Abstract
The innovation of energy storage technology and its solutions for energetic materials is an important direction in the current energy technology field. Hence, series of tetrazole-based ultra-high-energy-density high-nitrogen heterocyclic power compounds were designed and their energy characteristics and safety performances were evaluated by density functional theory (DFT). The results indicate that the type, number, and position of substituents have a significant effect on the comprehensive performance of these compounds. Research on electronic features shows that mono-substituents on the N atom connecting two tetrazole rings, substituents with more H atoms on the tetrazole ring, and less energetic substituents are beneficial for the stability of compounds. The discussion on energy characteristics and safety performance indicates that compounds B1(N-(1-nitro-1H-tetrazol-5-yl)-N-(1H-tetrazol-5-yl)nitramide), B7(N’-(1-nitro-1H-tetrazol-5-yl)-N’-(1H-tetrazol-5-yl)nitric hydrazide), B8(N-(1-(nitroamino)-1H-tetrazol-5-yl)-N-(1H-tetrazol-5-yl)nitramide), C1(5,5′-(hydrazine-1,1-diyl)bis(1-nitro-1H-tetrazole)), C4(N,N-bis(1-nitro-1H-tetrazol-5-yl)nitramide), and C6(N-(1-amino-1H-tetrazol-5-yl)-N-(1-nitro-1H-tetrazol-5-yl)nitramide) possess outstanding comprehensive performance concerning density, heat of formation, detonation heat, detonation velocity and pressure, oxygen balance, and impact sensitivity, and can be screened as candidates for high-energy-density compounds. The results are expected to provide new solutions for the innovation and progress of energy storage technologies in the energetic materials field.
1. Introduction
The advancement in energy storage technology is changing the global energy landscape today [1]. With the increasing emphasis on environmental protection and the demand for sustainable development, innovation in energy storage technology has become increasingly important, and innovation in energy storage solutions is an important direction in the current energy technology field [2,3,4,5,6,7]. Energetic materials are a special type of energy storage materials that rapidly undergo chemical reactions and release large amounts of energy to achieve destruction under specific external stimuli and energy excitations, which are widely used in various fields such as national defense, aerospace, and mining [8,9,10,11].
Designing new or improving old high-energy-density materials to achieve a good balance between their energy characteristics and safety performance is a focus of attention [12,13]. However, the conflicting demands for high energy and low impact sensitivity make the development of new high-energy-density materials a challenging problem. In the past few decades, many excellent energetic compounds have been discovered and widely used, such as 2,4,6-trinitrotoluene (TNT), 1,1-diamino-2,2-dinitroethylene (FOX-7), and so on [14,15,16]. Given the increasing demand for high-energy-density materials, it is evident that there is a need to continue designing and developing new candidate materials with good energy characteristics and acceptable safety performances. Molecular design utilizes relevant theoretical calculation methods to optimize the structure and predict the detonation performance of compounds, and summarizes the influence of different structures on the properties of energetic materials based on the obtained data, making new contributions to nitrogen-rich heterocyclic compound energy storage technology.
High-nitrogen heterocyclic compounds and their derivatives are important components of energetic materials, and they have become a research hotspot as a promising high-energy-density material. Among them, azole compounds have attracted widespread attention [17,18,19,20,21,22,23]. These high-nitrogen compounds generally have high heat of formation, mainly due to the differences in average bonding energy between a large number of different types of N-N and C-N bonds in the molecule [24]. Meanwhile, the proportion of carbon and hydrogen in their structure is relatively low, making it easier for them to achieve good oxygen balance. Moreover, these high-nitrogen compounds mainly produce nitrogen gas during the reaction process, which will not cause pollution to the environment and meets the requirements of environmental protection and sustainable development in new energy storage technologies [25,26,27].
High-nitrogen heterocyclic structures are mostly insensitive and have high thermal stability. In addition, their nitrogen content is usually higher, which means they have a higher heat of formation than ordinary energetic compounds. Therefore, in order to obtain a high-energy-density compound with excellent detonation performances, multiple high-nitrogen heterocyclic structures can be combined together. On the one hand, it can preserve the advantages of each constituent structure. On the other hand, the physical and chemical properties and detonation performances of the compound can be adjusted to a certain extent by modifying various substituents.
Based on the above discussions, we focus on the heterocyclic structure of tetrazole, with the core idea of constructing symmetrical bicyclic molecules and introducing various excellent energetic groups that help improve the overall properties of the molecule. We propose different series of molecular structure designs for energetic compounds composed of multiple tetrazole rings centered on nitrogen atoms. Based on density functional theory (DFT) and ab initio methods, a systematic study was conducted on their electronic structure, energy characteristics, and safety performance, which provides new solutions for the innovation and progress of energy storage technologies in the energetic materials field.
2. Computational Methods
The molecular structures of the designed power compounds were fully optimized by the DFT-B3LYP method, which was proven to predict the properties of many organic compounds successfully, and a 6-311G** base set [28,29], as implemented by the Gaussian 09 package [30], and subsequent analysis was performed by Multiwfn 3.8 [31]. The vibrational analysis results show that there were no imaginary frequencies, indicating stable configurations on their potential energy surfaces. The heat of formation (HOF, ΔfH298K(s)) of various compounds at room temperature was calculated by isodesmic reaction and atomization reaction methods, and the relevant equations are as follows [32]:
where X means the molecular skeleton; R means the substituted groups; and , , and mean the differences in total energy, zero-point energies (ZPEs), and thermal correction between products and reactants, respectively. HT was calculated by an executable file named T.exe, and the scaling for HT was 0.96. The theoretical density was obtained by the volume enclosed by a surface with an electron density of 0.001 e/Bohr3 based on the Monte Carlo method, and the Kamlet–Jacobs (K-J) equations were used to calculate the detonation velocity (D) and detonation pressure (P), which is suitable for predicting detonation properties of CHON system explosives. The equations are given as follows [33,34]:
where N means the moles of detonation gases per gram of compound, means the average molecular weight of gases, Q means the heat of detonation, and ρ means the density of the compounds. The Q was calculated by several empirical formulas determined by the inequality relationship between the number of C, H, and O atoms.
The safety performances of these power compounds can be evaluated by impact sensitivity (h50, cm), as shown in the following equation [35]:
where means the strengths of positive surface potentials, ν shows the equilibrium between positive potential and negative potential on an equipotential surface, and the coefficients α, β, and γ are −0.0064, 241.42, and −3.43, respectively.
