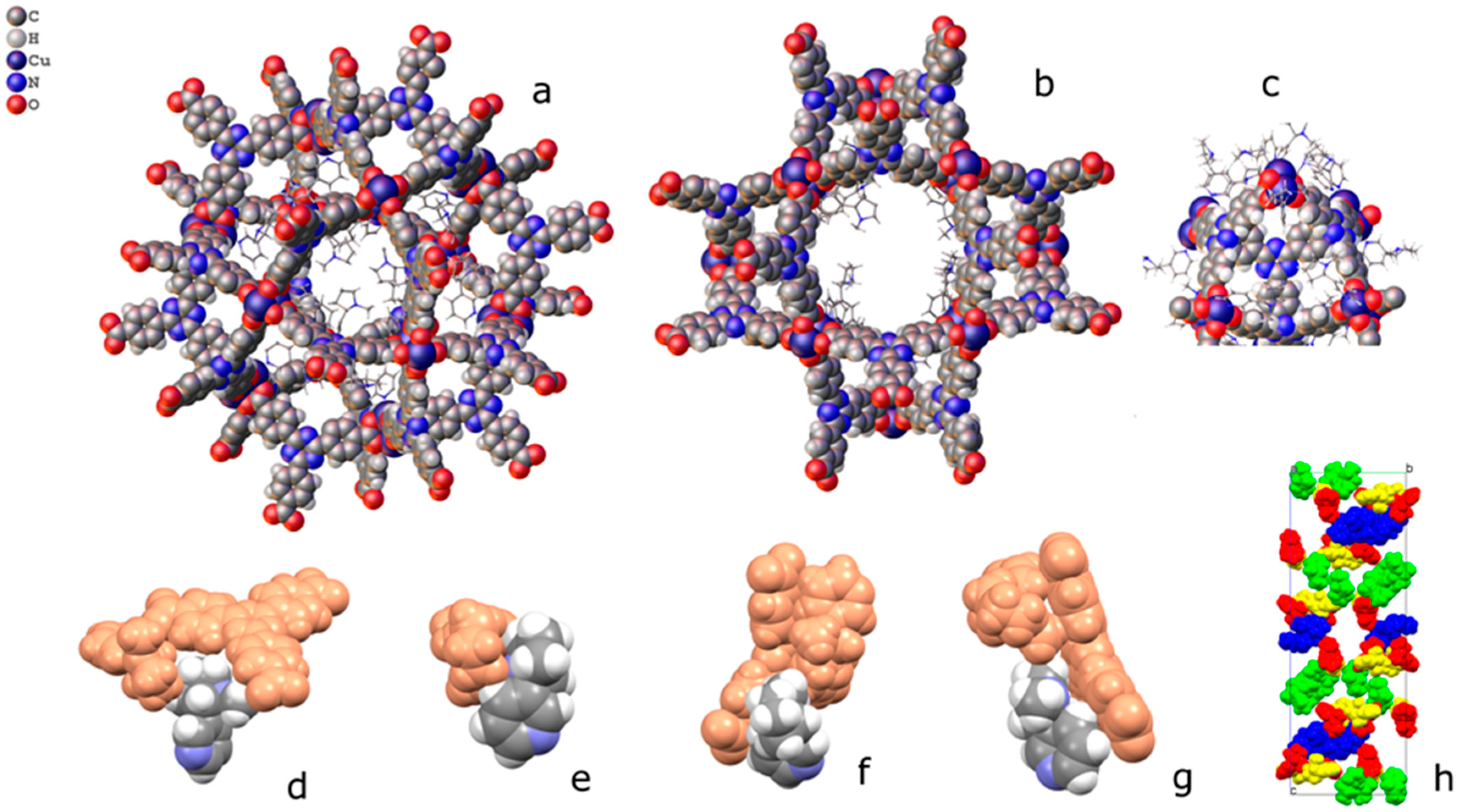
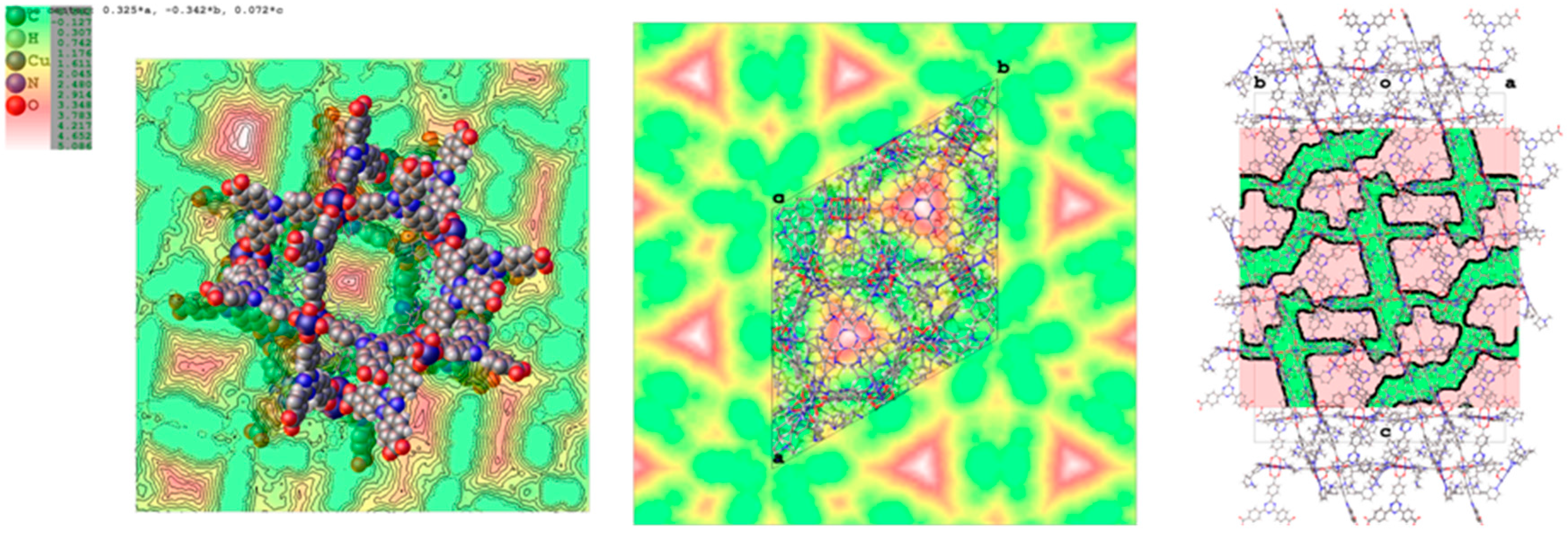
2.2. Structural Characterization of PCN-6@nicotine
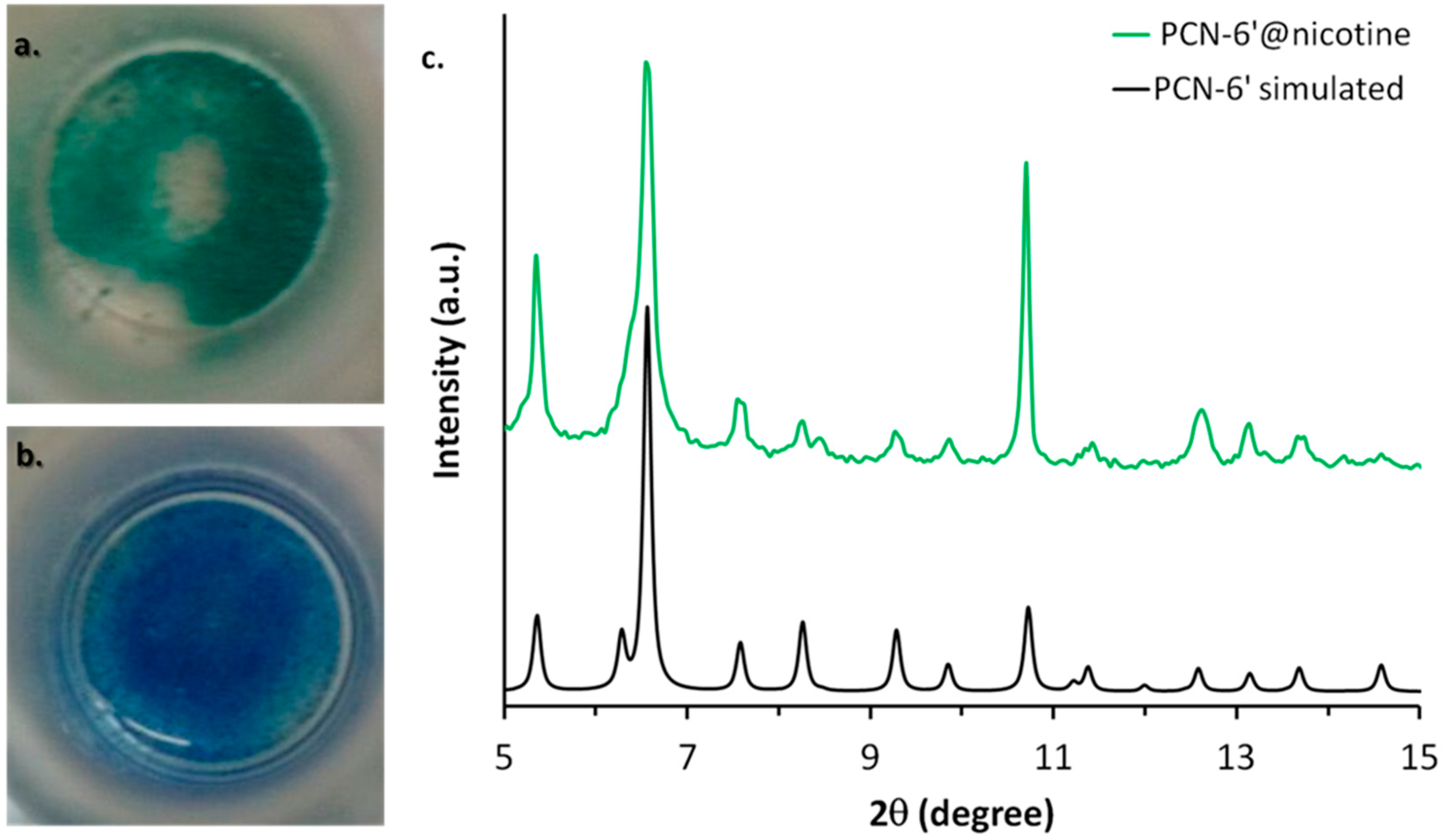
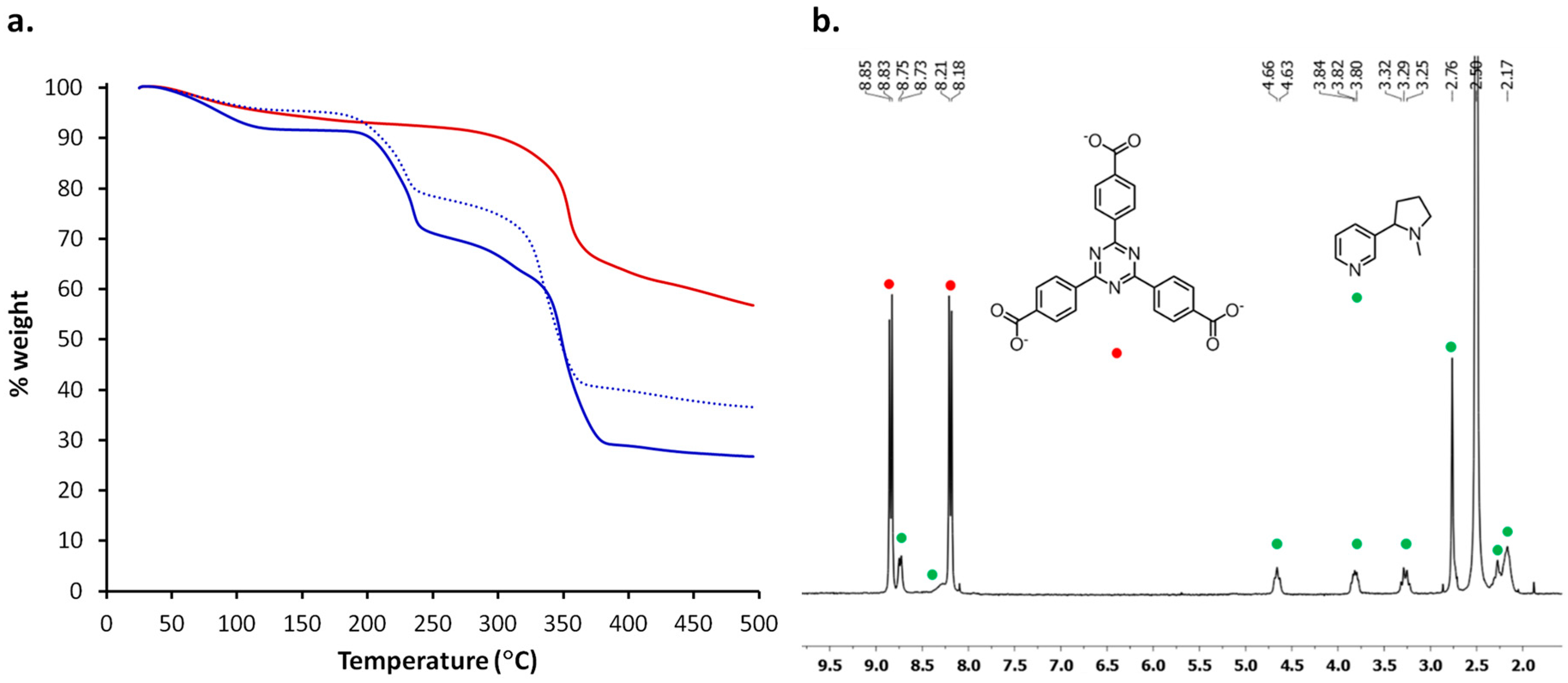
2.3. Characterization of Nicotine-Containing Material
3. Materials and Methods
3.1. ATB-4,4′,4″-(1,3,5-Triazine-2,4,6-Triyl)Tribenzoic Acid
3.2. Recrystallization Procedure
3.3. Synthesis of PCN-6′
3.4. Activation and Soaking with Neat Nicotine
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegmund, B.; Leitner, E.; Pfannhauser, W. Determination of the Nicotine Content of Various Edible Nightshades (Solanaceae) and Their Products and Estimation of the Associated Dietary Nicotine Intake. J. Agric. Food Chem. 1999, 47, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Casanova, H.; Araque, P.; Ortiz, C. Nicotine Carboxylate Insecticide Emulsions: Effect of the Fatty Acid Chain Length. J. Agric. Food Chem. 2005, 53, 9949–9953. