Probing Transition-Metal Silicides as PGM-Free Catalysts for Hydrogen Oxidation and Evolution in Acidic Medium


Abstract
:1. Introduction
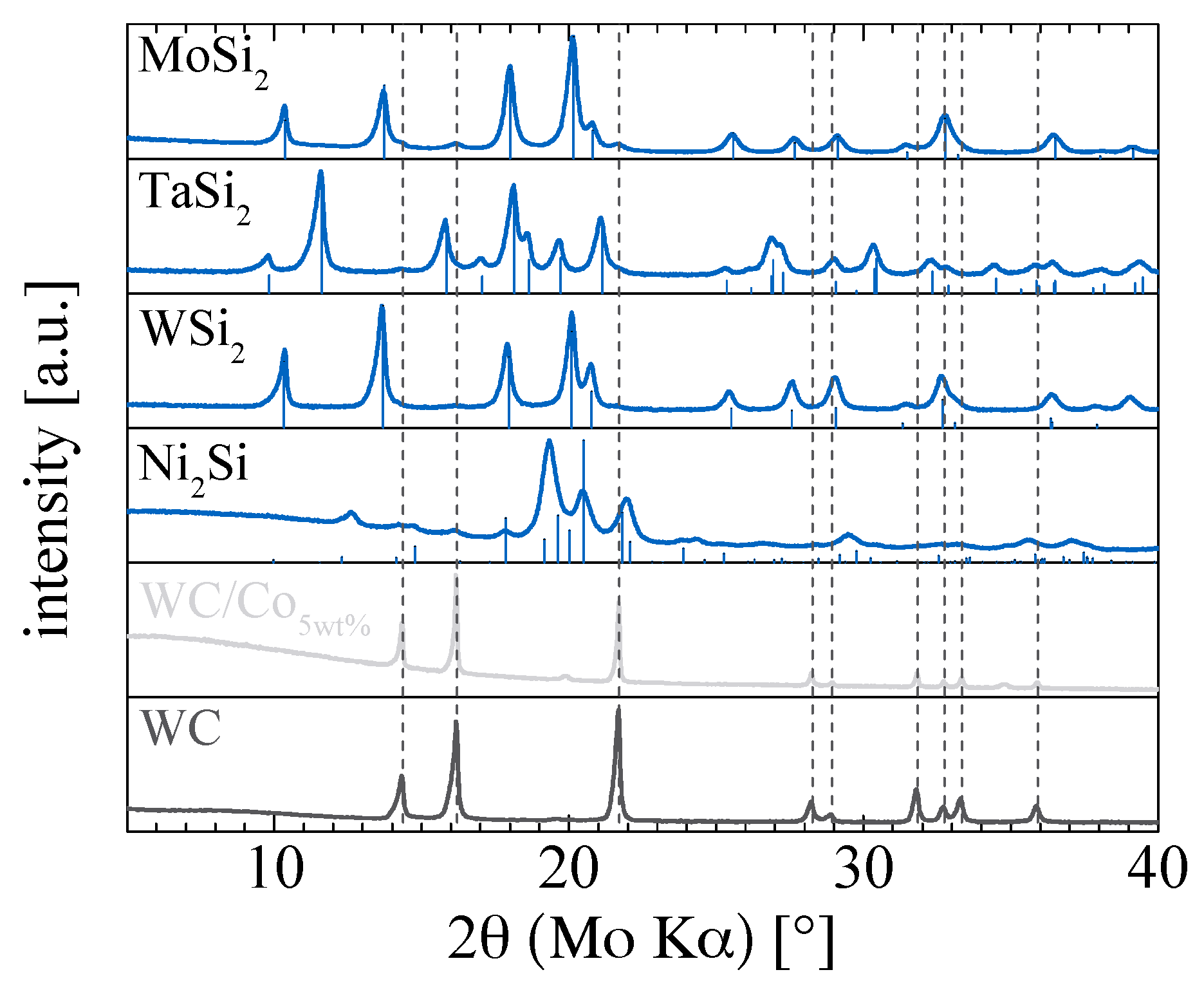
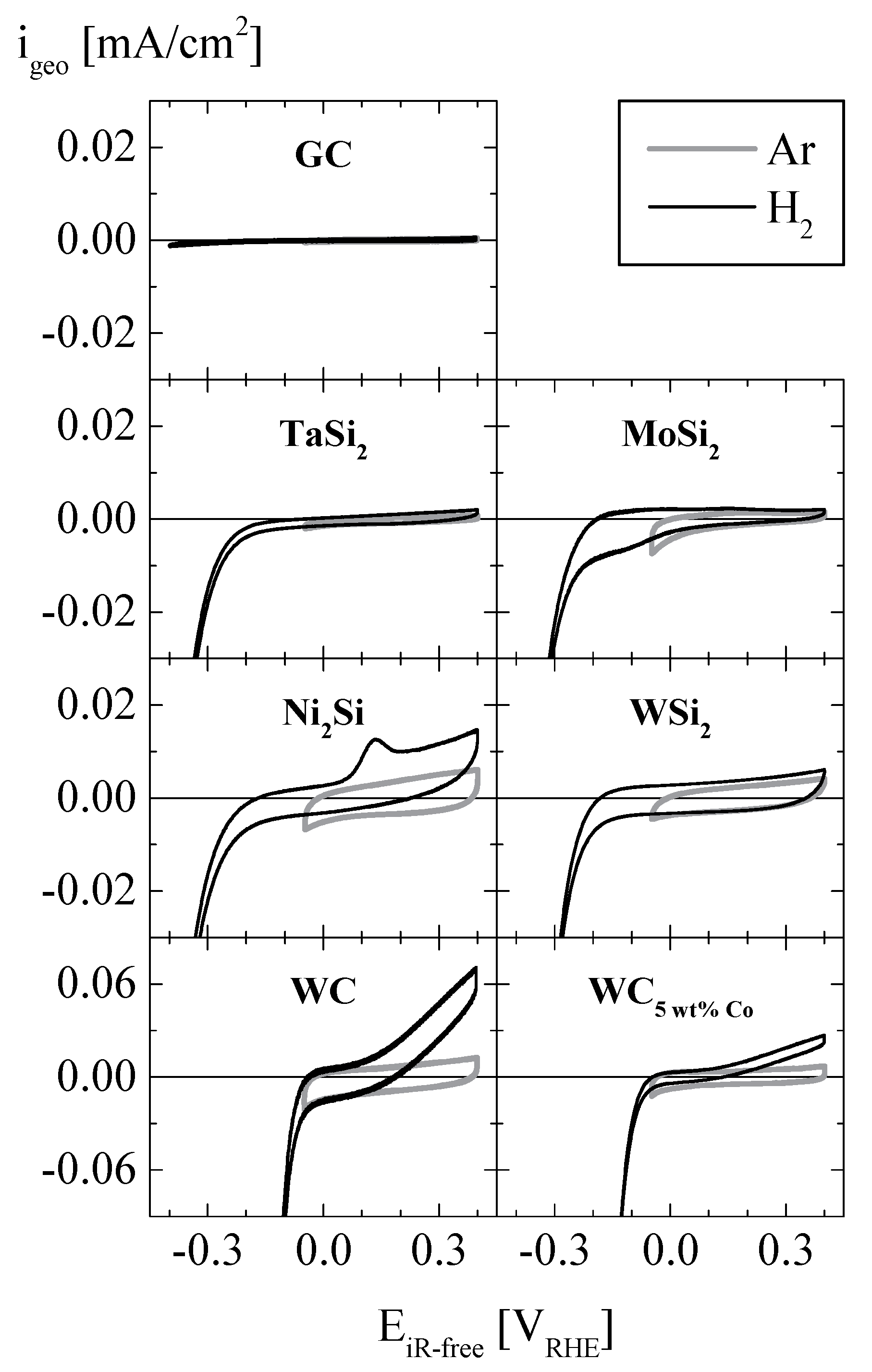
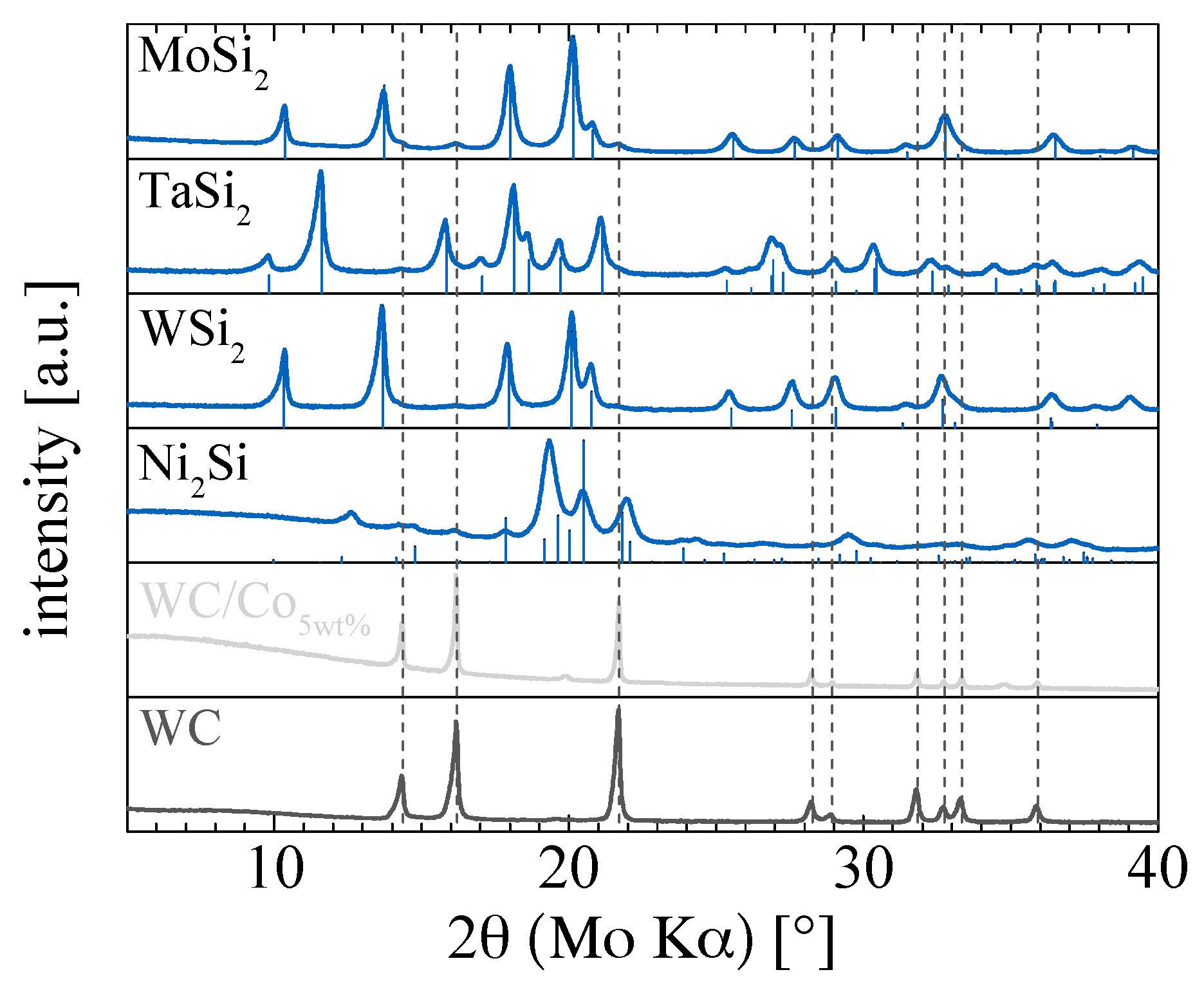
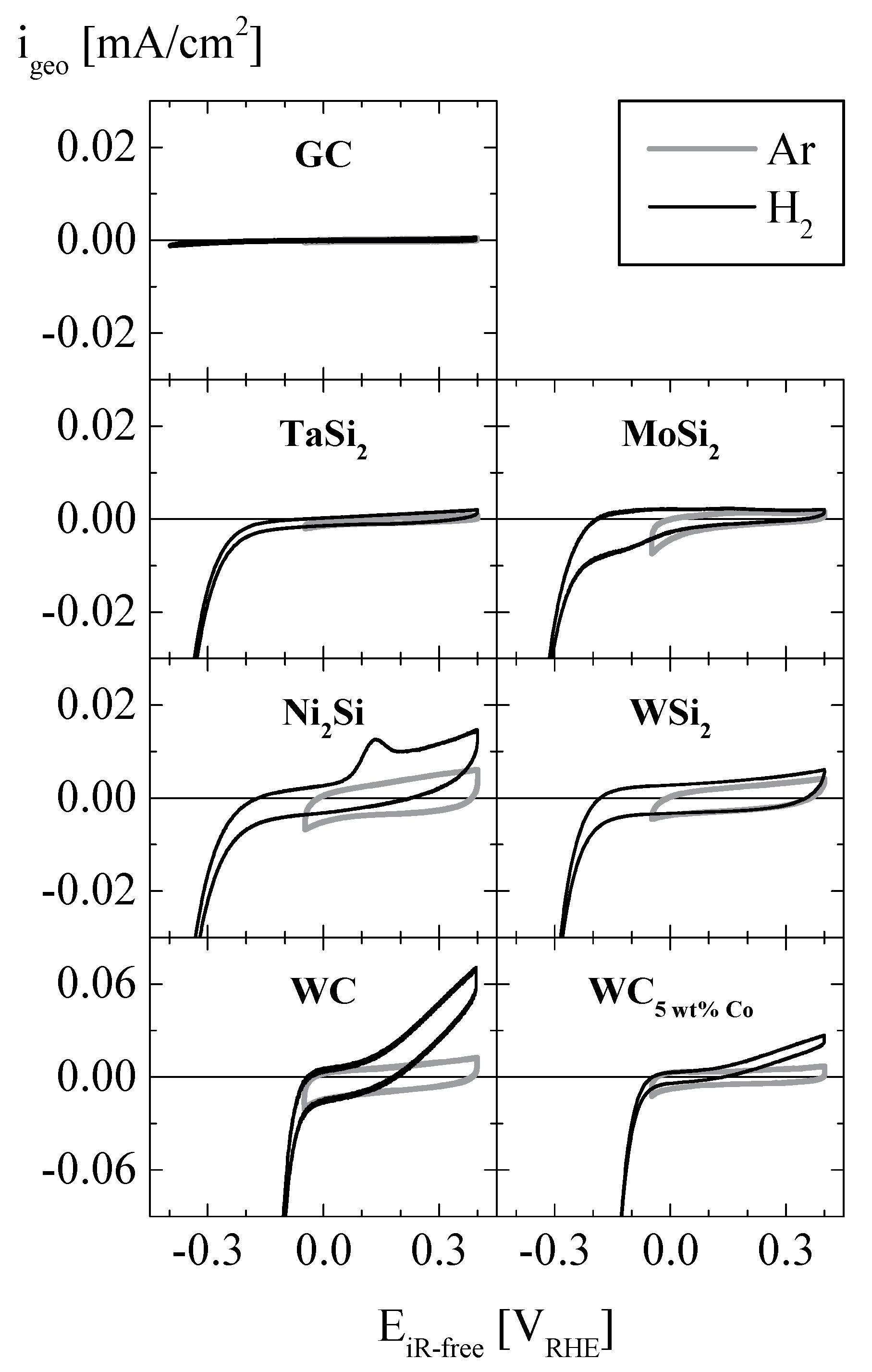
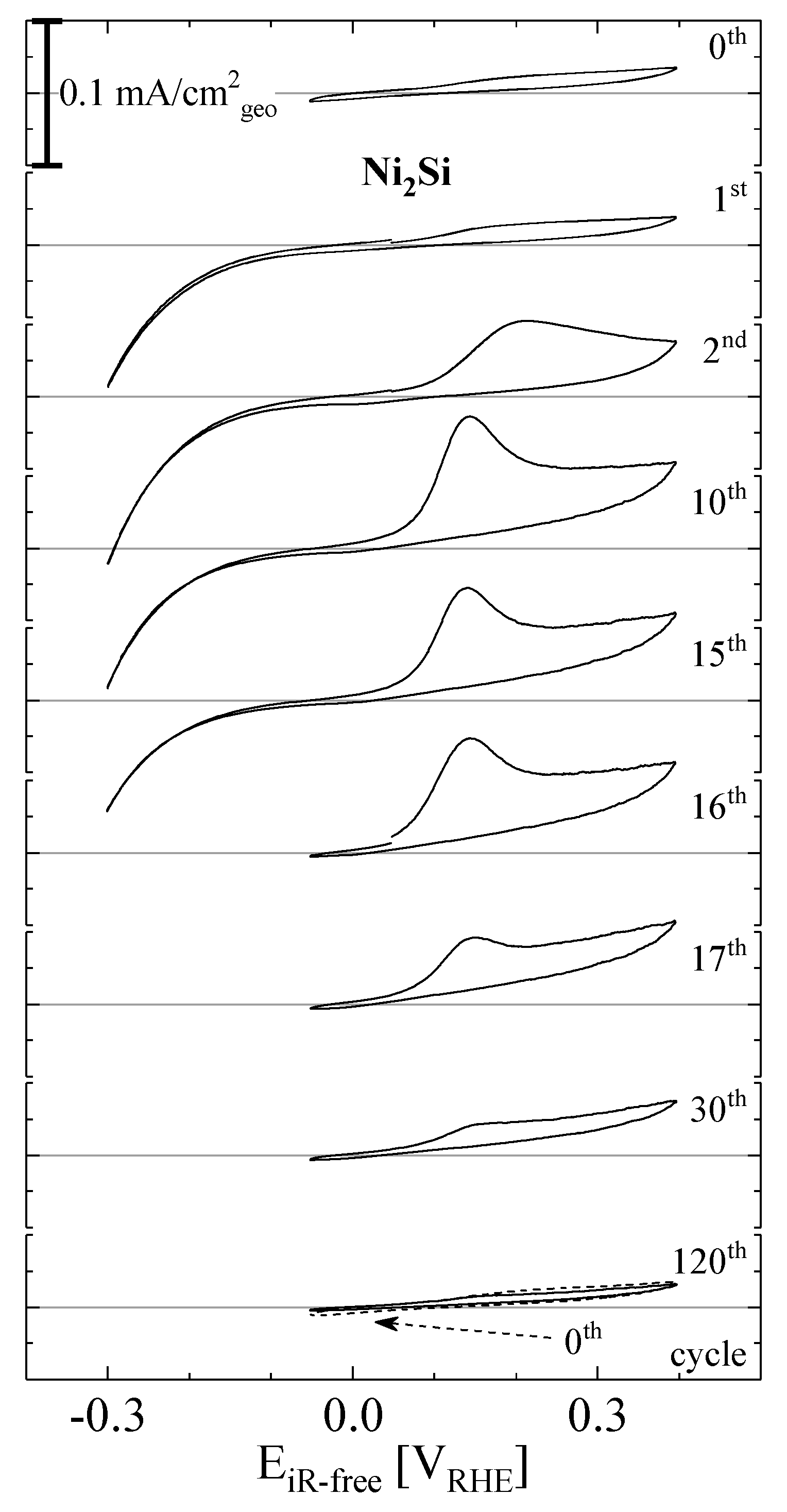
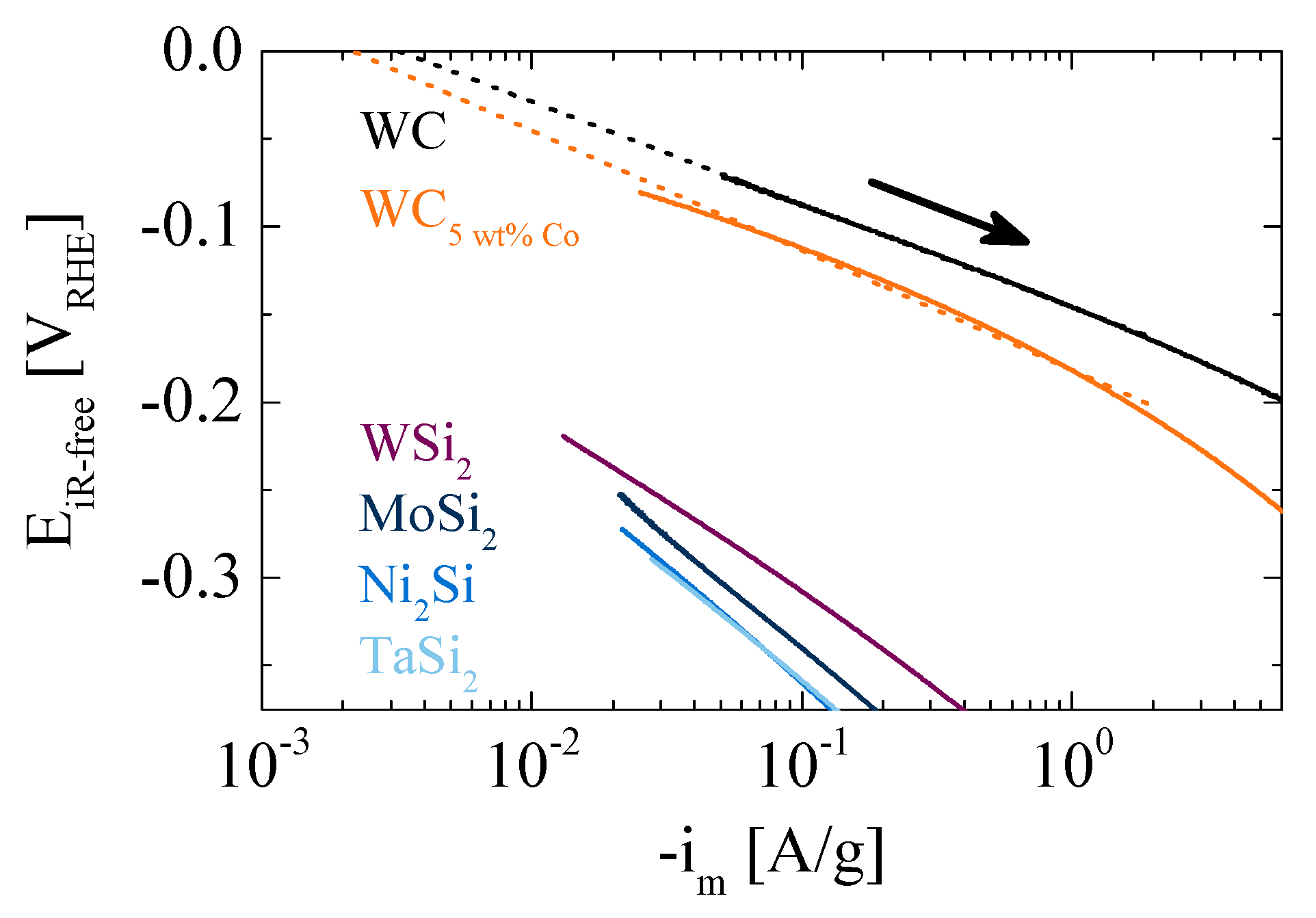
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Metal Silicide Nanopowders with Ball Milling
3.3. Characterization of the Metal Silicides Catalysts
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Durst, J.; Siebel, A.; Simon, C.; Hasche, F.; Herranz, J.; Gasteiger, H.A. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- Neyerlin, K.C.; Gu, W.; Jorne, J.; Gasteiger, H.A. Determination of Catalyst Unique Parameters for the Oxygen Reduction Reaction in a PEMFC. J. Electrochem. Soc. 2006, 153, A1955–A1963. [Google Scholar] [CrossRef]
- Kongkanand, A.; Mathias, M.F. The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2016, 7, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.J.; Burstein, G.T.; Kucernak, A.R.J.; Williams, K.R. Electrocatalytic activity of some carburized nickel, tungsten and molybdenum compounds. Electrochim. Acta 1997, 42, 2381–2388. [Google Scholar] [CrossRef]