3. Results and Discussion
3.1. Design Mentality
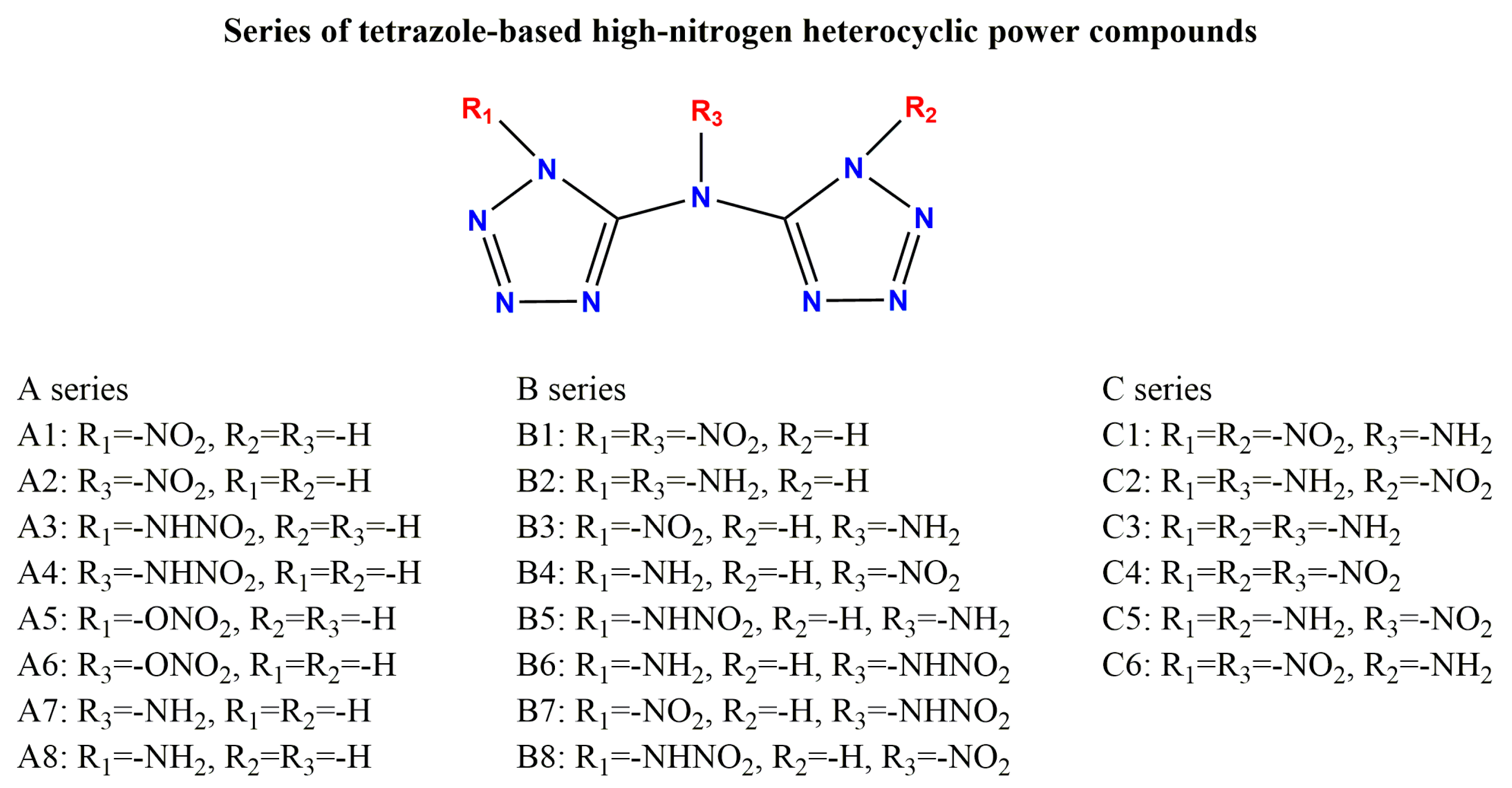
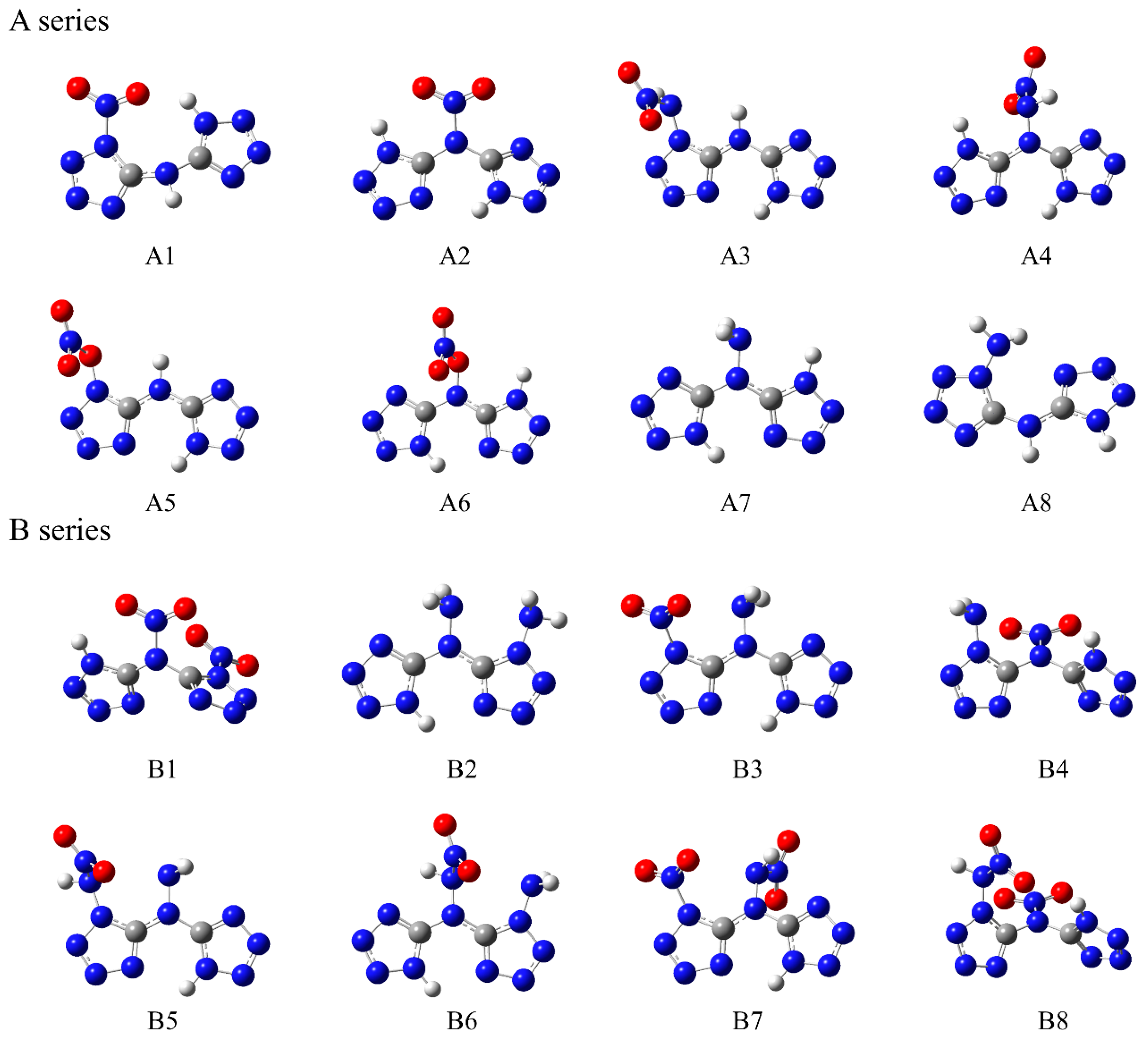
Based on a symmetrical bicyclic tetrazole skeleton centered on nitrogen atoms, the different type, number, and substitution positions of energetic groups (-NH2, -NO2, -ONO2, -NHNO2) were introduced to adjust the structure and properties of these compounds, and a series of novel bicyclic tetrazole heterocyclic power compounds was designed, taking both the detonation performance and the stability into account. Here, we divided these power compounds into A, B, and C series, with the number of substituents being one, two, and three, respectively, as shown in Figure 1. These power compounds are composed of a central nitrogen atom through two C-N bonds and two tetrazole rings, as well as abundant energetic groups. The compounds are all symmetrical structures, and the tetrazole and C-N bonds of all compounds are approximately in the same plane, as shown in Figure 2. As shown in Figure 2, except for compounds A1, A2, and A8, where all atoms are coplanar with the host structure, other compounds only have nitrogen or carbon atoms connected to the tetrazole ring coplanar. The substituent has undergone a certain degree of twisting due to the repulsive effect between electrons, which indicates that the effect of different substituents on the overall structure is relatively small.
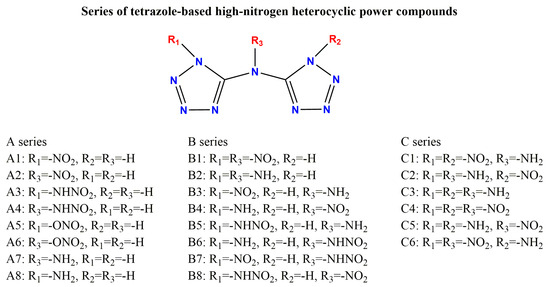
Figure 1.
Series of designed tetrazole-based high-nitrogen heterocyclic power compounds.
