
Recovery and Utilization of Lignin Monomers as Part of the Biorefinery Approach


Abstract
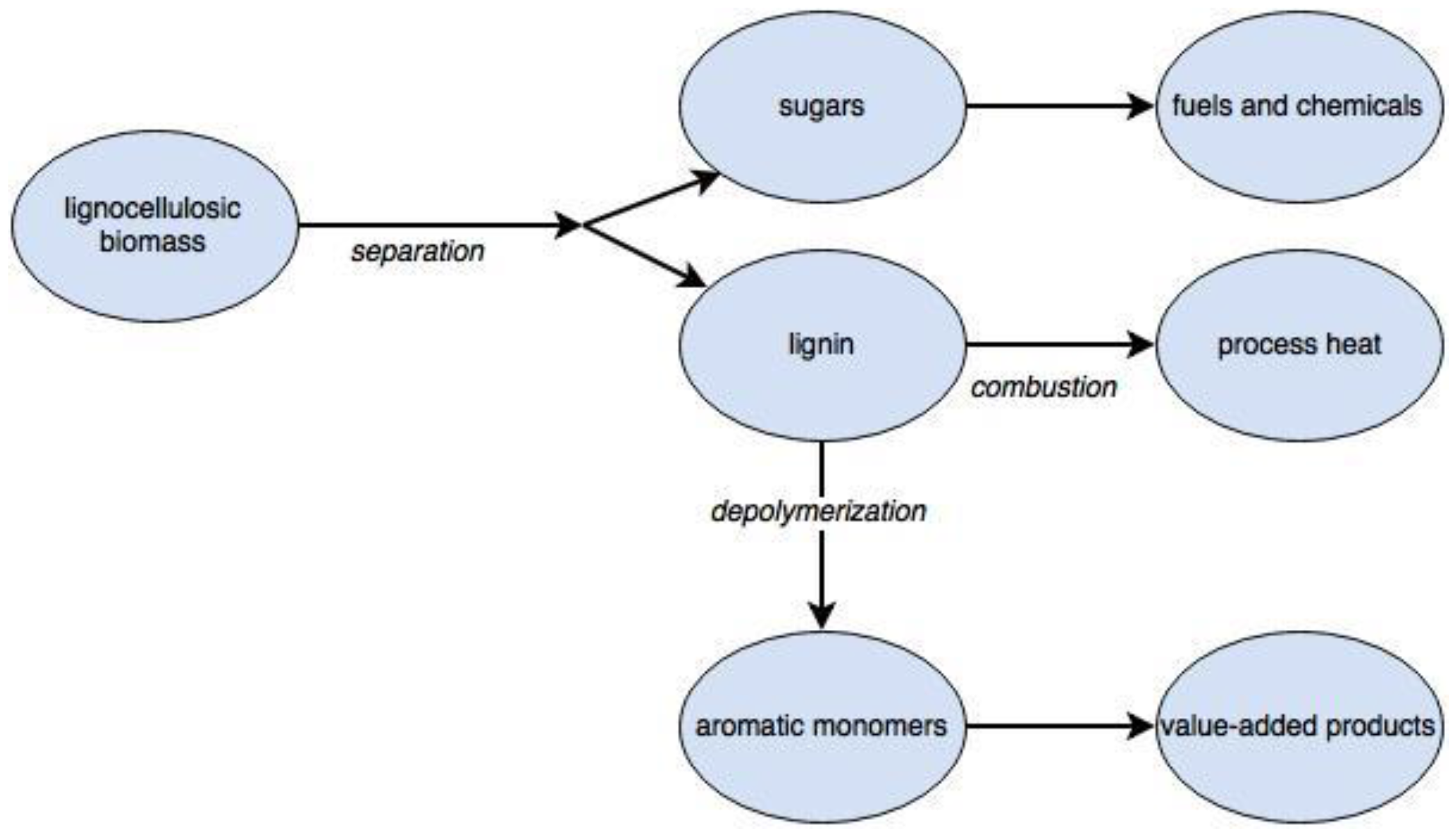
:1. Introduction
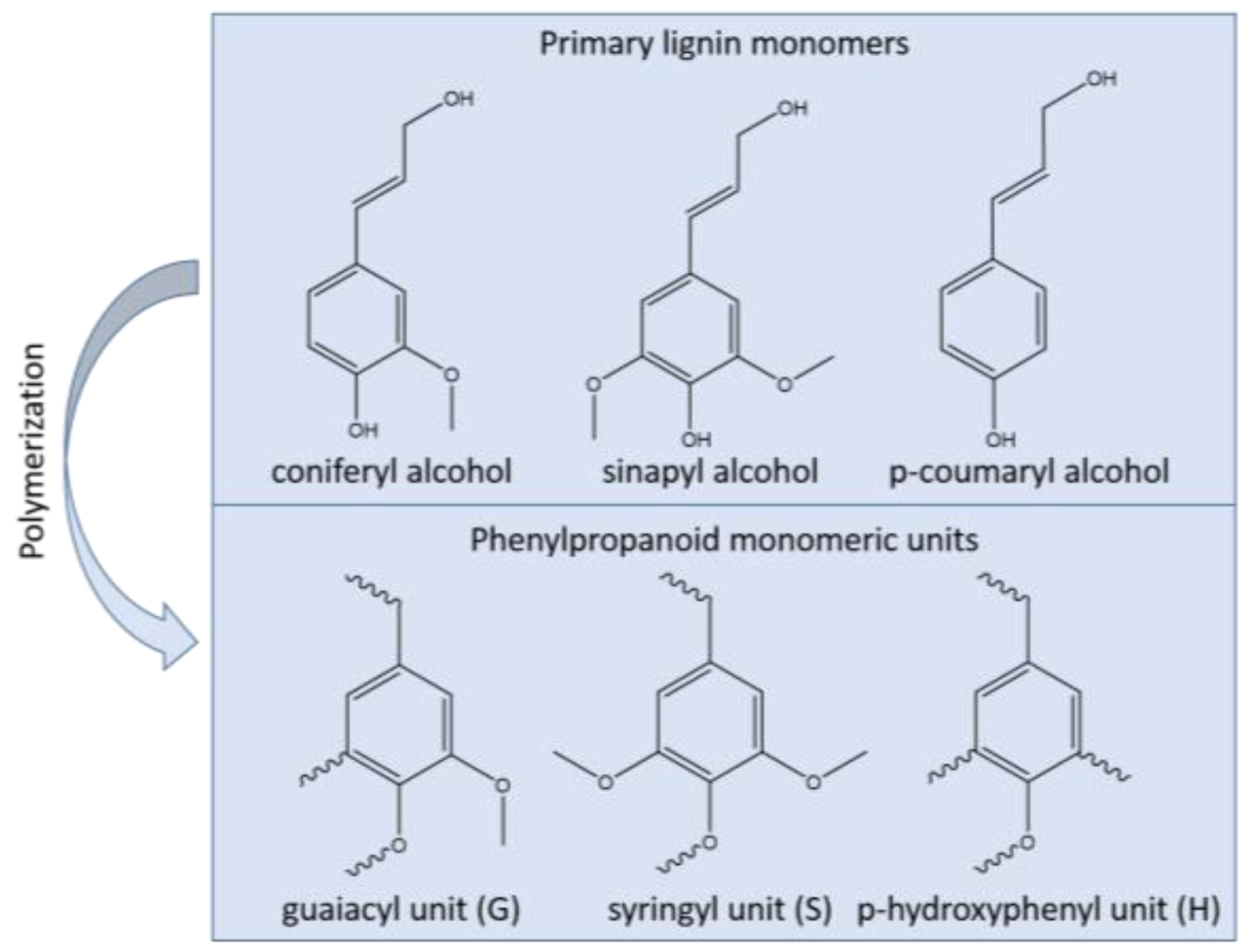
2. Lignin Structure and Abundance
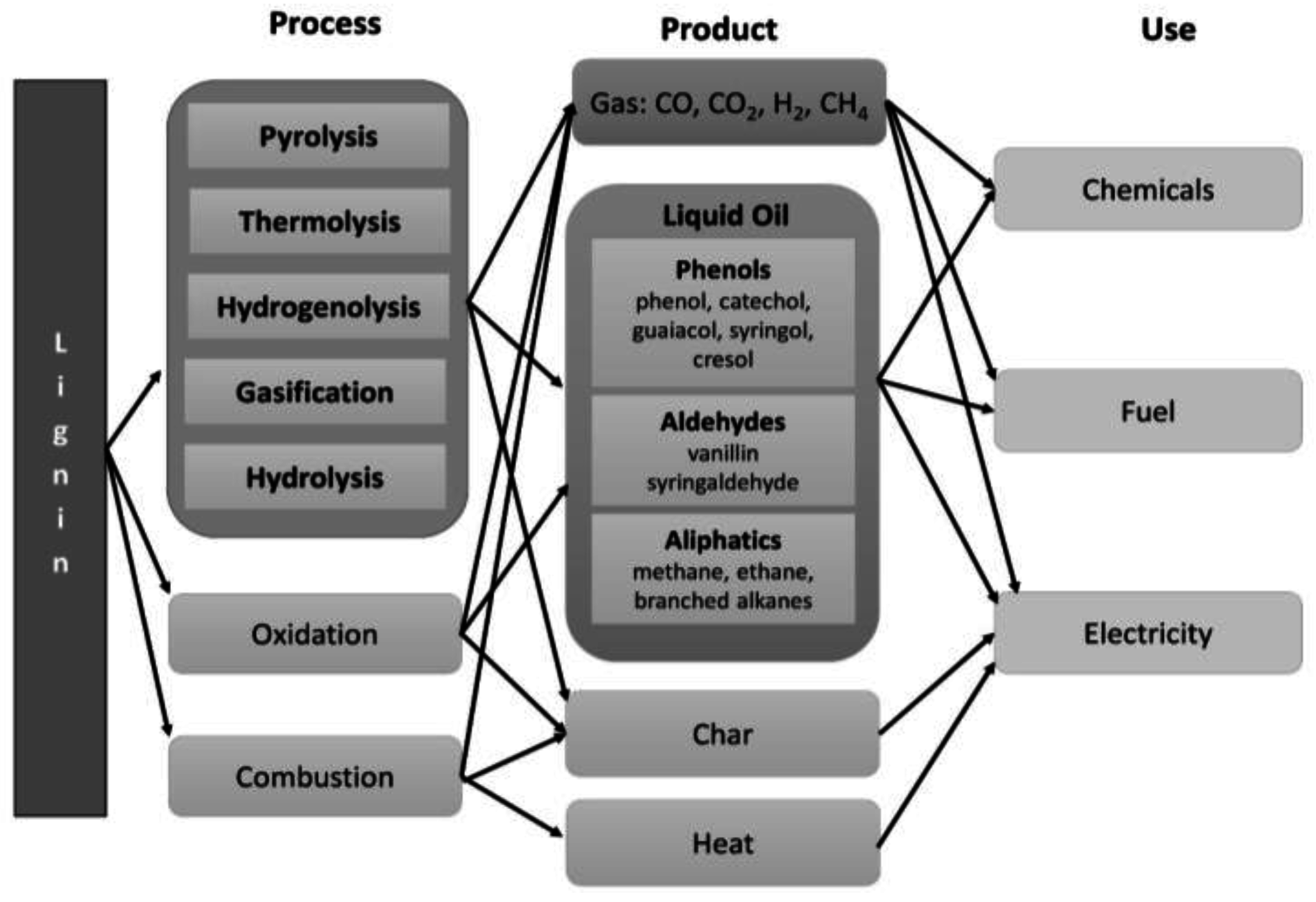
3. Challenges and Progress in Lignin Recovery
3.1. Pulping Processes
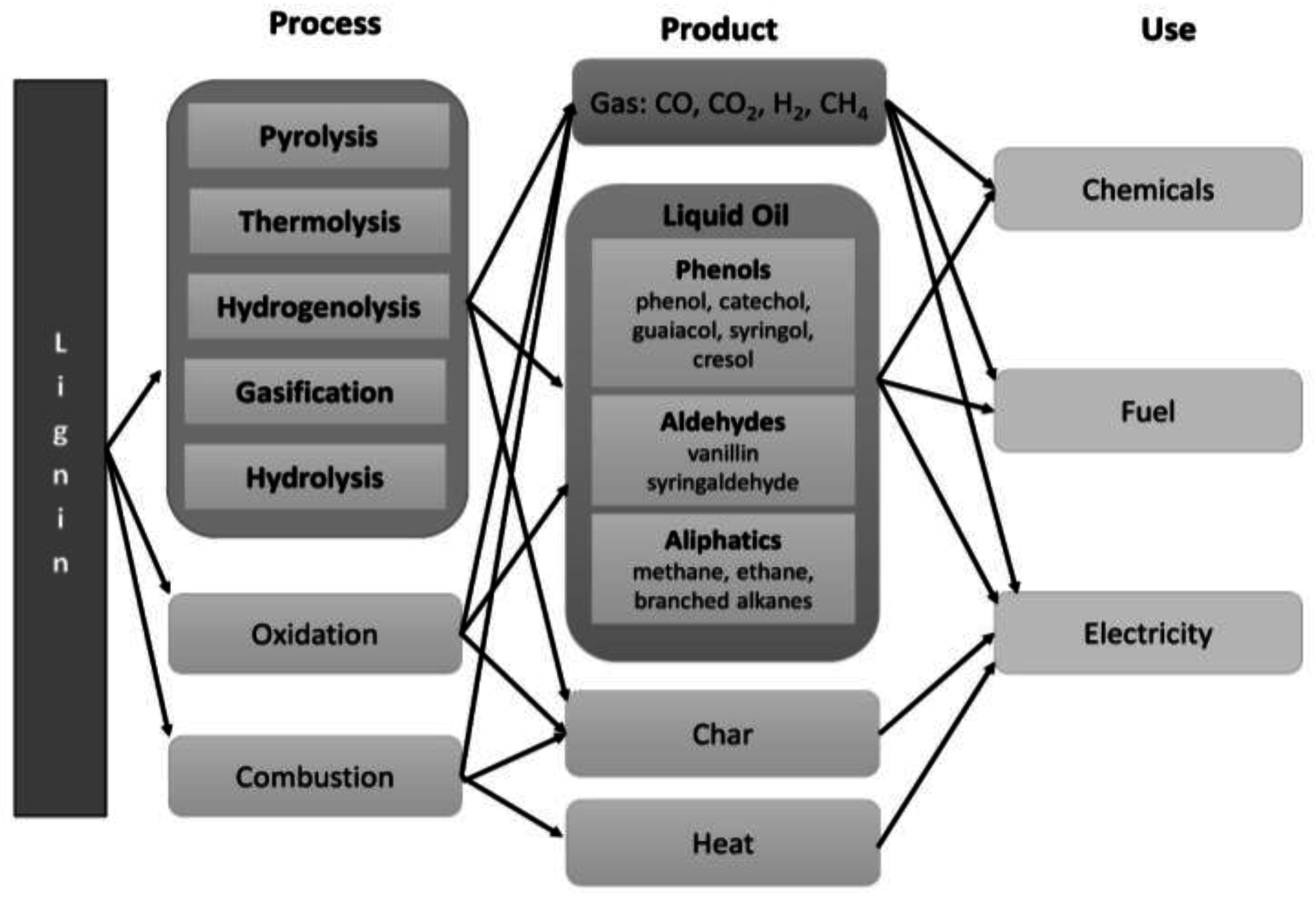
3.2. Thermochemical Depolymerization of Biomass
3.3. Dilute Acid Hydrolysis
3.4. Hydrothermal Fractionation
3.5. Biphasic Fractionation
3.6. Modeling of Lignin Isolation
4. Lignin Utilization in Nature
4.1. Lignin Degrading Enzymes
4.2. Bacterial and Fungal Pathways of Lignin Utilization
4.2.1. Aerobic Degradation
4.2.2. Anaerobic Conditions
4.3. Application Directed Studies of Lignin Degrading Microorganisms
5. Challenges and Progress in Depolymerization of Isolated Lignin
5.1. Pyrolysis of Isolated Lignin
5.2. Catalytic Pyrolysis of Isolated Lignin
5.3. Supercritical Water
5.4. Supercritical Solvents
5.5. Base-Catalyzed Depolymerization
6. Upgrading of Lignin Monomers
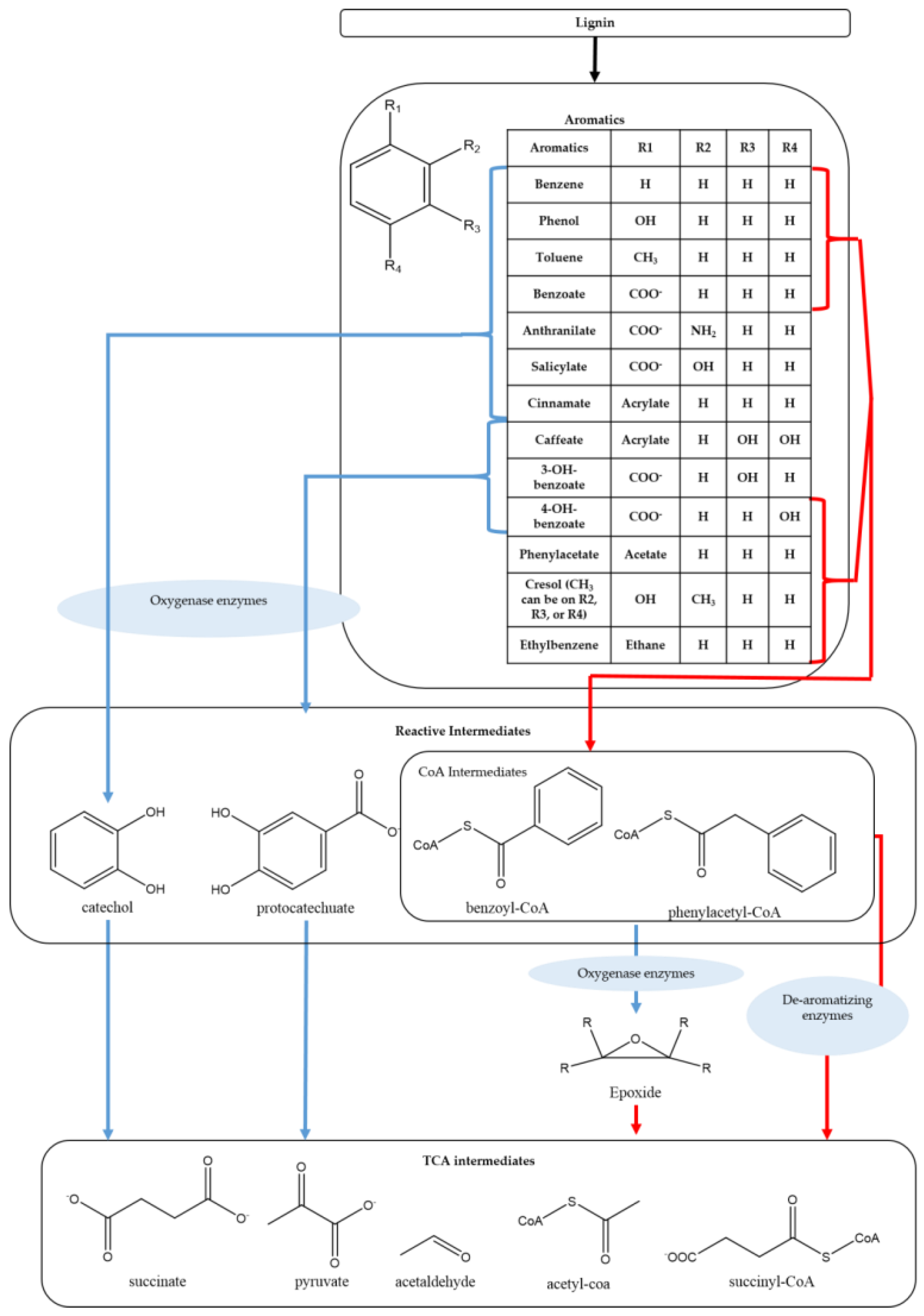
6.1. Progress in Biological Utilization of Depolymerized Lignin Monomers and Lignin Model Compounds
6.1.1. Biotransformation
6.1.2. Central Metabolism
6.2. Progress in Chemical Utilization
6.2.1. Cracking and Hydrolysis of Depolymerized Lignin
6.2.2. Reduction of Lignin Model Compounds and Depolymerized Lignin
6.2.3. Oxidation of Lignin Model Compounds and Depolymerized Lignin
7. Economic Analysis of Lignin Utilization Strategies
Acknowledgments
Conflicts of Interest
Abbreviations
| TCA | tricarboxylic acid cycle |
| TG-FTIR | thermogravimetric-fourier transform infrared spectroscopy |
| Py-GC/MS | pyrolysis-gas chromatography/mass spectrometry |
| PHA | polyhydroxyalkanoates |
References
- Brown, R.C.; Brown, T.R. Biorenewable Resources: Engineering New Products from Agriculture; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Rahmanpour, R. Enzymatic conversion of lignin into renewable chemicals. Curr. Opin. Chem. Biol. 2015, 29, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D. Lignin as a base material for materials applications: Chemistry, application and economics. Ind. Crops Prod. 2008, 27, 202–207. [Google Scholar] [CrossRef]
- Fisher, A.B.; Fong, S.S. Lignin biodegradation and industrial implications. AIMS Bioeng. 2014, 1, 92–112. [Google Scholar]
- Lora, J.H.; Glasser, W.G. Recent industrial applications of lignin: A sustainable alternative to nonrenewable materials. J. Polym. Environ. 2002, 10, 39–48. [Google Scholar] [CrossRef]
- Hamelinck, C.N.; van Hooijdonk, G.; Faaij, A.P.C. Ethanol from lignocellulosic biomass: Techno-economic performance in short-, middle- and long-term. Biomass Bioenergy 2005, 28, 384–410. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Boll, M.; Heider, J. Microbial degradation of aromatic compounds—From one strategy to four. Nat. Rev. Microbiol. 2011, 9, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Tuck, C.O.; Perez, E.; Horvath, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Serrano, L.; Labidi, J. Improving base catalyzed lignin depolymerization by avoiding lignin repolymerization. Fuel 2014, 116, 617–624. [Google Scholar] [CrossRef]
- Wong, D.W.S. Structure and action mechanism of ligninolytic enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Ragauskas, A. Extraction of hemicellulose from loblolly pine woodchips and subsequent kraft pulping. Ind. Eng. Chem. Res. 2013, 52, 1743–1749. [Google Scholar] [CrossRef]
- Sannigrahi, P.; Ragauskas, A.J.; Tuskan, G.A. Poplar as a feedstock for biofuels: A review of compositional characteristics. Biofuels Bioprod. Biorefin. 2010, 4, 209–226. [Google Scholar] [CrossRef]
- Nunes, C.A.; Lima, C.F.; Barbosa, L.C.D.A.; Colodette, J.L.; Fidêncio, P.H. Determination of chemical constituents in eucalyptus wood by Py-GC/MS and multivariate calibration: Comparison between artificial neural network and support vector machines. Quím. Nova 2011, 34, 279–283. [Google Scholar] [CrossRef]
- Brosse, N.; Dufour, A.; Meng, X.Z.; Sun, Q.N.; Ragauskas, A. Miscanthus: A fast-growing crop for biofuels and chemicals production. Biofuels Bioprod. Biorefin. 2012, 6, 580–598. [Google Scholar] [CrossRef]
- David, K.; Ragauskas, A.J. Switchgrass as an energy crop for biofuel production: A review of its ligno-cellulosic chemical properties. Energy Environ. Sci. 2010, 3, 1182–1190. [Google Scholar] [CrossRef]
- Saha, B.C.; Yoshida, T.; Cotta, M.A.; Sonomoto, K. Hydrothermal pretreatment and enzymatic saccharification of corn stover for efficient ethanol production. Ind. Crops Prod. 2013, 44, 367–372. [Google Scholar] [CrossRef]
- Sun, X.F.; Wang, H.H.; Zhang, G.C.; Fowler, P.; Rajaratnam, M. Extraction and characterization of lignins from maize stem and sugarcane bagasse. J. Appl. Polym. Sci. 2011, 120, 3587–3595. [Google Scholar] [CrossRef]
- Pat, R.; Kordsachi, O.; Süttinger, R. Pulp. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- Demirbaş, A. Pyrolysis and steam gasification processes of black liquor. Energy Convers. Manag. 2002, 43, 877–884. [Google Scholar] [CrossRef]
- Mimms, A.; Kocurek, M.; Pyatte, J.; Wright, E. Kraft Pulping Chemistry and Process; Tappi Press: Peach Tree Corners, GA, USA, 1989. [Google Scholar]
- El Mansouri, N.-E.; Salvadó, J. Structural characterization of technical lignins for the production of adhesives: Application to lignosulfonate, kraft, soda-anthraquinone, organosolv and ethanol process lignins. Ind. Crops Prod. 2006, 24, 8–16. [Google Scholar] [CrossRef]
- Sarkanen, K.V. Chemistry of solvent pulping. Tappi J. 1990, 73, 215–219. [Google Scholar]
- Holladay, J.E.; White, J.F.; Bozell, J.J.; Johnson, D. Top Value-Added Chemicals from Biomass-Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin; Pacific Northwest National Laboratory (PNNL): Richland, WA, USA, 2007.
- Hayes, D.J.; Fitzpatrick, S.; Hayes, M.H.B.; Ross, J.R.H. The biofine process—Production of levulinic acid, furfural, and formic acid from lignocellulosic feedstocks. In Biorefineries—Industrial Processes and Products: Status Quo and Future Directions; Kamm, B., Gruber, P.R., Kamm, M., Eds.; Wiley-VCH Verlag Gmbh: Weinheim, Germany, 2006; Volume 1, pp. 139–164. [Google Scholar]
- Azadi, P.; Carrasquillo-Flores, R.; Pagán-Torres, Y.J.; Gürbüz, E.I.; Farnood, R.; Dumesic, J.A. Catalytic conversion of biomass using solvents derived from lignin. Green Chem. 2012, 14, 1573–1576. [Google Scholar] [CrossRef]
- Yan, N.; Zhao, C.; Dyson, P.J.; Wang, C.; Liu, L.T.; Kou, Y. Selective degradation of wood lignin over noble-metal catalysts in a two-step process. ChemSusChem 2008, 1, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, H.; Kauppila, J. Engineering aspects and feasibility of a wood pulping process using Batelle-Geneva’s aqueous phenol process by Rintekno. In Proceedings of the World Pulp and Paper Week, Stockholm, Sweden, 10–13 April 1984.
- Scholze, B.; Meier, D. Characterization of the water-insoluble fraction from pyrolysis oil (pyrolytic lignin). Part I. Py-GC/MS, FTIR, and functional groups. J. Anal. Appl. Pyrolysis 2001, 60, 41–54. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Valle, B.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Pyrolytic lignin removal for the valorization of biomass pyrolysis crude bio-oil by catalytic transformation. J. Chem. Technol. Biotechnol. 2010, 85, 132–144. [Google Scholar] [CrossRef]