- Böhm, H. New Non-noble Metal Anode Catalysts for Acid Fuel Cells. Nature 1970, 227, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Durst, J.; Simon, C.; Hasché, F.; Gasteiger, H.A. Hydrogen Oxidation and Evolution Reaction Kinetics on Carbon Supported Pt, Ir, Rh, and Pd Electrocatalysts in Acidic Media. J. Electrochem. Soc. 2015, 162, F190–F203. [Google Scholar] [CrossRef]
- Highfield, J.G.; Claude, E.; Oguro, K. Electrocatalytic synergism in Ni/Mo cathodes for hydrogen evolution in acid medium: A new model. Electrochim. Acta 1999, 44, 2805–2814. [Google Scholar] [CrossRef]
- Martinez, S.; Metikoš-Huković, M.; Valek, L. Electrocatalytic properties of electrodeposited Ni–15Mo cathodes for the HER in acid solutions: Synergistic electronic effect. J. Mol. Catal. A Chem. 2006, 245, 114–121. [Google Scholar] [CrossRef]
- Vrubel, H.; Hu, X. Molybdenum boride and carbide catalyze hydrogen evolution in both acidic and basic solutions. Angew. Chem. Int. Ed. 2012, 51, 12703–12706. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Regmi, Y.N.; Leonard, B.M. Multiple phases of molybdenum carbide as electrocatalysts for the hydrogen evolution reaction. Angew. Chem. Int. Ed. 2014, 53, 6407–6410. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Ge, X.; Wang, H.; Liu, Z.; Fisher, A.; Wang, X. Novel molybdenum carbide–tungsten carbide composite nanowires and their electrochemical activation for efficient and stable hydrogen evolution. Adv. Funct. Mater. 2015, 25, 1520–1526. [Google Scholar] [CrossRef]