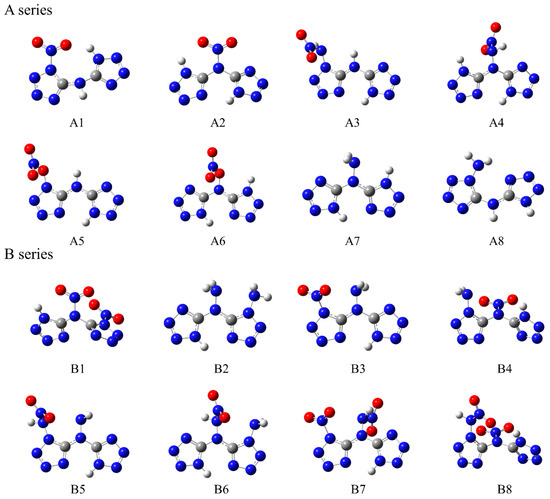
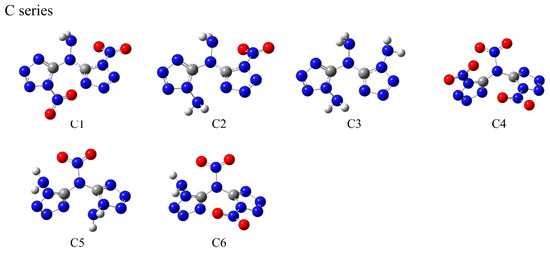
Figure 2.
Optimized structures of the designed tetrazole-based high-nitrogen heterocyclic power compounds.
3.2. Electronic Features
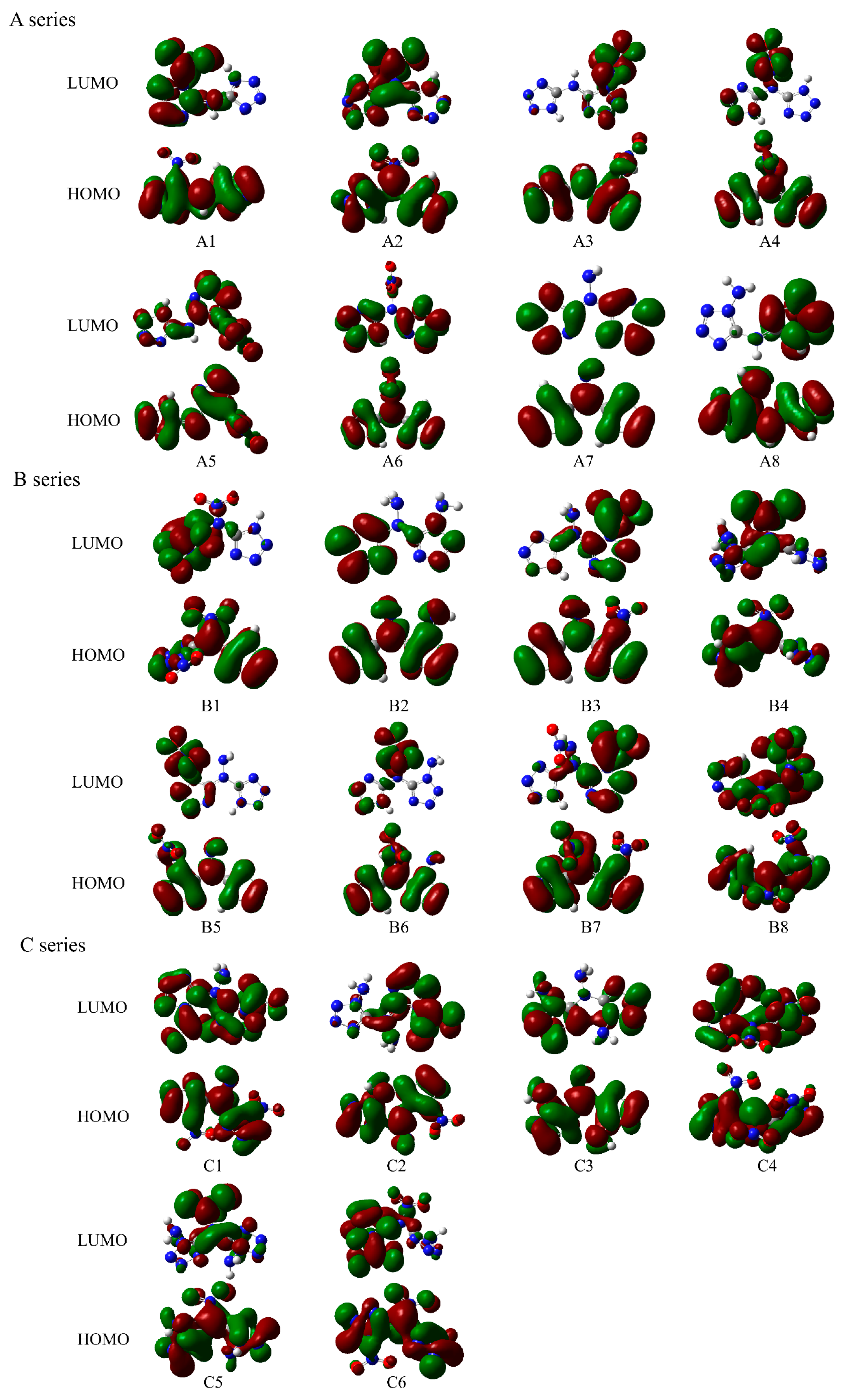
To reveal the chemical properties and reactivity of these power systems, electronic structural parameters, including frontier orbitals and surface electrostatic potential energy, were studied. Figure 3 shows the frontier orbitals of these compounds, including the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). As seen in the energy-level orbital diagrams, the red region shows the positive phase and the green region shows the negative one. For A series power compounds with one substituent, the LUMOs of compounds A1, A2, A3, A4, and A5 are mainly contributed by the substituent, with a small contribution from the tetrazole ring, and those of compounds A6, A7, and A8 are only contributed by the tetrazole ring, while the HOMOs of all these compounds are contributed by both the tetrazole ring and the substituents, indicating that the electron excitation of the A1–A5 molecules mainly occurs from the HOMOs of the whole molecule to the LUMO of the substituents, while the electron excitation of the A6–A8 molecules only occurs from the HOMO of the whole molecule to the LUMO of the tetrazole ring. For B series power compounds with two substituents, the LUMOs of compounds B1, B3, B4, B5, B6, and B7 are mainly contributed by