- Jiang, X.X.; Ellis, N.; Zhong, Z.P. Characterization of pyrolytic lignin extracted from bio-oil. Chin. J. Chem. Eng. 2010, 18, 1018–1022. [Google Scholar] [CrossRef]
- Deng, L.; Yan, Z.; Fu, Y.; Guo, Q.X. Green solvent for flash pyrolysis oil separation. Energy Fuels 2009, 23, 3337–3338. [Google Scholar] [CrossRef]
- Blackwell, P.; Riethmuller, G.; Collins, M. Biochar application to soil. In Biochar for Environmental Management: Science and Technology; Earthscan: London, UK, 2009. [Google Scholar]
- Jarboe, L.R.W.; Choi, Z.; Brown, D.; Robert, C. Hybrid themochemical processing: Fermentation of pyrolysis-derived bio-oil. Appl. Microbiol. Biotechnol. 2011, 91, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Funaoka, M.; Abe, I. Rapid separation of wood into carbohydrate and lignin with concentrated acid-phenol system. Tappi J. 1989, 72, 145–149. [Google Scholar]
- Vega, A.; Bao, M. Fractionation of lignocellulose materials with phenol and dilute hcl. Wood Sci. Technol. 1991, 25, 459–466. [Google Scholar] [CrossRef]
- Vega, A.; Bao, M.; Lamas, J. Application of factorial design to the modelling of organosolv delignification of Miscanthus sinensis (elephant grass) with phenol and dilute acid solutions. Bioresour. Technol. 1997, 61, 1–7. [Google Scholar] [CrossRef]
- Sakakibara, A.; Edashige, Y.; Sano, Y.; Takeyama, H. Solvolysis pulping with cresols-water system. Holzforschung 1984, 38, 159–165. [Google Scholar] [CrossRef]
- Skakibara, A.; Edashige, Y.; Sano, Y.; Takeyama, H. Solvolysis pulping with lignin degradation products. In Proceedings of the International Symposium on Wood and Pulping Chemistry, Tsukuba Science City, Japan, 23–27 May 1983.
- Willauer, H.D.; Huddleston, J.G.; Li, M.; Rogers, R.D. Investigation of aqueous biphasic systems for the separation of lignins from cellulose in the paper pulping process. J. Chromatogr. B 2000, 743, 127–135. [Google Scholar] [CrossRef]
- Guo, Z.; Li, M.; Willauer, H.D.; Huddleston, J.G.; April, G.C.; Rogers, R.D. Evaluation of polymer-based aqueous biphasic systems as improvement for the hardwood alkaline pulping process. Ind. Eng. Chem. Res. 2002, 41, 2535–2542. [Google Scholar] [CrossRef]
- Guo, Z.; April, G.C.; Li, M.; Willauer, H.D.; Huddleston, J.G.; Rogers, R.D. Peg-based aqueous biphasic systems as improvement for kraft hardwood pulping process. Chem. Eng. Commun. 2003, 190, 1155–1169. [Google Scholar] [CrossRef]
- Chen, J.; Spear, S.K.; Huddleston, J.G.; Rogers, R.D. Polyethylene glycol and solutions of polyethylene glycol as green reaction media. Green Chem. 2005, 7, 64–82. [Google Scholar]
- Vom Stein, T.; Grande, P.M.; Kayser, H.; Sibilla, F.; Leitner, W.; de Maria, P.D. From biomass to feedstock: One-step fractionation of lignocellulose components by the selective organic acid-catalyzed depolymerization of hemicellulose in a biphasic system. Green Chem. 2011, 13, 1772–1777. [Google Scholar] [CrossRef]
- Lee, R.A.; Berberi, V.; Labranche, J.; Lavoie, J.M. Lignin extraction—Reassessment of the severity factor with respect to hydroxide concentration. Bioresour. Technol. 2014, 169, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Overend, R.P.; Chornet, E. Fractionation of lignocellulosics by steam-aqueous pretreatments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1987, 321, 523–536. [Google Scholar] [CrossRef]
- Chum, H.L.; Johnson, D.K.; Black, S.K.; Overend, R.P. Pretreatment catalyst effects and the combined severity parameter. Appl. Biochem. Biotechnol. 1990, 24–25, 1–14. [Google Scholar] [CrossRef]
- Silverstein, R.A.; Chen, Y.; Sharma-Shivappa, R.R.; Boyette, M.D.; Osborne, J. A comparison of chemical pretreatment methods for improving saccharification of cotton stalks. Bioresour. Technol. 2007, 98, 3000–3011. [Google Scholar] [CrossRef] [PubMed]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, A.M.; Solbiati, J.O.; Cann, I.K.O. Insights into lignin degradation and its potential industrial applications. In Advances in Applied Microbiology; Sariaslani, S., Gadd, G.M., Eds.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2013; Volume 82, pp. 1–28. [Google Scholar]
- Johnson, C.W.; Beckham, G.T. Aromatic catabolic pathway selection for optimal production of pyruvate and lactate from lignin. Metab. Eng. 2015, 28, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.H.; Ahmad, M.; Hardiman, E.M.; Singh, R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr. Opin. Biotechnol. 2011, 22, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Sugano, Y.; Muramatsu, R.; Ichiyanagi, A.; Sato, T.; Shoda, M. Dyp, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family. J. Biol. Chem. 2007, 282, 36652–36658. [Google Scholar] [CrossRef] [PubMed]
- Tien, M.; Kirk, T.K. Lignin-degrading enzyme from Phanerochaete chrysosporium purification, characterization, and catalytic properties of a unique H2O2-requiring oxygenase. Proc. Natl. Acad. Sci. USA 1984, 81, 2280–2284. [Google Scholar] [CrossRef] [PubMed]
- Miki, K.; Renganathan, V.; Gold, M.H. Mechanism of beta-aryl ether dimeric lignin model-compound oxidation by lignin peroxidase of Phanerochaete chrysosporium. Biochemistry 1986, 25, 4790–4796. [Google Scholar] [CrossRef]
- Paszczynski, A.; Huynh, V.B.; Crawford, R. Enzymatic-activities of an extracellular, manganese-dependent peroxidase from Phanerochaete chrysosporium. FEMS Microbiol. Lett. 1985, 29, 37–41. [Google Scholar] [CrossRef]
- Perez-Boada, M.; Ruiz-Duenas, F.J.; Pogni, R.; Basosi, R.; Choinowski, T.; Martinez, M.J.; Piontek, K.; Martinez, A.T. Versatile peroxidase oxidation of high redox potential aromatic compounds: Site-directed mutagenesis, spectroscopic and crystallographic investigation of three long-range electron transfer pathways. J. Mol. Biol. 2005, 354, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Kersten, P.J.; Kirk, T.K. Involvement of a new enzyme, glyoxal oxidase, in extracellular H2O2 production by Phanerochaete chrysosporium. J. Bacteriol. 1987, 169, 2195–2201. [Google Scholar] [PubMed]
- Guillen, F.; Martinez, M.J.; Munoz, C.; Martinez, A.T. Quinone redox cycling in the ligninolytic fungus Pleurotus eryngii leading to extracellular production of superoxide anion radical. Arch. Biochem. Biophys. 1997, 339, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Thurston, C.F. The structure and function of fungal laccases. Microbiology 1994, 140, 19–26. [Google Scholar] [CrossRef]
- Have, R.; Teunissen, P.J.M. Oxidative mechanisms involved in lignin degradation by white-rot fungi. Chem. Rev. 2001, 101, 3397–3413. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.O.; Anderson, A.M.; Berhow, M.A. Laccase-mediator catalyzed conversion of model lignin compounds. Biocatal. Agric. Biotechnol. 2016, 5, 111–115. [Google Scholar] [CrossRef]
- Zimmermann, W. Degradation of lignin by bacteria. J. Biotechnol. 1990, 13, 119–130. [Google Scholar] [CrossRef]
- Hayaishi, O. From oxygenase to sleep. J. Biol. Chem. 2008, 283, 19165–19175. [Google Scholar] [CrossRef] [PubMed]
- Dagley, S.; Evans, W.C.; Ribbons, D.W. New pathways in the oxidative metabolism of aromatic compounds by micro-organisms. Nature 1960, 188, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Dagley, S. Degradation of benzene nucleus by bacteria. Sci. Prog. 1965, 53, 381–392. [Google Scholar] [CrossRef]