- Esposito, D.V.; Hunt, S.T.; Stottlemyer, A.L.; Dobson, K.D.; McCandless, B.E.; Birkmire, R.W.; Chen, J.G. Low-cost hydrogen-evolution catalysts based on monolayer platinum on tungsten monocarbide substrates. Angew. Chem. Int. Ed. 2010, 49, 9859–9862. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.V.; Hunt, S.T.; Kimmel, Y.C.; Chen, J.G. A new class of electrocatalysts for hydrogen production from water electrolysis: Metal monolayers supported on low-cost transition metal carbides. J. Am. Chem. Soc. 2012, 134, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.F.; Wang, C.H.; Sasaki, K.; Marinkovic, N.; Xu, W.; Muckerman, J.T.; Zhu, Y.; Adzic, R.R. Highly active and durable nanostructured molybdenum carbide electrocatalysts for hydrogen production. Energy Environ. Sci. 2013, 6, 943–951. [Google Scholar] [CrossRef]
- Chen, W.F.; Iyer, S.; Iyer, S.; Sasaki, K.; Wang, C.H.; Zhu, Y.; Muckerman, J.T.; Fujita, E. Biomass-derived electrocatalytic composites for hydrogen evolution. Energy Environ. Sci. 2013, 6, 1818–1826. [Google Scholar] [CrossRef]
- Hunt, S.T.; Nimmanwudipong, T.; Román-Leshkov, Y. Engineering non-sintered, metal-terminated tungsten carbide nanoparticles for catalysis. Angew. Chem. Int. Ed. 2014, 53, 5131–5136. [Google Scholar]
- Liao, L.; Wang, S.; Xiao, J.; Bian, X.; Zhang, Y.; Scanlon, M.D.; Hu, X.; Tang, Y.; Liu, B.; Girault, H.H. A nanoporous molybdenum carbide nanowire as an electrocatalyst for hydrogen evolution reaction. Energy Environ. Sci. 2014, 7, 387–392. [Google Scholar] [CrossRef]