the substituents, with a small contribution from the tetrazole ring, and the LUMO of compound B2 is only contributed by the tetrazole ring and that of compound B8 is contributed by both the tetrazole ring and the substituents, while the HOMOs of all these compounds are contributed by both the tetrazole ring and the substituents, which suggests that the electron excitation for the B1, B3, B4, B5, B6, and B7 molecules mainly occurs from the HOMOs of the whole molecule to the LUMO of the substituent; the electron excitation for the B2 molecules only occurs from the HOMO of the whole molecule to the LUMO of the tetrazole ring, and that of B8 occurs from the HOMO of the whole molecule to the LUMO of both the tetrazole ring and the substituents. For C series power compounds with three substituents, the LUMOs of all these compounds are mainly contributed by both the tetrazole ring and the nitro group substituents, while the HOMOs of these compounds are contributed by both the tetrazole ring and the substituents, denoting that the electron excitation for all these power compounds mainly occurs from the HOMOs of the whole molecule to the LUMO of both the tetrazole ring and the nitro group substituents. The molecular skeleton of all compounds involving both HOMO and LUMO denotes a strengthened or weakened skeleton due to the electron transfer between HOMO and LUMO.
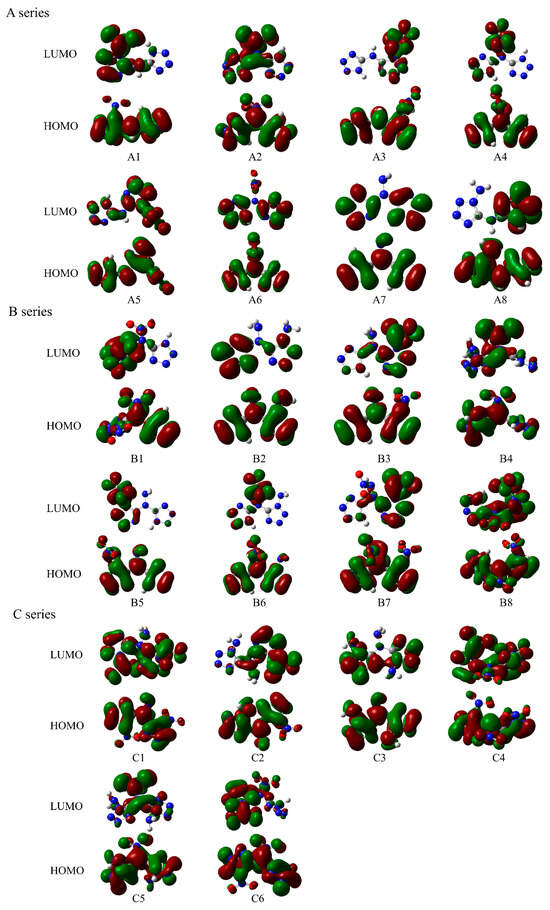
Figure 3.
HOMO and LUMO of the designed tetrazole-based high-nitrogen heterocyclic power compounds.