- Dagley, S. Catabolism of aromatic compounds by microorganisms. Adv. Microb. Physiol. 1971, 6, 1–46. [Google Scholar] [PubMed]
- Ornston, L.N.; Stanier, R.Y. Conversion of catechol and protocatechuate to beta-ketoadipate by Pseudomonas putida. J. Biol. Chem. 1966, 241, 3776–3786. [Google Scholar] [PubMed]
- Stanier, R.Y.; Ornston, L.N. The beta-ketoadipate pathway. Adv. Microb. Physiol. 1973, 9, 89–151. [Google Scholar] [PubMed]
- Harwood, C.S.; Burchhardt, G.; Herrmann, H.; Fuchs, G. Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol. Rev. 1998, 22, 439–458. [Google Scholar] [CrossRef]
- Harayama, S.; Kok, M.; Neidle, E.L. Functional and evolutionary relationships among diverse oxygenases. Ann. Rev. Microbiol. 1992, 46, 565–601. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.S.; Mason, J.R. Structure-function analysis of the bacterial aromatic ring-hydroxylating dioxygenases. Adv. Microb. Physiol. 1997, 38, 47–84. [Google Scholar] [PubMed]
- Gibson, D.T.; Parales, R.E. Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 2000, 11, 236–243. [Google Scholar] [CrossRef]
- Leahy, J.G.; Batchelor, P.J.; Morcomb, S.M. Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 2003, 27, 449–479. [Google Scholar] [CrossRef]
- Vaillancourt, F.H.; Bolin, J.T.; Eltis, L.D. The ins and outs of ring-cleaving dioxygenases. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 241–267. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, J.D. Mechanism of extradiol aromatic ring-cleaving dioxygenases. Curr. Opin. Struct. Biol. 2008, 18, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Rather, L.J.; Knapp, B.; Haehnel, W.; Fuchs, G. Coenzyme A-dependent aerobic metabolism of benzoate via epoxide formation. J. Biol. Chem. 2010, 285, 20615–20624. [Google Scholar] [CrossRef] [PubMed]
- Rather, L.J.; Bill, E.; Ismail, W.; Fuchs, G. The reducing component BoxA of benzoyl-coenzyme A epoxidase from Azoarcus evansii is a 4Fe–4S protein. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Rather, L.J.; Weinert, T.; Demmer, U.; Bill, E.; Ismail, W.; Fuchs, G.; Ermler, U. Structure and mechanism of the diiron benzoyl-coenzyme A epoxidase BoxB. J. Biol. Chem. 2011, 286, 29241–29248. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.E.-S.; Zaar, A.; Ebenau-Jehle, C.; Fuchs, G. Reinvestigation of a new type of aerobic benzoate metabolism in the proteobacterium Azoarcus evansii. J. Bacteriol. 2001, 183, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Zaar, A.; Eisenreich, W.; Bacher, A.; Fuchs, G. A novel pathway of aerobic benzoate catabolism in the bacteria Azoarcus evansii and Bacillus stearothermophilus. J. Biol. Chem. 2001, 276, 24997–25004. [Google Scholar] [CrossRef] [PubMed]
- Zaar, A.; Gescher, J.; Eisenreich, W.; Bacher, A.; Fuchs, G. New enzymes involved in aerobic benzoate metabolism in Azoarcus evansii. Mol. Microbiol. 2004, 54, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Gescher, J.; Zaar, A.; Mohamed, M.; Schagger, H.; Fuchs, G. Genes coding for a new pathway of aerobic benzoate metabolism in Azoarcus evansii. J. Bacteriol. 2002, 184, 6301–6315. [Google Scholar] [CrossRef] [PubMed]
- Gescher, J.; Eisenreich, W.; Worth, J.; Bacher, A.; Fuchs, G. Aerobic benzoyl-CoA catabolic pathway in Azoarcus evansii: Studies on the non-oxygenolytic ring cleavage enzyme. Mol. Microbiol. 2005, 56, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Gescher, J.; Ismail, W.; Olgeschlager, E.; Eisenreich, W.; Worth, J.; Fuchs, G. Aerobic benzoyl-Coenzyme a (CoA) catabolic pathway in Azoarcus evansii: Conversion of ring cleavage product by 3,4-dehydroadipyl-coa semialdehyde dehydrogenase. J. Bacteriol. 2006, 188, 2919–2927. [Google Scholar] [CrossRef] [PubMed]
- Ismail, W. Benzoyl-coenzyme a thioesterase of Azoarcus evansii: Properties and function. Arch. Microbiol. 2008, 190, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.; Leon, R.; Boulanger, M.J. Structural and biophysical characterization of BoxC from Burkholderia xenovorans LB400 a novel ring-cleaving enzyme in the crotonase superfamily. J. Biol. Chem. 2009, 284, 16377–16385. [Google Scholar] [CrossRef] [PubMed]
- Heider, J.; Fuchs, G. Microbial anaerobic aromatic metabolism. Anaerobe 1997, 3, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Heider, J.; Fuchs, G. Anaerobic metabolism of aromatic compounds. Eur. J. Biochem. 1997, 243, 577–596. [Google Scholar] [CrossRef] [PubMed]
- Schink, B.; Philipp, B.; Muller, J. Anaerobic degradation of phenolic compounds. Naturwissenschaften 2000, 87, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Boll, M.; Fuchs, G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism—ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur. J. Biochem. 1995, 234, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.W.; Loffler, C.; Dorner, K.; Heintz, D.; Gallien, S.; van Dorsselaer, A.; Friedrich, T.; Boll, M. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc. Natl. Acad. Sci. USA 2009, 106, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, K.M.; Fortney, J.L.; Borglin, S.; Silver, W.L.; Simmons, B.A.; Hazen, T.C. Anaerobic decomposition of switchgrass by tropical soil-derived feedstock-adapted consortia. mBio 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, K.M.; D’Haeseleer, P.; Chivian, D.; Fortney, J.L.; Khudyakov, J.; Simmons, B.; Woo, H.; Arkin, A.P.; Davenport, K.W.; Goodwin, L.; et al. Complete genome sequence of Enterobacter lignolyticus SCF1. Stand. Genom. Sci. 2011, 5, 69–85. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, K.M.; Sharma, D.; Varney, R.; Simmons, B.; Isern, N.G.; Markilllie, L.M.; Nicora, C.; Norbeck, A.D.; Taylor, R.C.; Aldrich, J.T.; et al. Evidence supporting dissimilatory and assimilatory lignin degradation in Enterobacter lignolyticus SCF1. Front. Microbiol. 2013, 4, 280. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, F.; Luginbuhl, P.; Ivanova, N.; Ghassemian, M.; Richardson, T.H.; Stege, J.T.; Cayouette, M.; McHardy, A.C.; Djordjevic, G.; Aboushadi, N.; et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Kozaki, S.; Sakuranaga, M. Degradation of lignin compounds by bacteria from termite guts. Biotechnol. Lett. 1998, 20, 459–462. [Google Scholar] [CrossRef]
- Geib, S.M.; Filley, T.R.; Hatcher, P.G.; Hoover, K.; Carlson, J.E.; Jimenez-Gasco, M.D.; Nakagawa-Izumi, A.; Sleighter, R.L.; Tien, M. Lignin degradation in wood-feeding insects. Proc. Natl. Acad. Sci. USA 2008, 105, 12932–12937. [Google Scholar] [CrossRef] [PubMed]
- Strachan, C.R.; Singh, R.; VanInsberghe, D.; Ievdokymenko, K.; Budwill, K.; Mohn, W.W.; Eltis, L.D.; Hallam, S.J. Metagenomic scaffolds enable combinatorial lignin transformation. Proc. Natl. Acad. Sci. USA 2014, 111, 10143–10148. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Duenas, F.J.; Martinez, A.T. Microbial degradation of lignin: How a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microb. Biotechnol. 2009, 2, 164–177. [Google Scholar] [CrossRef] [PubMed]