- Jaramillo, T.F.; Jørgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Bonde, J.; Moses, P.G.; Jaramillo, T.F.; Nørskov, J.K.; Chorkendorff, I. Hydrogen evolution on nano-particulate transition metal sulfides. Faraday Discuss. 2009, 140, 219–231. [Google Scholar] [CrossRef]
- Chen, Z.; Cummins, D.; Reinecke, B.N.; Clark, E.; Sunkara, M.K.; Jaramillo, T.F. Core-shell MoO3-MoS2 nanowires for hydrogen evolution: A functional design for electrocatalytic materials. Nano Lett. 2011, 11, 4168–4175. [Google Scholar] [CrossRef] [PubMed]
- Benck, J.D.; Chen, Z.; Kuritzky, L.Y.; Forman, A.J.; Jaramillo, T.F. Amorphous molybdenum sulfide catalysts for electrochemical hydrogen production: Insights into the origin of their catalytic activity. ACS Catal. 2012, 2, 1916–1923. [Google Scholar] [CrossRef]
- Chen, Z.; Forman, A.J.; Jaramillo, T.F. Bridging the gap between bulk and nanostructured photoelectrodes: The impact of surface states on the electrocatalytic and photoelectrochemical properties of MoS2. J. Phys. Chem. C 2013, 117, 9713–9722. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, C.; Grauer, D.C.; Yano, J.; Long, J.R.; Yang, P.; Chang, C.J. Electrodeposited cobalt-sulfide catalyst for electrochemical and photoelectrochemical hydrogen generation from water. J. Am. Chem. Soc. 2013, 135, 17699–17702. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, L.; Li, J.; Lin, X.; Li, X.; Fang, Y.; Huang, J.; Li, W.; Tian, M.; Jin, J.; Li, R. Synthesis of Cu–MoS2/rGO hybrid as non-noble metal electrocatalysts for the hydrogen evolution. J. Power Sources 2015, 292, 15–22. [Google Scholar] [CrossRef]