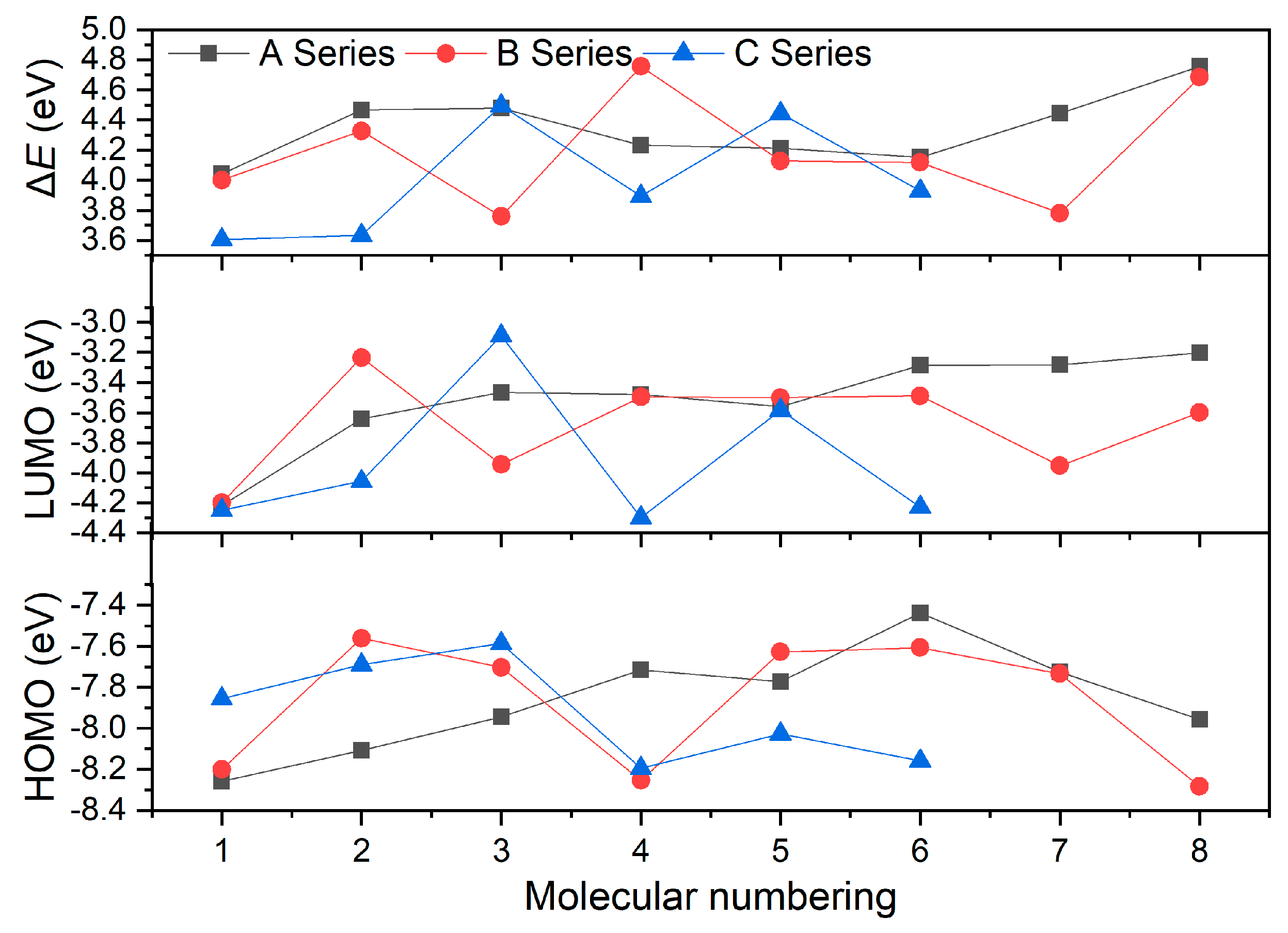
Energy gaps (ΔE), the difference between HOMO and LUMO energy levels, were calculated. Figure 4 presents the HOMO and LUMO energy levels of these compounds, as well as their ΔE. Generally, the larger the ΔE value, the more difficult it is for the valence electrons to transition from HOMO to LUMO, and the more stable the molecule is. It was found that the order of contribution to ΔE of the compounds with the substituent located on the N atom connecting two tetrazole rings is –NO2 > –NH2 > –NHNO2 > –ONO2, while that of the compounds with the substituent located on the tetrazole ring is –NH2 > –NHNO2 > –ONO2 > –NO2. This indicates that mono-substituents –NO2 and –NH2 on the N atom connecting two tetrazole rings and the substituents of –NH2 and –NHNO2 with more H atoms on the tetrazole ring will increase the ΔE value of the compound, thereby improving its relative stability. Moreover, it can be observed that the more energetic substituents there are, the smaller the value of ΔE, indicating that the compound is more unstable. As show in Table 1, the order of ΔE values of A series power compounds with one substituent is A8 > A3 > A2 > A7 > A4 > A5 > A6 > A1, and those of B and C series power compounds with two and three substituents are B4 > B8 > B2 > B5 > B6 > B1 > B7 > B3 and C3 > C5 > C6 > C4 > C2 > C1, respectively. On the one hand, substituents simultaneously increase or decrease the HOMO and LUMO energy levels of these power compounds. Specifically, the HOMO energy-level range of A series compounds is from −8.26 eV to −7.44 eV, and the LUMO energy-level range is from −4.22 eV to −3.20 eV. The HOMO energy-level range of B series compounds is from −8.28 eV to −7.56 eV, and the LUMO energy-level range is from −4.20 eV to −3.23 eV. The HOMO energy-level range of C series compounds is from −8.19 eV to −7.59 eV, and the LUMO energy-level range is from −4.30 eV to −3.09 eV. It was found that the introduction of –NO2 and –ONO2 leads to a decrease in the HOMO and LUMO energy levels of the compounds, while the introduction of –NH2 and –NH2NO2 leads to an increase in the HOMO and LUMO energy levels of the molecule, reflecting the influence of the type, number, and relative position of the substituents on the molecular structures. On the other hand, ΔE values of these compounds with different type, number, and relative position of substituents are also different. The ΔE value ranges of A, B, and C series compounds are from 4.04 eV to 4.76 eV, from 3.76 eV to 4.76 eV, and from 3.61 eV to 4.50 eV, respectively. Among them, power compounds A8, B4, and C3 have higher ΔE values and A1, B3, and C1 have lower ones, which reflects the relative stability of them.
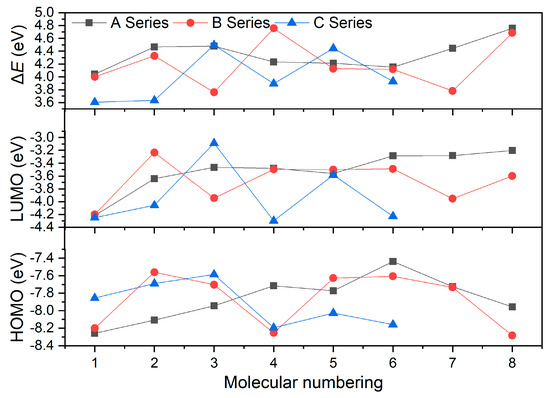
Figure 4.
ΔE of the designed tetrazole-based high-nitrogen heterocyclic power compounds.

Table 1.
HOMO, LUMO, and ΔE values of the designed tetrazole-based high-nitrogen heterocyclic power compounds.