- He, S.M.; Ivanova, N.; Kirton, E.; Allgaier, M.; Bergin, C.; Scheffrahn, R.H.; Kyrpides, N.C.; Warnecke, F.; Tringe, S.G.; Hugenholtz, P. Comparative metagenomic and metatranscriptomic analysis of hindgut paunch microbiota in wood- and dung-feeding higher termites. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, C.M.; Harmann, C.H.; Takasuka, T.E.; Hura, G.L.; Dyer, K.; Fox, B.G. Fusion of dioxygenase and lignin-binding domains in a novel secreted enzyme from cellulolytic Streptomyces sp. SirexAA-E. J. Biol. Chem. 2013, 288, 18574–18587. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Lukk, T.; Solbiati, J.O.; Bauer, S.; Nair, S.K.; Cronan, J.E.; Gerlt, J.A. Roles of small laccases from Streptomyces in lignin degradation. Biochemistry 2014, 53, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Heider, J. Adding handles to unhandy substrates: Anaerobic hydrocarbon activation mechanisms. Curr. Opin. Chem. Biol. 2007, 11, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Chakraborty, D. Biotechnological and molecular approaches for vanillin production: A review. Appl. Biochem. Biotechnol. 2013, 169, 1353–1372. [Google Scholar] [CrossRef] [PubMed]
- Walton, N.J.; Mayer, M.J.; Narbad, A. Molecules of interest—Vanillin. Phytochemistry 2003, 63, 505–515. [Google Scholar] [CrossRef]
- Jackson, M.A.; Compton, D.L.; Boateng, A.A. Screening heterogeneous catalysts for the pyrolysis of lignin. J. Anal. Appl. Pyrolysis 2009, 85, 226–230. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Catalytic Pyrolysis-GC/MS of lignin from several sources. Fuel Process. Technol. 2010, 91, 1446–1458. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Troussard, E.; van Bokhoven, J.A. Controlling the selectivity to chemicals from lignin via catalytic fast pyrolysis. Appl. Catal. A Gen. 2012, 423, 130–136. [Google Scholar] [CrossRef]
- Choi, H.S.; Meier, D.; Windt, M. Rapid screening of catalytic pyrolysis reactions of organosolv lignins with the vti-mini fast pyrolyzer. Environ. Prog. Sustain. Energy 2012, 31, 240–244. [Google Scholar] [CrossRef]
- Sharma, R.K.; Wooten, J.B.; Baliga, V.L.; Lin, X.H.; Chan, W.G.; Hajaligol, M.R. Characterization of chars from pyrolysis of lignin. Fuel 2004, 83, 1469–1482. [Google Scholar] [CrossRef]
- Meier, D.; Ante, R.; Faix, O. Catalytic hydropyrolysis of lignin: Influence of reaction conditions on the formation and composition of liquid products. Bioresour. Technol. 1992, 40, 171–177. [Google Scholar] [CrossRef]
- Meier, D.; Berns, J.; Grünwald, C.; Faix, O. Analytical pyrolysis and semicontinuous catalytic hydropyrolysis of organocell lignin. J. Anal. Appl. Pyrolysis 1993, 25, 335–347. [Google Scholar] [CrossRef]
- Balagurumurthy, B.; Oza, T.S.; Bhaskar, T.; Adhikari, D.K. Renewable hydrocarbons through biomass hydropyrolysis process: Challenges and opportunities. J. Mater. Cycles Waste Manag. 2013, 15, 9–15. [Google Scholar] [CrossRef]
- Linck, M.; Felix, L.; Marker, T.; Roberts, M. Integrated biomass hydropyrolysis and hydrotreating: A brief review. Wiley Interdiscip. Rev. Energy Environ. 2014, 3, 575–581. [Google Scholar] [CrossRef]
- Li, C.Z.; Zhao, X.C.; Wang, A.Q.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Saisu, M.; Sato, T.; Watanabe, M.; Adschiri, T.; Arai, K. Conversion of lignin with supercritical water-phenol mixtures. Energy Fuels 2003, 17, 922–928. [Google Scholar] [CrossRef]
- Matsumura, Y.; Sasaki, M.; Okuda, K.; Takami, S.; Ohara, S.; Umetsu, M.; Adschiri, T. Supercritical water treatment of biomass for energy and material recovery. Combust. Sci. Technol. 2006, 178, 509–536. [Google Scholar] [CrossRef]
- Okuda, K.; Man, X.; Umetsu, M.; Takami, S.; Adschiri, T. Efficient conversion of lignin into single chemical species by solvothermal reaction in water-p-cresol solvent. J. Phys. Condens. Matter 2004, 16, S1325–S1330. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Yagi, T.; Shinohara, S.; Fukunaga, T.; Nakasaka, Y.; Tago, T.; Masuda, T. Production of phenols from lignin via depolymerization and catalytic cracking. Fuel Process. Technol. 2013, 108, 69–75. [Google Scholar] [CrossRef]
- Roberts, V.M.; Stein, V.; Reiner, T.; Lemonidou, A.; Li, X.B.; Lercher, J.A. Towards quantitative catalytic lignin depolymerization. Chem. A Eur. J. 2011, 17, 5939–5948. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.E.; Evans, L.; Littlewolf, A.; Trudell, D.E. Batch microreactor studies of lignin and lignin model compound depolymerization by bases in alcohol solvents. Fuel 1999, 78, 1363–1366. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, Y.; Guo, Q.X. Catalytic hydrocracking of pyrolytic lignin to liquid fuel in supercritical ethanol. Ind. Eng. Chem. Res. 2010, 49, 2040–2046. [Google Scholar] [CrossRef]
- Nagy, M.; David, K.; Britovsek, G.J.P.; Ragauskas, A.J. Catalytic hydrogenolysis of ethanol organosolv lignin. Holzforschung 2009, 63, 513–520. [Google Scholar] [CrossRef]
- Cheng, S.N.; Wilks, C.; Yuan, Z.S.; Leitch, M.; Xu, C.B. Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water-ethanol co-solvent. Polym. Degrad. Stab. 2012, 97, 839–848. [Google Scholar] [CrossRef]
- Tsujino, J.; Kawamoto, H.; Saka, S. Reactivity of lignin in supercritical methanol studied with various lignin model compounds. Wood Sci. Technol. 2003, 37, 299–307. [Google Scholar] [CrossRef]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640–1647. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; Teunissen, W.; van Dam, J.E.G.; de Jong, E.; Gellerstedt, G.; Scott, E.L.; Sanders, J.P.M. Lignin depolymerisation in supercritical carbon dioxide/acetone/water fluid for the production of aromatic chemicals. Bioresour. Technol. 2012, 106, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Erdocia, X.; Prado, R.; Corcuera, M.A.; Labidi, J. Base catalyzed depolymerization of lignin: Influence of organosolv lignin nature. Biomass Bioenergy 2014, 66, 379–386. [Google Scholar] [CrossRef]
- Asmadi, M.; Kawamoto, H.; Saka, S. Pyrolysis reactions of Japanese cedar and Japanese beech woods in a closed ampoule reactor. J. Wood Sci. 2010, 56, 319–330. [Google Scholar] [CrossRef]
- Shen, D.K.; Liu, G.F.; Zhao, J.; Xue, J.T.; Guan, S.P.; Xiao, R. Thermo-chemical conversion of lignin to aromatic compounds: Effect of lignin source and reaction temperature. J. Anal. Appl. Pyrolysis 2015, 112, 56–65. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Understanding the fast pyrolysis of lignin. ChemSusChem 2011, 4, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.L.; Kim, K.H.; Brown, R.C.; Dalluge, E.; Hutchinson, C.; Lee, Y.J.; Dalluge, D. Formation of phenolic oligomers during fast pyrolysis of lignin. Fuel 2014, 128, 170–179. [Google Scholar] [CrossRef]
- Ben, H.X.; Ragauskas, A.J. NMR characterization of pyrolysis oils from kraft lignin. Energy Fuels 2011, 25, 2322–2332. [Google Scholar] [CrossRef]
- Chu, S.; Subrahmanyam, A.V.; Huber, G.W. The pyrolysis chemistry of a Beta-O-4 type oligomeric lignin model compound. Green Chem. 2013, 15, 125–136. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, S.; Bai, X. Role of hydrogen transfer during catalytic copyrolysis of lignin and tetralin over HZSM-5 and HY zeolite catalysts. ACS Sustain. Chem. Eng. 2016, 4, 4237–4250. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Xiao, R.; Wang, D.H.; He, G.Y.; Shao, S.S.; Zhang, J.B.; Zhong, Z.P. Biomass fast pyrolysis in a fluidized bed reactor under N2, CO2, CO, Ch4 and H2 atmospheres. Bioresour. Technol. 2011, 102, 4258–4264. [Google Scholar] [CrossRef] [PubMed]
- De Wild, P.; van der Laan, R.; Kloekhorst, A.; Heeres, E. Lignin valorisation for chemicals and (transportation) fuels via (catalytic) pyrolysis and hydrodeoxygenation. Environ. Prog. Sustain. Energy 2009, 28, 461–469. [Google Scholar] [CrossRef]