- Tian, J.; Liu, Q.; Asiri, A.M.; Sun, X. Self-supported nanoporous cobalt phosphide nanowire arrays: An efficient 3D hydrogen-evolving cathode over the wide range of pH 0–14. J. Am. Chem. Soc. 2014, 136, 7587–7590. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, J.; Zhou, X.; Wang, W.; Zuo, J.; Yang, Q. Alternative synthesis of cobalt monophosphide@C core–shell nanocables for electrochemical hydrogen production. J. Power Sources 2015, 286, 464–469. [Google Scholar] [CrossRef]
- Gao, S.; Liu, Y.; Li, G.D.; Guo, Y.; Zou, Y.; Zou, X. General urea-assisted synthesis of carbon-coated metal phosphide nanoparticles for efficient hydrogen evolution electrocatalysis. Electrochim. Acta 2016, 199, 99–107. [Google Scholar] [CrossRef]
- Gupta, S.; Patel, N.; Miotello, A.; Kothari, D.C. Cobalt-boride: An efficient and robust electrocatalyst for hydrogen evolution reaction. J. Power Sources 2015, 279, 620–625. [Google Scholar] [CrossRef]
- Hu, C.C.; Weng, C.Y. Hydrogen evolving activity on nickel–molybdenum deposits using experimental strategies. J. Appl. Electrochem. 2000, 30, 499–506. [Google Scholar] [CrossRef]
- Brown, D.E.; Mahmood, M.N.; Man, M.C.M.; Turner, A.K. Preparation and characterization of low overvoltage transition metal alloy electrocatalysts for hydrogen evolution in alkaline solutions. Electrochim. Acta 1984, 29, 1551–1556. [Google Scholar] [CrossRef]