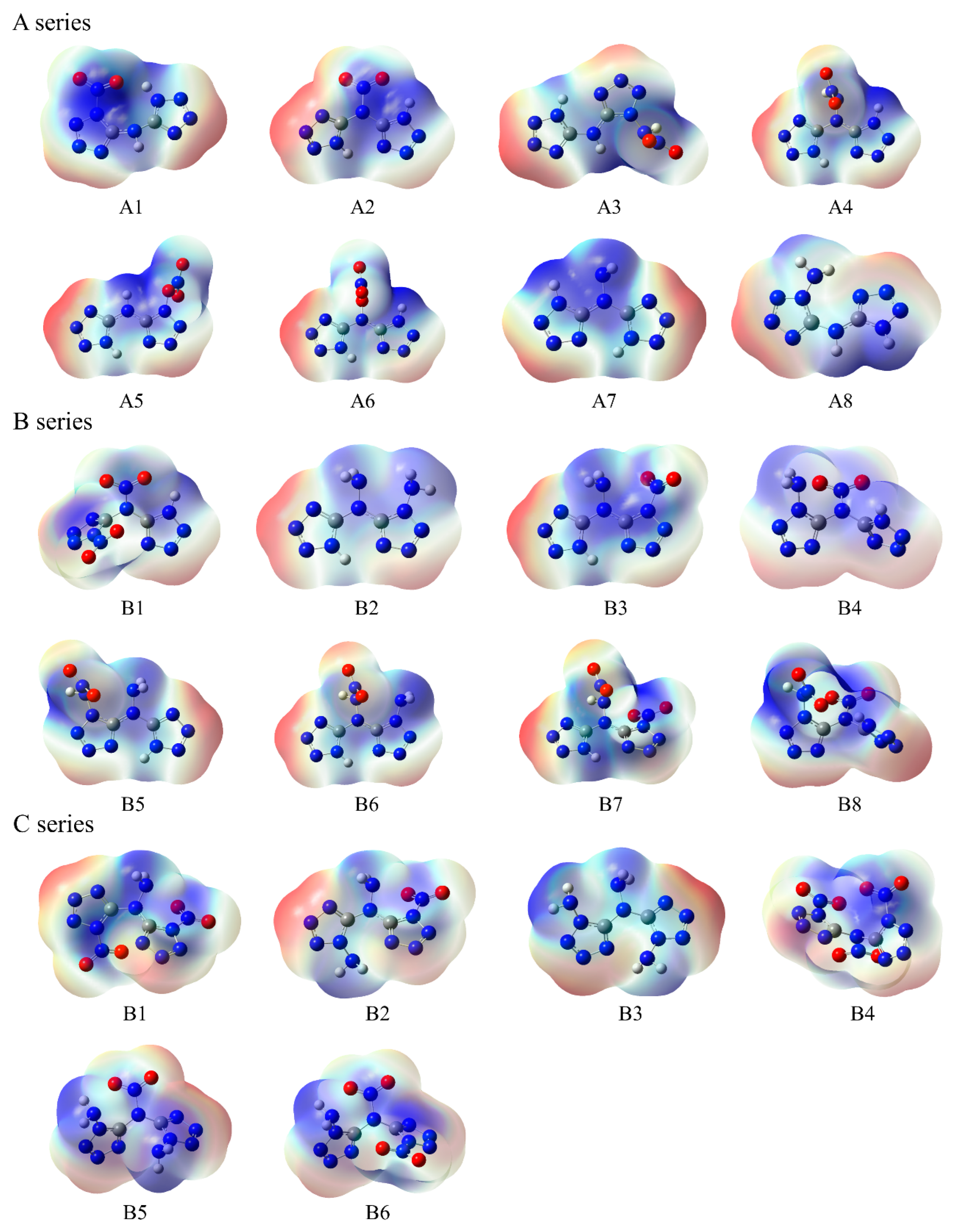
The molecular surface electrostatic potential of these compounds is shown in Figure 5, with local maximum points indicated in the red region and minimum points indicated in the blue region. For all series power compounds, it was found that the extreme points of negative ESP in compounds are mainly distributed near the N atom on the tetrazole ring. The minimum ESP values of compounds A8, B2, and C3 are relatively small, while the minimum ESP values of compounds A5, B1, and C4 are relatively large. This is mainly due to the electron-donating properties of –NH2 and the electron-withdrawing properties of –NO2, which change the electron density distribution of the tetrazole ring. The extreme points of positive ESP in compounds are close to the position of the substituent on the outer side of the ring. The maximum global ESP of compounds containing H atoms in substituents is concentrated near the H atom. The compounds with larger maximum ESP values are B8 and C4, while compounds A7 and C3 have relatively smaller maximum ESP values.
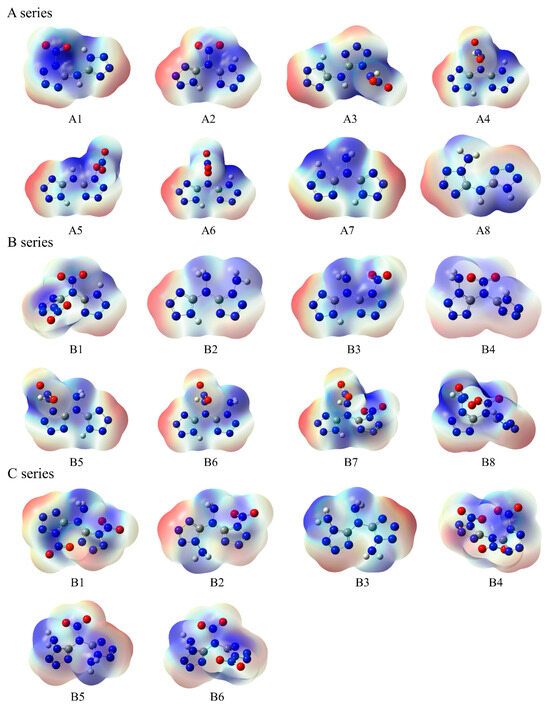
Figure 5.
ESP of the designed tetrazole-based high-nitrogen heterocyclic power compounds. Red, blue, and white regions represent negative charge, positive charge, and neutral state in the molecule.