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Casid, J.E. Neonicotinoid Insecticide Toxicology: Mechanisms of Selective Action. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.J.; Denk, L.D.; Wax, P.M. Catastrophic brain injury after nicotine insecticide ingestion. J. Emerg. Med. 2004, 26, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Majumder, P.; Gupta, T. Pharmacological Intervention of Nicotine Dependence. BioMed Res. Int. 2013, 278392. [Google Scholar] [CrossRef] [PubMed]
- Powledge, T.M. Nicotine as Therapy. PLoS Biol. 2004, 2, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 495, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Kaskel, S. The Chemistry of Metal-Organic-Frameworks, 1st ed.; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal–organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mat. 2010, 9, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Serre, C.; Vallet-Reg, M.; Sebban, M.; Taulelle, F.; Férey, G. Metal–Organic Frameworks as Efficient Materials for Drug Delivery. Angew. Chem. Int. Ed. 2006, 45, 5974–5978. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.; Ben Yahia, M.; Hall, S.; Miller, S.R.; Chevreau, H.; Elkaïm, E.; Maurin, G.; Horcajada, P.; Serre, C. Rationale of Drug Encapsulation and Release from Biocompatible Porous Metal–Organic Frameworks. Chem. Mater. 2013, 25, 2767–2776. [Google Scholar] [CrossRef]
- Liedana, N.; Galve, A.; Rubio, C.; Téllez, C.; Coronas, J. CAF@ZIF-8: One-Step Encapsulation of Caffeine in MOF. ACS Appl. Mater. Interfaces 2012, 4, 5016–5021. [Google Scholar] [CrossRef] [PubMed]
- Sanna, E.; Escudero Adán, E.C.; Bauzá, A.; Ballester, P.; Frontera, A.; Rotger, C.; Costa, A. A crystalline sponge based on dispersive forces for X-ray structure determination of included molecular guests. Chem. Sci. 2015, 6, 5466–5472. [Google Scholar] [CrossRef]
- Fukawa, K.; Harada, S.; Kasai, N.; Toda, M.; Mori, K.; Toda, F. The Crystal and Molecular Structure of 2:2 Complex of 1,1,6,6-Tetraphenyl-2,4-hexadiyne-1,6-diol (DD) with 1-Methyl-2-(3-pyridyl)pyrrolidine (Nicotine). Bull. Chem. Soc. Jpn. 1989, 62, 2714–2716. [Google Scholar] [CrossRef]
- Bacchi, A.; Balestri, D.; Capucci, D.; Mazzeo, M.; Pelagatti, P. Liquid nicotine tamed in solid forms by cocrystallization. Manuscript in preparation.
- Basiewicz, B.; Hoffmann, M.; Gasowska, A.; Jastrząb, R.; Malczewska-Jaskóła, K. Spectroscopic, Potentiometric and Quantum-Mechanical Studies of S-(-)-Nicotine Complexes with Cu(II) Ion. Acta Chim. Slov. 2014, 61, 137–141. Available online: http://acta-arhiv.chem-soc.si/61/Graph/acta-61(1)-GA.htm (accessed on 5 May 2017).