- Jakšić, J.M.; Vojnović, M.V.; Krstajić, N.V. Kinetic analysis of hydrogen evolution at Ni-Mo alloy electrodes. Electrochim. Acta 2000, 45, 4151–4158. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Douglas, A.F. The anodic oxidation of the binary compounds of the transition elements in sulphuric acid. J. Appl. Electrochem. 1972, 2, 143–149. [Google Scholar] [CrossRef]
- Gottlieb, U.; Nava, F.; Affronte, M.; Laborde, O.; Madar, R. Properties of Metal Silicides, 1st ed.; Maex, K., Van Rossum, M., Eds.; INSPEC, the Institution of Electrical Engineers: London, UK, 1995; pp. 188–204. [Google Scholar]
- Maex, K.; Van Rossum, M.; Reader, A. Properties of Metal Silicides, 1st ed.; Maex, K., Van Rossum, M., Eds.; INSPEC, the Institution of Electrical Engineers: London, UK, 1995; pp. 3–14. [Google Scholar]
- Colgan, E.G.; Mäenpää, M.; Finetti, M.; Nicolet, M.A. Electrical characteristics of thin Ni2Si, NiSi, and NiSi2 layers grown on silicon. J. Electron. Mater. 1983, 12, 413–422. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 86th ed.; CRC Press: Boca Raton, FL, USA, 2005; Section 4–37; pp. 34–96. [Google Scholar]
- Bachmann, K.; Williams, W.S. Low-temperature electrical resistivity and Hall effect of single-crystal α-tungsten carbide. J. Appl. Phys. 1971, 42, 4406–4407. [Google Scholar] [CrossRef]
- Morozkin, A.V.; Klyamkin, S.N.; Verbetsky, V.N.; Lushnikov, S.N.; Portnoy, V.K.; Movlaev, E.A.; Chernavskii, A.P.; Tarasov, A.V. Hydrogen sorption in homologous lanthanum and cerium nickel silicides. J. Alloys Compd. 2000, 309, 197–200. [Google Scholar] [CrossRef]