3.3. Energy Characteristics
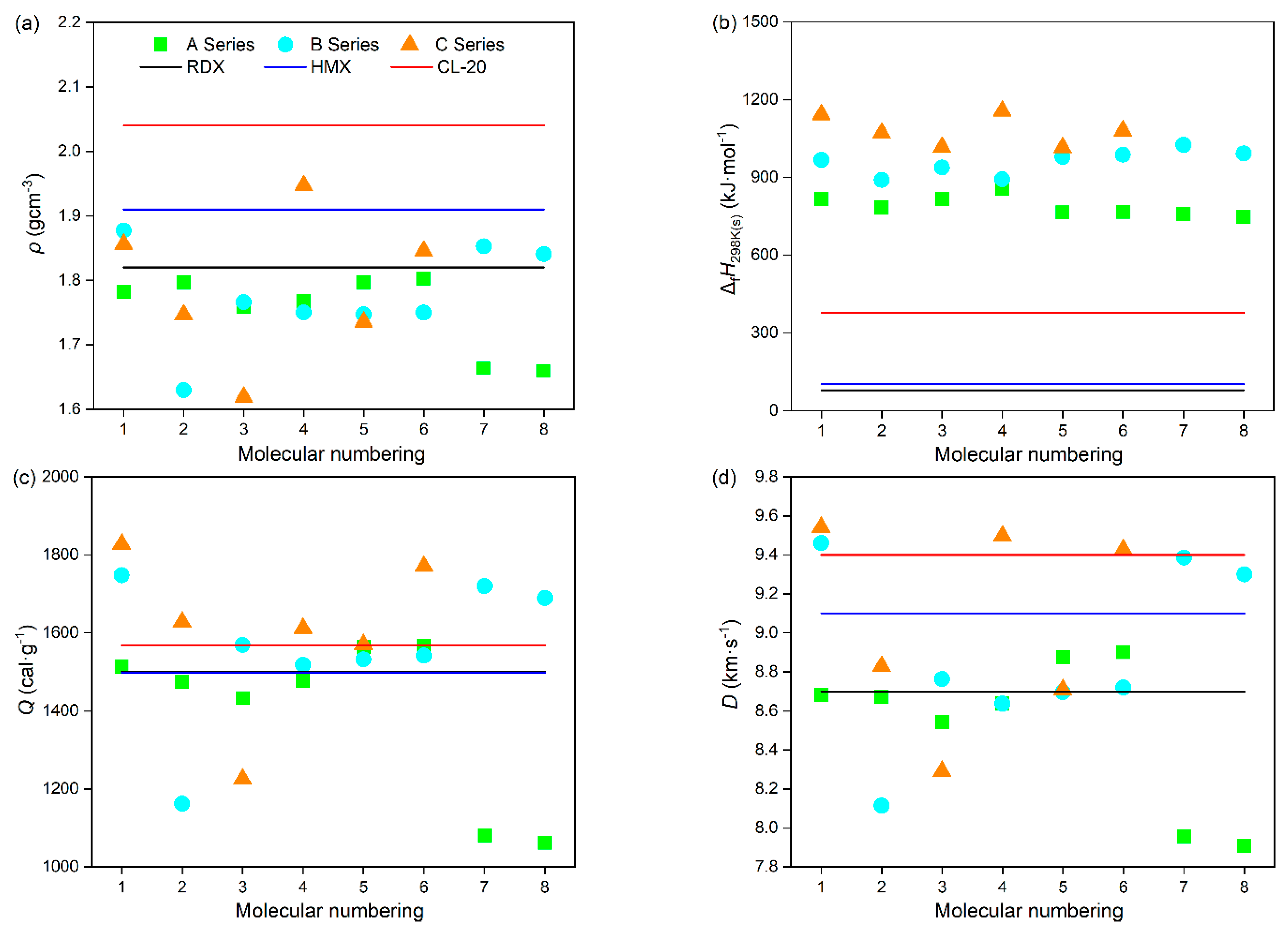
Energy characteristics are one of the important features of power molecular systems. Table 2 lists the density (ρ), heat of formation (ΔfH298K(s)), detonation heat (Q), detonation velocity (D), detonation pressure (P), and oxygen balance (OB) of these power compounds, while listing several common energetic compounds for comparison. As shown in Figure 6a, it was found that the type, number, and position of substituents have a significant impact on the ρ of compounds. Overall, the more substituents there are, the higher the ρ of the compound. For A series power compounds with one substituent, the ρ of all compounds is no more than 1.80 g·cm−3, while there are three compounds with a ρ value of higher than 1.80 g·cm−3 for both B and C series power compounds with two and three substituents, with compound C4 possessing a ρ value as high as 1.95 g·cm−3, which is more than that of 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX) [33]. From the ρ values of these power compounds, it was inferred that the order of contribution to ρ of compounds is –ONO2 > –NO2 > –NHNO2 > –NH2, namely, introducing –NO2 is beneficial for significantly increasing the ρ value of compounds, while the introduction of –NH2 has no obvious increase in the ρ of compounds. Moreover, it was also found that the substituent located on the N atom connecting two tetrazole rings is more advantageous for increasing the ρ of the compound than it is when located on the tetrazole ring. For example, compounds A1 and A3, containing one substituent located on the tetrazole ring, have lower ρ values than those of compounds A2 and A4, containing one substituent located on the N atom connecting two tetrazole rings. Overall, the results are of great significance for the design of high-energy-density compounds, among which compounds B1, B7, B8, C1, C4, and C6 have densities exceeding 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX) [33] and can be considered candidates for high-energy-density compounds.
Table 2.
The ρ, ΔfH298K(s), Q, D, P, h50, and OB values of the designed tetrazole-based high-nitrogen heterocyclic power compounds.