- Kloekhorst, A.; Heeres, H.J. Catalytic hydrotreatment of alcell lignin using supported Ru, Pd, and Cu catalysts. ACS Sustain. Chem. Eng. 2015, 3, 1905–1914. [Google Scholar] [CrossRef]
- Jan, O.; Marchand, R.; Anjos, L.C.A.; Seufitelli, G.V.S.; Nikolla, E.; Resende, F.L.P. Hydropyrolysis of lignin using Pd/HZSM-5. Energy Fuels 2015, 29, 1793–1800. [Google Scholar] [CrossRef]
- Ye, Y.Y.; Zhang, Y.; Fan, J.; Chang, J. Selective production of 4-ethylphenolics from lignin via mild hydrogenolysis. Bioresour. Technol. 2012, 118, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Brown, R.C.; Kieffer, M.; Bai, X.L. Hydrogen-donor-assisted solvent liquefaction of lignin to short-chain alkylphenols using a micro reactor/gas chromatography system. Energy Fuels 2014, 28, 6429–6437. [Google Scholar] [CrossRef]
- Kleinert, M.; Barth, T. Towards a lignincellulosic biorefinery: Direct one-step conversion of lignin to hydrogen-enriched biofuel. Energy Fuels 2008, 22, 1371–1379. [Google Scholar] [CrossRef]
- Xu, W.Y.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid. ChemSusChem 2012, 5, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Wang, F.; Cai, J.Y.; Wang, Y.H.; Zhang, J.J.; Yu, W.Q.; Xu, J. Lignin depolymerization (ldp) in alcohol over nickel-based catalysts via a fragmentation-hydrogenolysis process. Energy Environ. Sci. 2013, 6, 994–1007. [Google Scholar] [CrossRef]
- Vardon, D.R.; Franden, M.A.; Johnson, C.W.; Karp, E.M.; Guarnieri, M.T.; Linger, J.G.; Salm, M.J.; Strathmann, T.J.; Beckham, G.T. Adipic acid production from lignin. Energy Environ. Sci. 2015, 8, 617–628. [Google Scholar] [CrossRef]
- Salvachua, D.; Karp, E.M.; Nimlos, C.T.; Vardon, D.R.; Beckham, G.T. Towards lignin consolidated bioprocessing: Simultaneous lignin depolymerization and product generation by bacteria. Green Chem. 2015, 17, 4951–4967. [Google Scholar] [CrossRef]
- Linger, J.G.; Vardon, D.R.; Guarnieri, M.T.; Karp, E.M.; Hunsinger, G.B.; Franden, M.A.; Johnson, C.W.; Chupka, G.; Strathmann, T.J.; Pienkos, P.T.; et al. Lignin valorization through integrated biological funneling and chemical catalysis. Proc. Natl. Acad. Sci. USA 2014, 111, 12013–12018. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Steinbuchel, A.; Priefert, H. Highly efficient biotransformation of eugenol to ferulic acid and further conversion to vanillin in recombinant strains of Escherichia coli. Appl. Environ. Microbiol. 2003, 69, 6569–6576. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Seo, J.; Unno, T.; Ahn, J.H.; Yan, T.; Sadowsky, M.J.; Hur, H.G. Isoeugenol monooxygenase and its putative regulatory gene are located in the eugenol metabolic gene cluster in Pseudomonas nitroreducens Jin1. Arch. Microbiol. 2010, 192, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Plaggenborg, R.; Overhage, J.; Loos, A.; Archer, J.A.C.; Lessard, P.; Sinskey, A.J.; Steinbuchel, A.; Priefert, H. Potential of Rhodococcus strains for biotechnological vanillin production from ferulic acid and eugenol. Appl. Microbiol. Biotechnol. 2006, 72, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Steinbuchel, A.; Priefert, H. Harnessing eugenol as a substrate for production of aromatic compounds with recombinant strains of Amycolatopsis sp. hr167. J. Biotechnol. 2006, 125, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Priefert, H.; Rabenhorst, J.; Steinbuechel, A. Construction of Production Strains for Producing Substituted Phenols by Specifically Inactivating Genes of the Eugenol and Ferulic Acid Catabolism. Patent CA2348962 A1, 11 May 2000. [Google Scholar]
- Srivastava, S.; Luqman, S.; Khan, F.; Chanotiya, C.S.; Darokar, M.P. Metabolic pathway reconstruction of eugenol to vanillin bioconversion in Aspergillus niger. Bioinformation 2010, 4, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Ashengroph, M.; Nahvi, I.; Zarkesh-Esfahani, H.; Momenbeik, F. Conversion of isoeugenol to vanillin by Psychrobacter sp. Strain CSW4. Appl. Biochem. Biotechnol. 2012, 166, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shimoni, E.; Ravid, U.; Shoham, Y. Isolation of a Bacillus sp capable of transforming isoeugenol to vanillin. J. Biotechnol. 2000, 78, 1–9. [Google Scholar] [CrossRef]
- Furukawa, H.; Morita, H.; Yoshida, T.; Nagasawa, T. Conversion of isoeugenol into vanillic acid by Pseudomonas putida 158 cells exhibiting high isoeugenol-degrading activity. J. Biosci. Bioeng. 2003, 96, 401–403. [Google Scholar] [CrossRef]
- Shimoni, E.; Baasov, T.; Ravid, U.; Shoham, Y. Biotransformations of propenylbenzenes by an Arthrobacter sp. and its t-anethole blocked mutants. J. Biotechnol. 2003, 105, 61–70. [Google Scholar] [CrossRef]
- Zhao, L.Q.; Sun, Z.H.; Zheng, P.; Zhu, L.L. Biotransformation of isoeugenol to vanillin by a novel strain of Bacillus fusiformis. Biotechnol. Lett. 2005, 27, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Xu, P.; Han, S.; Yan, H.Q.; Ma, C.Q. Metabolism of isoeugenol via isoeugenol-diol by a newly isolated strain of Bacillus subtilis hs8. Appl. Microbiol. Biotechnol. 2006, 73, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Q.; Sun, Z.H.; Zheng, P.; He, J.Y. Biotransformation of isoeugenol to vanillin by Bacillus fusiformis CGMCC1347 with the addition of resin HD-8. Process Biochem. 2006, 41, 1673–1676. [Google Scholar] [CrossRef]
- Kasana, R.C.; Sharma, U.K.; Sharma, N.; Sinha, A.K. Isolation and identification of a novel strain of Pseudomonas chlororaphis capable of transforming isoeugenol to vanillin. Curr. Microbiol. 2007, 54, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.L.; Ma, C.Q.; Lin, S.; Song, L.F.; Deng, Z.X.; Maomy, Z.R.; Zhang, Z.B.; Yu, B.; Xu, P. Biotransformation of isoeugenol to vanillin by a newly isolated Bacillus pumilus strain: Identification of major metabolites. J. Biotechnol. 2007, 130, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Okada, Y.; Yoshida, T.; Nagasawa, T. Biotransformation of isoeugenol to vanillin by Pseudomonas putida IE27 cells. Appl. Microbiol. Biotechnol. 2007, 73, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Unno, T.; Kim, S.J.; Kanaly, R.A.; Ahn, J.H.; Kang, S.I.; Hur, H.G. Metabolic characterization of newly isolated Pseudomonas nitroreducens Jin1 growing on eugenol and isoeugenol. J. Agric. Food Chem. 2007, 55, 8556–8561. [Google Scholar] [CrossRef] [PubMed]
- Ashengroph, M.; Nahvi, I.; Zarkesh-Esfahani, H.; Momenbeik, F. Candida galli strain PGO6: A novel isolated yeast strain capable of transformation of isoeugenol into vanillin and vanillic acid. Curr. Microbiol. 2011, 62, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Achterholt, S.; Priefert, H.; Steinbuchel, A. Identification of Amycolatopsis sp strain hr167 genes, involved in the bioconversion of ferulic acid to vanillin. Appl. Microbiol. Biotechnol. 2000, 54, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Plaggenborg, R.; Overhage, J.; Steinbuchel, A.; Priefert, H. Functional analyses of genes involved in the metabolism of ferulic acid in Pseudomonas putida KT2440. Appl. Microbiol. Biotechnol. 2003, 61, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Li, C.; Lee, Y.M.; Lee, S.H.; Kim, S.H.; Choi, M.S.; Seo, W.T.; Yang, J.K.; Kim, J.Y.; Kim, S.W. Production of vanillin from ferulic acid using recombinant strains of Escherichia coli. Biotechnol. Bioprocess Eng. 2005, 10, 378–384. [Google Scholar] [CrossRef]
- Cheetham, P.S.J.; Gradley, M.L.; Sime, J.T. Flavour/Aroma Materials and Their Preparation. Patent WO2000050622 A1, 31 August 2000. [Google Scholar]