- Levy, R.B.; Boudart, M. Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.F.; Bonde, J.; Zhang, J.; Ooi, B.L.; Andersson, K.; Ulstrup, J.; Chorkendorff, I. Hydrogen evolution on supported incomplete cubane-type [Mo3S4]4+ electrocatalysts. J. Phys. Chem. C 2008, 112, 17492–17498. [Google Scholar] [CrossRef]
- Sheng, W.; Gasteiger, H.A.; Yang, S.H. Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs. Alkaline Electrolytes. J. Electrochem. Soc. 2010, 157, B1529–B1536. [Google Scholar] [CrossRef]
- Mittermeier, T.; Weiß, A.; Hasché, F.; Gasteiger, H.A. Activity, stability and degradation of carbon supported palladium (Pd/C) fuel cell electrocatalysts for the oxygen reduction. ECS Trans. 2015, 69, 303–313. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Size [nm] | ABET [m²/g] | σ20 °C [kS/cm] | ρ20 °C [g/cm³] | ||
|---|---|---|---|---|---|---|
| Number | XRD | BET | ||||
| MoSi2 | 138 | 13 | 48 | 20 | 5.9–7.9 [33] | 6.3 [34] |
| TaSi2 | 79 | 11 | 37 | 18 | 2.5–5.0 [33] | 9.1 [34] |
| WSi2 | 129 | 15 | 61 | 10 | 7.8–8.4 [33] | 9.9 [34] |
| Ni2Si | 865 | 10 | 152 | 5 | 4.2 [35] | 7.9 [34] |
| WC5 wt % Co | 76 | 42 | ≈192 | 2 | -- | ≈15.6 [36] |
| WC | 313 | 22 | 192 | 2 | 7.0 [37] | 15.6 [36] |
| Material | Electrolyte | i0m [mA/g] | Surface Area Via | i0s [µA/cm²] | Ref. |
|---|---|---|---|---|---|
| WC5 wt % Co | 0.1 M HClO4 | 2 | BET | 0.11 | t. s. |