- Jana, S.; Cormack, P.A.G.; Kennedy, A.R.; Sherrington, D.C. Synthesis of main chain chiral methacrylate copolymers via chirality transfer from polymerizable chiral metal complexes. J. Mater. Chem. 2009, 19, 3427–3442. [Google Scholar] [CrossRef]
- Ma, S.; Sun, D.; Ambrogio, M.; Fillinger, J.A.; Parkin, S.; Zhou, H.C. Framework-Catenation Isomerism in Metal-Organic Frameworks and Its Impact in Hydrogen Uptake. J. Am. Chem. Soc. 2007, 129, 1858–1859. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kapustin, E.A.; Yaghi, O.M. Coordinative alignment of molecules in chiral metal-organic frameworks. Science 2016, 353, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Chui, S.S.Y.; Lo, S.M.F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Ma, S.; Ke, Y.; Collins, D.J.; Zhou, H.-C. An Interweaving MOF with High Hydrogen Uptake. J. Am. Chem. Soc. 2006, 128, 3896–3897. [Google Scholar] [CrossRef] [PubMed]
- Mondloch, J.E.; Katz, M.J.; Planas, N.; Semrouni, D.; Gagliardi, L.; Hupp, J.T.; Farha, O.K. Are Zr6-based MOFs water stable? Linker hydrolysis vs. capillary-force-driven channel collapse. Chem. Commun. 2014, 50, 8944–8946. [Google Scholar] [CrossRef] [PubMed]
- Burtch, N.C.; Jasuia, H.; Walton, K.S. Water Stability and Adsorption in Metal−Organic Frameworks. Chem. Rev. 2014, 114, 10575–10612. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Simis, K.; Lei, M.; Lu, A.T.; Sharma, K.C.; Hale, R.L.; Timmons, R.; Cassella, J. Nicotine Aerosol Generation from Thermally Reversible Zinc Halide Complexes Using the Staccato® System. Drug Dev. Ind. Pharm. 2008, 34, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Go, Y.B.; Ko, N.; Park, Y.K.; Romo, F.J.U.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Isoreticular Expansion of Metal–Organic Frameworks with Triangular and Square Building Units and the Lowest Calculated Density for Porous Crystals. Inorg. Chem. 2011, 50, 9147–9152. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Sicker, D. (Eds.) For a comparison with NMR spectra of free nicotine. In Classics in Spectroscopy; WILEY-CH, Verlag GmbH &Co, KGaA: Weinheim, Germany, 2009; pp. 3–24. ISBN 978-3-527-32516-0. [Google Scholar]
- Lausi, A.; Polentarutti, M.; Onesti, S.; Plaisier, J.R.; Busetto, E.; Bais, G.; Barba, L.; Cassetta, A.; Campi, G.; Lamba, D.; et al. Status of the crystallography beamline at Elettra. Eur. Phys. J. Plus 2015, 130, 1–8. [Google Scholar] [CrossRef]
- Oxford Diffraction, CrysAlis CCD, and CrysAlis RED; Oxford Diffraction Ltd.: Abingdon, UK, 2008.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Macromolecular phasing with SHELXE. Z. Crystallogr. 2002, 217, 644–650. [Google Scholar] [CrossRef]
- Rees, B.; Jenner, L.; Yusupov, M. Bulk-solvent correction in large macromolecular structures. Acta Cryst. 2005, D61, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.G.M.; Farwell, D.W.; Rose, S.J.; Smith, D.N. Vibrational spectra of copper(II) oxalate dehydrate, CuC2O4∙2H2O, and dipotassium bis-oxalato copper(II) tetrahydrate, K2Cu(C2O4)2∙4H2O. J. Mol. Struct. 1991, 249, 233–243. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balestri, D.; Capucci, D.; Demitri, N.; Bacchi, A.; Pelagatti, P. Coordination Driven Capture of Nicotine Inside a Mesoporous MOF. Materials 2017, 10, 727. https://doi.org/10.3390/ma10070727
Balestri D, Capucci D, Demitri N, Bacchi A, Pelagatti P. Coordination Driven Capture of Nicotine Inside a Mesoporous MOF. Materials. 2017; 10(7):727. https://doi.org/10.3390/ma10070727
Chicago/Turabian StyleBalestri, Davide, Davide Capucci, Nicola Demitri, Alessia Bacchi, and Paolo Pelagatti. 2017. "Coordination Driven Capture of Nicotine Inside a Mesoporous MOF" Materials 10, no. 7: 727. https://doi.org/10.3390/ma10070727
APA StyleBalestri, D., Capucci, D., Demitri, N., Bacchi, A., & Pelagatti, P. (2017). Coordination Driven Capture of Nicotine Inside a Mesoporous MOF. Materials, 10(7), 727. https://doi.org/10.3390/ma10070727