- Alvarado, I.E.; Lomascolo, A.; Navarro, D.; Delattre, M.; Asther, M.; Lesage-Meessen, L. Evidence of a new biotransformation pathway of p-coumaric acid into p-hydroxybenzaldehyde in Pycnoporus cinnabarinus. Appl. Microbiol. Biotechnol. 2001, 57, 725–730. [Google Scholar]
- Lesage-Meessen, L.; Lomascolo, A.; Bonnin, E.; Thibault, J.F.; Buleon, A.; Roller, M.; Asther, M.; Record, E.; Ceccaldi, B.C. A biotechnological process involving filamentous fungi to produce natural crystalline vanillin from maize bran. Appl. Biochem. Biotechnol. 2002, 102, 141–153. [Google Scholar] [CrossRef]
- Agrawal, R.; Seetharam, Y.N.; Kelamani, R.C.; Jyothishwaran, G. Biotransformation of ferulic acid to vanillin by locally isolated bacterial cultures. Indian J. Biotechnol. 2003, 2, 610–612. [Google Scholar]
- Brunati, M.; Marinelli, F.; Bertolini, C.; Gandolfi, R.; Daffonchio, D.; Molinari, F. Biotransformations of cinnamic and ferulic acid with actinomycetes. Enzym. Microb. Technol. 2004, 34, 3–9. [Google Scholar] [CrossRef]
- Torre, P.; de Faveri, D.; Perego, P.; Ruzzi, M.; Barghini, P.; Gandolfi, R.; Converti, A. Bioconversion of ferulate into vanillin by Escherichia coli strain JM109/pBB1 in an immobilized-cell reactor. Ann. Microbiol. 2004, 54, 517–527. [Google Scholar]
- Martinez-Cuesta, M.D.; Payne, J.; Hanniffy, S.B.; Gasson, M.J.; Narbad, A. Functional analysis of the vanillin pathway in a vdh-negative mutant strain of Pseudomonas fluorescens AN103. Enzym. Microb. Technol. 2005, 37, 131–138. [Google Scholar] [CrossRef]
- Bloem, A.; Bertrand, A.; Lonvaud-Funel, A.; de Revel, G. Vanillin production from simple phenols by wine-associated lactic acid bacteria. Lett. Appl. Microbiol. 2007, 44, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.L.; Ma, C.Q.; Song, L.F.; Lin, S.; Zhang, Z.B.; Deng, Z.X.; Xu, P. Enhanced vanillin production from ferulic acid using adsorbent resin. Appl. Microbiol. Biotechnol. 2007, 74, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Barghini, P.; Di Gioia, D.; Fava, F.; Ruzzi, M. Vanillin production using metabolically engineered Escherichia coli under non-growing conditions. Microb. Cell Factories 2007, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Yoon, S.H.; Das, A.; Lee, S.H.; Li, C.; Kim, J.Y.; Choi, M.S.; Oh, D.K.; Kim, S.W. Directing vanillin production from ferulic acid by increased acetyl-CoA consumption in recombinant Escherichia coli. Biotechnol. Bioeng. 2009, 102, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Calisti, C.; Ficca, A.G.; Barghini, P.; Ruzzi, M. Regulation of ferulic catabolic genes in Pseudomonas fluorescens BF13: Involvement of a MarR family regulator. Appl. Microbiol. Biotechnol. 2008, 80, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Ruzzi, M.; Luziatelli, F. Genetic engineering of Escherichia coli to enhance biological production of vanillin from ferulic acid. Bull. Univ. Agric. Sci. Vet. Med. Cluj-Napoca Anim. Sci. Biotechnol. 2008, 65, 4–8. [Google Scholar]
- Sarangi, P.K.; Sahoo, H.P. Enhancing the rate of ferulic acid bioconversion utilizing glucose as carbon source. Science 2010, 6, 115–117. [Google Scholar]
- Tilay, A.; Bule, M.; Annapure, U. Production of biovanillin by one-step biotransformation using fungus Pycnoporous cinnabarinus. J. Agric. Food Chem. 2010, 58, 4401–4405. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, E.D.; Perrone, D.; Vendramini, A.L.D.; Leite, S.G.F. Vanillin production by Phanerochaete chrysosorium grown on green coconut agro-industrial husk in solid state fermentation. Bioresources 2008, 3, 1042–1050. [Google Scholar]
- Macfarlane, A.L.; Mai, M.; Kadla, J.F. Bio-based chemicals from biorefining: Lignin conversion and utilisation. In Advances in Biorefineries: Biomass and Waste Supply Chain Management; Woodhead Publishing: Cambridge, UK, 2014; pp. 659–692. [Google Scholar]
- Ouyang, X.P.; Zhu, G.D.; Huang, X.Z.; Qiu, X.Q. Microwave assisted liquefaction of wheat straw alkali lignin for the production of monophenolic compounds. J. Energy Chem. 2015, 24, 72–76. [Google Scholar] [CrossRef]
- Lavoie, J.M.; Bare, W.; Bilodeau, M. Depolymerization of steam-treated lignin for the production of green chemicals. Bioresour. Technol. 2011, 102, 4917–4920. [Google Scholar] [CrossRef] [PubMed]
- Bridgwater, A.V.; Meier, D.; Radlein, D. An overview of fast pyrolysis of biomass. Org. Geochem. 1999, 30, 1479–1493. [Google Scholar] [CrossRef]
- Liu, C.; Hu, J.; Zhang, H.Y.; Xiao, R. Thermal conversion of lignin to phenols: Relevance between chemical structure and pyrolysis behaviors. Fuel 2016, 182, 864–870. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Kuhire, S.S.; Avadhani, C.V.; Wadgaonkar, P.P. New poly(ether urethane)s based on lignin derived aromatic chemicals via a-b monomer approach: Synthesis and characterization. Eur. Polym. J. 2015, 71, 547–557. [Google Scholar] [CrossRef]
- Laurichesse, S.; Averous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Ten, E.; Vermerris, W. Recent developments in polymers derived from industrial lignin. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Harvey, B.G.; Guenthner, A.J.; Lai, W.W.; Meylemans, H.A.; Davis, M.C.; Cambrea, L.R.; Reams, J.T.; Lamison, K.R. Effects of o-methoxy groups on the properties and thermal stability of renewable high-temperature cyanate ester resins. Macromolecules 2015, 48, 3173–3179. [Google Scholar] [CrossRef]
- Meylemans, H.A.; Groshens, T.J.; Harvey, B.G. Synthesis of renewable bisphenols from creosol. ChemSusChem 2012, 5, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Pion, F.; Ducrot, P.H.; Allais, F. Renewable alternating aliphatic-aromatic copolyesters derived from biobased ferulic acid, diols, and diacids: Sustainable polymers with tunable thermal properties. Macromol. Chem. Phys. 2014, 215, 431–439. [Google Scholar] [CrossRef]
- Firdaus, M.; Meier, M.A.R. Renewable co-polymers derived from vanillin and fatty acid derivatives. Eur. Polym. J. 2013, 49, 156–166. [Google Scholar] [CrossRef]
- Mialon, L.; Vanderhenst, R.; Pemba, A.G.; Miller, S.A. Polyalkylenehydroxybenzoates (pahbs): Biorenewable aromatic/aliphatic polyesters from lignin. Macromol. Rapid Commun. 2011, 32, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Mialon, L.; Pemba, A.G.; Miller, S.A. Biorenewable polyethylene terephthalate mimics derived from lignin and acetic acid. Green Chem. 2010, 12, 1704–1706. [Google Scholar] [CrossRef]
- Aouf, C.; Lecomte, J.; Villeneuve, P.; Dubreucq, E.; Fulcrand, H. Chemo-enzymatic functionalization of gallic and vanillic acids: Synthesis of bio-based epoxy resins prepolymers. Green Chem. 2012, 14, 2328–2336. [Google Scholar] [CrossRef]
- Fache, M.; Boutevin, B.; Caillol, S. Vanillin, a key-intermediate of biobased polymers. Eur. Polym. J. 2015, 68, 488–502. [Google Scholar] [CrossRef]
- Fache, M.; Darroman, E.; Besse, V.; Auvergne, R.; Caillol, S.; Boutevin, B. Vanillin, a promising biobased building-block for monomer synthesis. Green Chem. 2014, 16, 1987–1998. [Google Scholar] [CrossRef]
- Thring, R.W.; Breau, J. Hydrocracking of solvolysis lignin in a batch reactor. Fuel 1996, 75, 795–800. [Google Scholar] [CrossRef]
- Sharma, R.K.; Bakhshi, N.N. Upgrading of wood-derived bio-oil over HZSM-5. Bioresour. Technol. 1991, 35, 57–66. [Google Scholar] [CrossRef]