| WC | 0.1 M HClO4 | 3 | BET | 0.16 | t. s. |
| WC | 0.5 M H2SO4 | 0.53 | -- | -- | [11] |
| Mo2C | 0.5 M H2SO4 | 4.5 | -- | -- | [11] |
| Mo2C/CNT | 0.1 M HClO4 | 6.9 | -- | 0.046 | [14] |
| MoS2/C | 0.5 M H2SO4 | -- | CV irrev. Ox. | 1.2 | [19] |
| [Mo3S4]4+/HOPG | 0.5 M H2SO4 | -- | STM (rf = 1 *) | 0.22 | [40] |
| Pt/C | PEMFC | 2.6 ± 0.7 × 108 | Hupd | 2.2 ± 0.5 × 105 | [1] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mittermeier, T.; Madkikar, P.; Wang, X.; Gasteiger, H.A.; Piana, M. Probing Transition-Metal Silicides as PGM-Free Catalysts for Hydrogen Oxidation and Evolution in Acidic Medium. Materials 2017, 10, 661. https://doi.org/10.3390/ma10060661
Mittermeier T, Madkikar P, Wang X, Gasteiger HA, Piana M. Probing Transition-Metal Silicides as PGM-Free Catalysts for Hydrogen Oxidation and Evolution in Acidic Medium. Materials. 2017; 10(6):661. https://doi.org/10.3390/ma10060661
Chicago/Turabian StyleMittermeier, Thomas, Pankaj Madkikar, Xiaodong Wang, Hubert A. Gasteiger, and Michele Piana. 2017. "Probing Transition-Metal Silicides as PGM-Free Catalysts for Hydrogen Oxidation and Evolution in Acidic Medium" Materials 10, no. 6: 661. https://doi.org/10.3390/ma10060661
APA StyleMittermeier, T., Madkikar, P., Wang, X., Gasteiger, H. A., & Piana, M. (2017). Probing Transition-Metal Silicides as PGM-Free Catalysts for Hydrogen Oxidation and Evolution in Acidic Medium. Materials, 10(6), 661. https://doi.org/10.3390/ma10060661