- Sharma, R.K.; Bakhshi, N.N. Catalytic upgrading of fast pyrolysis oil over HZSM-5. Can. J. Chem. Eng. 1993, 71, 383–391. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakhshi, N.N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil.1. Conversion over various catalysts. Fuel Process. Technol. 1995, 45, 161–183. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Bakhshi, N.N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil.2. Comparative catalyst performance and reaction pathways. Fuel Process. Technol. 1995, 45, 185–202. [Google Scholar] [CrossRef]
- Chantal, P.; Kaliaguine, S.; Grandmaison, J.L.; Mahay, A. Production of hydrocarbons from aspen poplar pyrolytic oils over HZSM-5. Appl. Catal. 1984, 10, 317–332. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Aguado, R.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols. Ind. Eng. Chem. Res. 2004, 43, 2610–2618. [Google Scholar] [CrossRef]
- Sheu, Y.H.E.; Anthony, R.G.; Soltes, E.J. Kinetic-studies of upgrading pine pyrolytic oil by hydrotreatment. Fuel Process. Technol. 1988, 19, 31–50. [Google Scholar] [CrossRef]
- Bredenberg, J.B.S.; Huuska, M.; Raty, J.; Korpio, M. Hydrogenolysis and hydrocracking of the carbon oxygen bond 1. Hydrocracking of some simple aromatic o-compounds. J. Catal. 1982, 77, 242–247. [Google Scholar] [CrossRef]
- Gevert, B.S.; Otterstedt, J.E.; Massoth, F.E. Kinetics of the HDO of methyl-substituted phenols. Appl. Catal. 1987, 31, 119–131. [Google Scholar] [CrossRef]
- Hurff, S.J.; Klein, M.T. Reaction pathway analysis of thermal and catalytic lignin fragmentation by use of model compounds. Ind. Eng. Chem. Fundam. 1983, 22, 426–430. [Google Scholar] [CrossRef]
- Kallury, R.; Restivo, W.M.; Tidwell, T.T.; Boocock, D.G.B.; Crimi, A.; Douglas, J. Hydrodeoxygenation of hydroxy, methoxy, and methyl phenols with molybdenum oxide nickel-oxide alumina catalyst. J. Catal. 1985, 96, 535–543. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Study of the hydrodeoxygenation of carbonyl, carboxylic and guaiacyl groups over sulfided como/gamma-Al2O3 and nimo/gamma-Al2O3 catalyst.2. Influence of water, ammonia and hydrogen-sulfide. Appl. Catal. A Gen. 1994, 109, 97–115. [Google Scholar] [CrossRef]
- De la Puente, G.; Gil, A.; Pis, J.J.; Grange, P. Effects of support surface chemistry in hydrodeoxygenation reactions over como/activated carbon sulfided catalysts. Langmuir 1999, 15, 5800–5806. [Google Scholar] [CrossRef]
- Petrocelli, F.P.; Klein, M.T. Chemical modeling analysis of the yields of single-ring phenolics from lignin liquefaction. Ind. Eng. Chem. Prod. Res. Dev. 1985, 24, 635–641. [Google Scholar] [CrossRef]
- Koyama, M. Hydrocracking of lignin-related model dimers. Bioresour. Technol. 1993, 44, 209–215. [Google Scholar] [CrossRef]
- Ratcliff, M.A.; Johnson, D.K.; Posey, F.L.; Chum, H.L. Hydrodeoxygenation of lignins and model compounds. Appl. Biochem. Biotechnol. 1988, 17, 151–160. [Google Scholar] [CrossRef]
- Yakovlev, V.A.; Khromova, S.A.; Sherstyuk, O.V.; Dundich, V.O.; Ermakov, D.Y.; Novopashina, V.M.; Lebedev, M.Y.; Bulavchenko, O.; Parmon, V.N. Development of new catalytic systems for upgraded bio-fuels production from bio-crude-oil and biodiesel. Catal. Today 2009, 144, 362–366. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical developments in hydroprocessing bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Zhao, C.; Kou, Y.; Lemonidou, A.A.; Li, X.; Lercher, J.A. Highly selective catalytic conversion of phenolic bio-oil to alkanes. Angew. Chem. Int. Ed. 2009, 48, 3987–3990. [Google Scholar] [CrossRef] [PubMed]
- Filley, J.; Roth, C. Vanadium catalyzed guaiacol deoxygenation. J. Mol. Catal. A Chem. 1999, 139, 245–252. [Google Scholar] [CrossRef]
- Dabo, P.; Cyr, A.; Lessard, J.; Brossard, L.; Menard, H. Electrocatalytic hydrogenation of 4-phenoxyphenol on active powders highly dispersed in a reticulated vitreous carbon electrode. Can. J. Chem./Rev. Can. Chim. 1999, 77, 1225–1229. [Google Scholar] [CrossRef]
- Hu, T.Q.; James, B.R.; Rettig, S.J.; Lee, C.L. Stereoselective hydrogenation of lignin degradation model compounds. Can. J. Chem./Revue Canadienne Chimie 1997, 75, 1234–1239. [Google Scholar] [CrossRef]
- Crestini, C.; Pro, P.; Neri, V.; Saladino, R. Methyltrioxorhenium: A new catalyst for the activation of hydrogen peroxide to the oxidation of lignin and lignin model compounds. Bioorg. Med. Chem. 2005, 13, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Crestini, C.; Caponi, M.C.; Argyropoulos, D.S.; Saladino, R. Immobilized methyltrioxo rhenium (mto)/H2O2 systems for the oxidation of lignin and lignin model compounds. Bioorg. Med. Chem. 2006, 14, 5292–5302. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.A.; Weskamp, T.; Zoller, J.P.; Fischer, R.W. Methyltrioxorhenium: Oxidative cleavage of cc-double bonds and its application in a highly efficient synthesis of vanillin from biological waste. J. Mol. Catal. A Chem. 2000, 153, 49–52. [Google Scholar] [CrossRef]
- Bhargava, S.; Jani, H.; Tardio, J.; Akolekar, D.; Hoang, M. Catalytic wet oxidation of ferulic acid (a model lignin compound) using heterogeneous copper catalysts. Ind. Eng. Chem. Res. 2007, 46, 8652–8656. [Google Scholar] [CrossRef]
- Pardini, V.L.; Vargas, R.R.; Viertler, H.; Utley, J.H.P. Anodic cleavage of lignin model dimers in methanol. Tetrahedron 1992, 48, 7221–7228. [Google Scholar] [CrossRef]
- Habe, T.; Shimada, M.; Higuchi, T. Biomimetic approach to lignin degradation.1. H2O2-dependent c-c bond-cleavage of the lignin model compounds with a natural iron porphyrin and imidazole complex. Mokuzai Gakkaishi 1985, 31, 54–55. [Google Scholar]
- Crestini, C.; Pastorini, A.; Tagliatesta, P. Metalloporphyrins immobilized on motmorillonite as biomimetic catalysts in the oxidation of lignin model compounds. J. Mol. Catal. A Chem. 2004, 208, 195–202. [Google Scholar] [CrossRef]
- Amaral Labat, G.A.; Goncalves, A.R. Oxidation in acidic medium of lignins from agricultural residues. Appl. Biochem. Biotechnol. 2008, 148, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.M.; Ford, W.T. Oxidation of lignin model compounds in water with dioxygen and hydrogen-peroxide catalyzed by metallophthalocyanines. J. Mol. Catal. 1993, 78, 367–378. [Google Scholar] [CrossRef]
- Artaud, I.; Benaziza, K.; Mansuy, D. Iron porphyrin-catalyzed oxidation of 1,2-dimethoxyarenes—A discussion of the different reactions involved and the competition between the formation of methoxyquinones or muconic dimethyl esters. J. Org. Chem. 1993, 58, 3373–3380. [Google Scholar] [CrossRef]
- Robinson, M.J.; Wright, L.J.; Suckling, I.D. Fe(tspc)-catalysed benzylic oxidation and subsequent dealkylation of a non-phenolic lignin model. J. Wood Chem. Technol. 2000, 20, 357–373. [Google Scholar] [CrossRef]
- Dicosimo, R.; Szabo, H.C. Oxidation of lignin model compounds using single-electron-transfer catalysts. J. Org. Chem. 1988, 53, 1673–1679. [Google Scholar] [CrossRef]
- Hocking, M. Vanillin: Synthetic flavoring from spent sulfite liquor. J. Chem. Educ. 1997, 74, 1055. [Google Scholar] [CrossRef]
- Davis, R.; Tao, L.; Tan, E.C.D.; Biddy, M.J.; Beckham, G.T.; Scarlata, C.; Jacobson, J.; Cafferty, K.; Ross, J.; Lukas, J.; et al. Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbons: Dilute-Acid and Enzymatic Deconstruction of Biomass to Sugars and Biological Conversion of Sugars to Hydrocarbons; National Renewable Energy Laboratory: Golden, CO, USA, 2013.
- Kautto, J.; Realff, M.J.; Ragauskas, A.J.; Kassi, T. Economic analysis of an organosolv process for bioethanol production. Bioresources 2014, 9, 6041–6072. [Google Scholar] [CrossRef]
- Oleskowicz-Popiel, P.; Klein-Marcuschamer, D.; Simmons, B.A.; Blanch, H.W. Lignocellulosic ethanol production without enzymes—Technoeconomic analysis of ionic liquid pretreatment followed by acidolysis. Bioresour. Technol. 2014, 158, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fu, X. Industrial technologies for bioethanol production from lignocellulosic biomass. Renew. Sustain. Energy Rev. 2016, 57, 468–478. [Google Scholar] [CrossRef]
- Petrou, E.C.; Pappis, C.P. Sustainability of systems producing ethanol, power, and lignosulfonates or lignin from corn stover: A comparative assessment. ACS Sustain. Chem. Eng. 2014, 2, 2527–2535. [Google Scholar] [CrossRef]
- Pourhashem, G.; Adler, P.R.; McAloon, A.J.; Spatari, S. Cost and greenhouse gas emission tradeoffs of alternative uses of lignin for second generation ethanol. Environ. Res. Lett. 2013, 8, 025021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
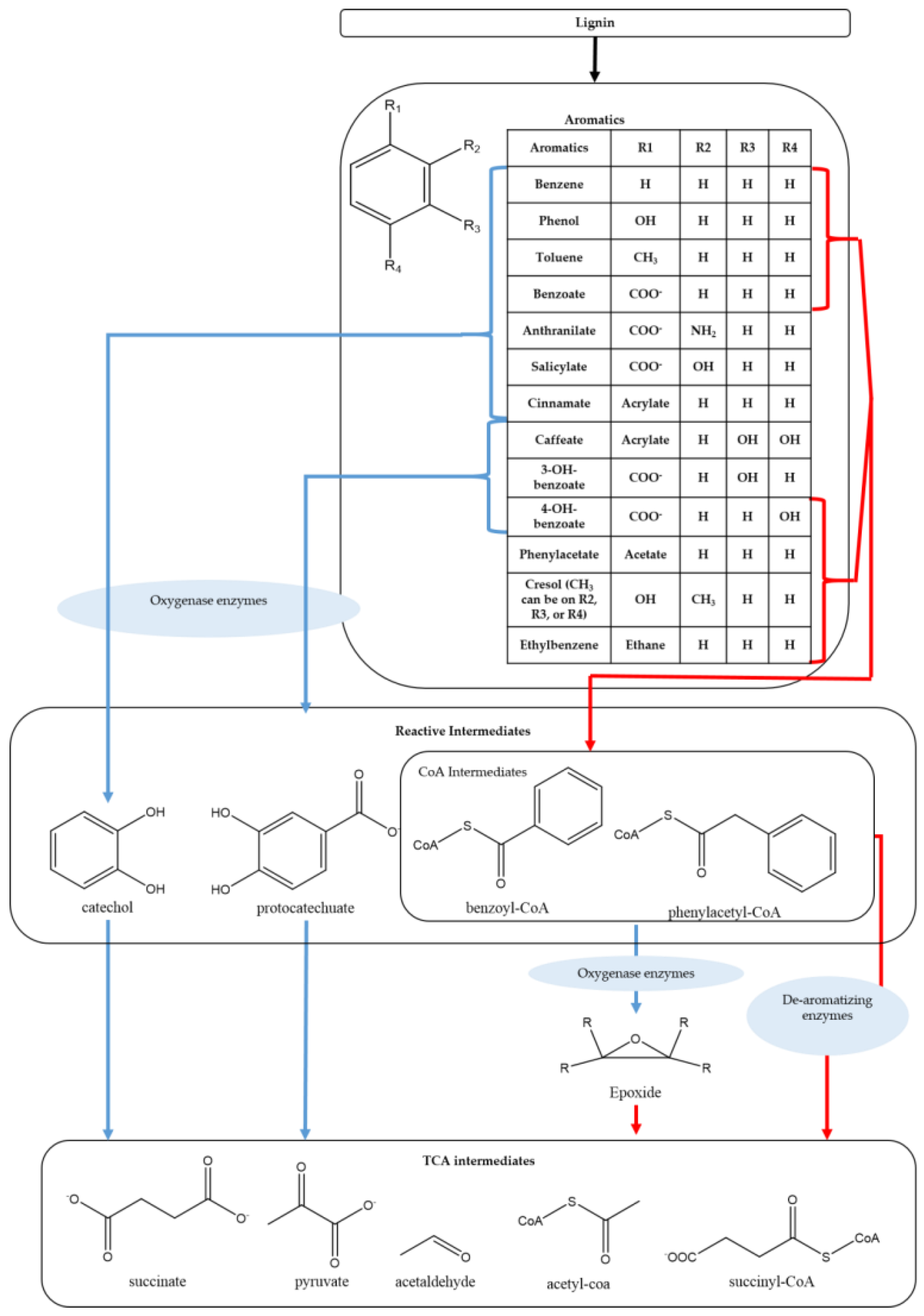
| Biomass Category | Biomass Type | Lignin Content (wt%) |
|---|---|---|
| Softwood | Pine | 28 [14] |
| Hardwood | Poplar | 21–27 [15] |
| Eucalyptus | 29–32 [16] | |
| Herbaceous | Miscanthus | 9–13 [17] |
| Switchgrass | 17–18 [18] | |
| Corn Stover | 18 [19] | |
| Bagasse | 20 [20] |
| Recovery Methods | Benefits | Challenges | Products |
|---|---|---|---|
| Kraft [23] and sulfite pulping [21] | Well-developed | Harsh chemicals | Cellulose, hemicellulose/lignin |
| Sulfur free alkali (soda) pulping [24] | Sulfur-free | Lower lignin removal rate | Solid polysaccharides, lignin-rich liquid |
| Organosolv pulping [25] | Sulfur-free | Has not been adapted to production scale | Varies by process, some organosolv processes can essentially isolate cellulose, hemicellulose, and lignin |
| Fast pyrolysis [26] | Fast | Undesired char formation | Solid (bio-char), Liquid (bio-oil), and gas |
| Dilute acid hydrolysis [27] | Highly advanced | Solid product is acid insoluble | Monomeric sugars, Biofine ligneous char (high heating value) |
| Hydrothermal Fractionation [28,29] | High product selectivity, produces monomeric products | Separation of hydrogen catalyst from the wood residue is challenging | Aromatic monomers, hydrolyzed hemicellulose |
| Biphasic fractionation [30] | Lower temperatures, near atmospheric pressure | Toxic solvents used in some cases | Hemicellulose degradation products (such as C5 oligomers, furfural), Cellulose solid, and lignin fragments |
| Enzyme | Function |
|---|---|
| Ligninolytic peroxidase (LiP) | Oxidizes molecules with high redox potential, including moderately activated non-phenolic aromatics (up to 90% of lignin polymer) [5,56,57] |
| Manganese-dependent peroxidase (MnP) | Oxidizes phenolic compounds [5,58] |
| Versatile peroxidase (VP) | Oxidizes both non-phenolic and phenolic compounds [5,59] |
| Dye-decolorizing peroxidase (DyP) | Oxidizes hydroxyl-free antraquinone and peroxidase substrates [55] |
| Lacasse | Oxidize aromatics and phenols, take action on smaller molecules in lignin such as ABTS and HBT in order to oxidize non-phenolic aromatics [54,62,63,64] |
| Recovery Methods | Benefits | Challenges | Products |
|---|---|---|---|
| Pyrolysis of isolated lignin [109,110,111,112,113] | Simple process | Selectivity for specific aromatic compounds is very low; char formation | Aromatic and non-aromatic molecules, char, and light gasses |
| Catalytic pyrolysis [114,115,116,117,118] | Products are less oxygenated and more stable | Coke deposits on catalysts | Aromatic hydrocarbon containing liquid, char, coke, light hydrocarbons, and oxygenate gasses |
| Supercritical water [119,120,121,122,123] | Lower concentration of lignin means lower chance of condensation reactions | High cost for process heat; only one-third of lignin product is low molecular weight | Aromatic hydrocarbon containing liquid, char |
| Supercritical solvent [124,125,126,127,128,129,130] | Products have a lower boiling point allowing for easier separation | Mid-high pressure High temperature | Primary product is monomeric substituted cyclohexyl derivatives, negligible aromatics, little to no char |
| Base-catalyzed depolymerization [12,131] | Oil contains low molecular weight species | Produces around 20% (wt/wt) desired oil product compared to the total weight of the products (oil, residual lignin, and coke) | Coke (undesired), oil (desired) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, K.M.; Rover, M.; Brown, R.C.; Bai, X.; Wen, Z.; Jarboe, L.R. Recovery and Utilization of Lignin Monomers as Part of the Biorefinery Approach. Energies 2016, 9, 808. https://doi.org/10.3390/en9100808
Davis KM, Rover M, Brown RC, Bai X, Wen Z, Jarboe LR. Recovery and Utilization of Lignin Monomers as Part of the Biorefinery Approach. Energies. 2016; 9(10):808. https://doi.org/10.3390/en9100808
Chicago/Turabian StyleDavis, Kirsten M., Marjorie Rover, Robert C. Brown, Xianglan Bai, Zhiyou Wen, and Laura R. Jarboe. 2016. "Recovery and Utilization of Lignin Monomers as Part of the Biorefinery Approach" Energies 9, no. 10: 808. https://doi.org/10.3390/en9100808
APA StyleDavis, K. M., Rover, M., Brown, R. C., Bai, X., Wen, Z., & Jarboe, L. R. (2016). Recovery and Utilization of Lignin Monomers as Part of the Biorefinery Approach. Energies, 9(10), 808. https://doi.org/10.3390/en9100808