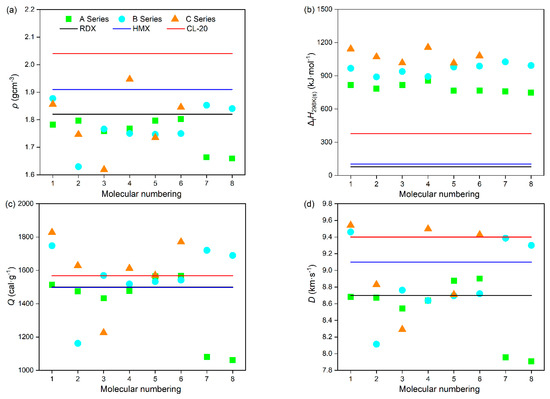
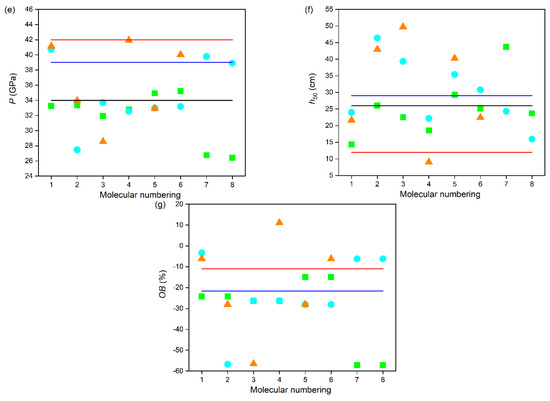
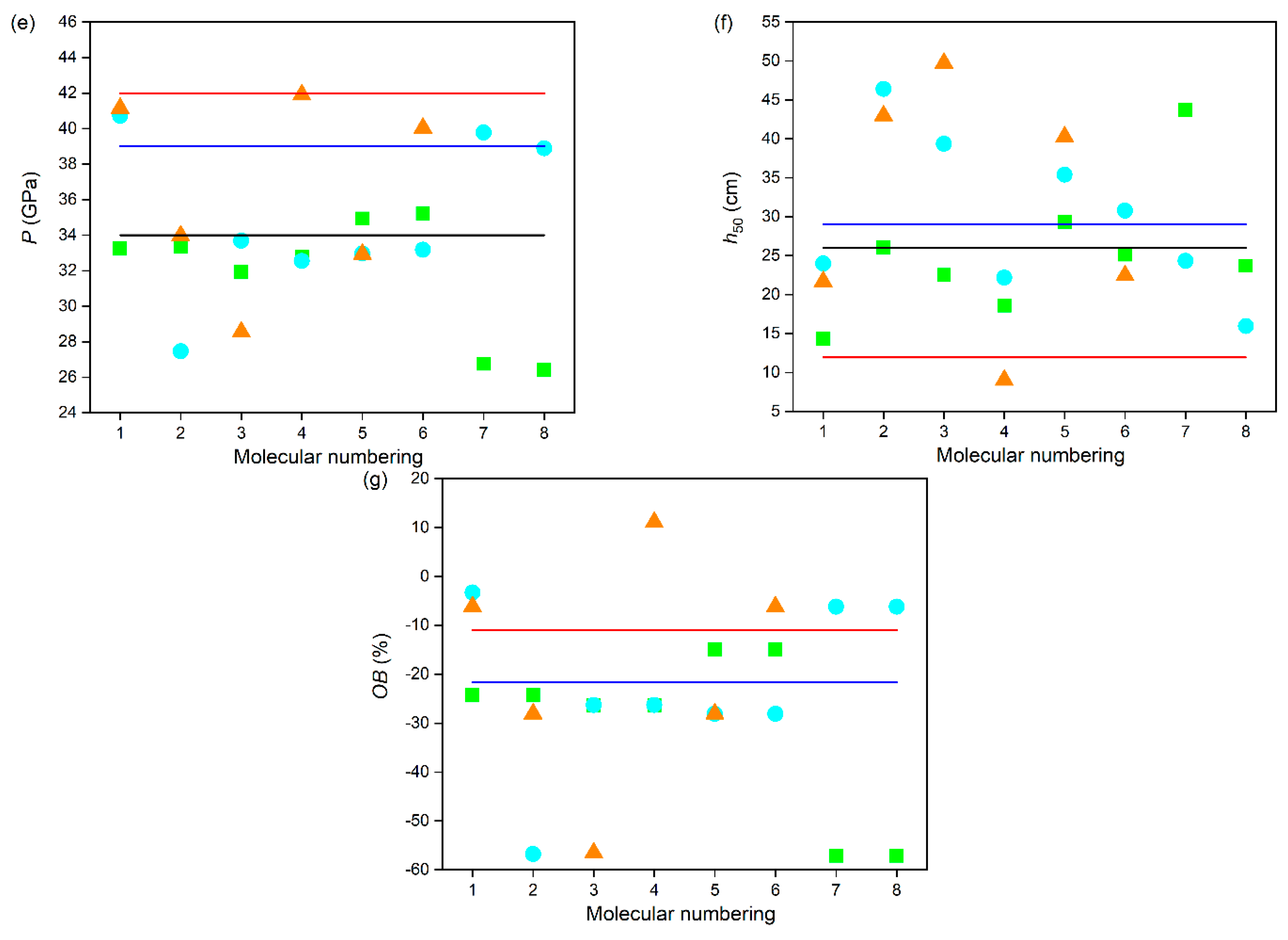
Figure 6.
The (a) ρ, (b) ΔfH298K(s), (c) Q, (d) D, (e) P, (f) h50, and (g) OB of the designed tetrazole-based high-nitrogen heterocyclic power compounds.
Figure 6b shows the ΔfH298K(s) of these power compounds. It is obvious that all these compounds possess much higher ΔfH298K(s) values than that of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) [33], although there exists an effect of the type, number, and position of substituents on ΔfH298K(s), indicating that these power compounds possess a certain degree of excellent detonation performance. Meanwhile, their Q values were also evaluated and are presented in Figure 6c. It can be seen that most compounds possess high Q values, except for compounds A7, A8, B2, and C3. Among them, nine compounds—B1, B3, B7, B8, C1, C2, C4, C5, and C6—possess higher Q values than that of CL-20, and compound C1 possesses the highest Q value of 1828 J·g−1. The Q values of A series power compounds with one substituent are obviously lower than those of B and C series power compounds with two and three substituents, suggesting that the introduction of more energetic groups is beneficial for improving the Q value. Moreover, it was also found from A series compounds that the introduction of –NO2 is beneficial for obviously increasing the Q value of compounds, while the introduction of –NH2 leads to no obvious increase in the Q value of compounds, and the order of contribution to Q value of compounds is–ONO2 > –NO2 > –NHNO2 > –NH2. For example, compound A8, with one amino substituted, possesses the lowest Q value, while compound C1, with one amino and two nitro substituted, possesses the highest Q value. What is more, it was found from compounds A1 and A2, B3 and B4, and C1 and C4 that –NO2 located on the N atom connecting two tetrazole rings is more conducive to increasing the Q value of the power compound than on the N atom connecting two tetrazole rings, while –NH2 is the opposite, namely, –NH2 located on the N atom connecting two tetrazole rings is more conducive to increasing the Q value of power compounds than on the tetrazole ring.
To further evaluate the detonation performances of these power compounds, D and P values were calculated and are displayed in Figure 6d,e. The results show that the type, number, and position of the substituents have significant effects on the D and P values of these power compounds. Undoubtedly, the more energetic groups there are, the better the detonation performance of the compound, which is consistent with the experimental data that the D and P values of C series compounds are generally higher than those of B series compounds, and even higher than those of A series compounds. On the whole, most of these power compounds exhibit relatively excellent detonation performance, except for compounds A7, A8, B2, and C3. Specifically, for A series power compounds with one substituent, except for compounds A7 and A8, the D and P values of all compounds are more than 8.0 km·s−1 and 30 GPa, respectively. For B series power compounds with two substituents, except for compound B2, the D and P values of all compounds are more than 8.5 km·s−1 and 30 GPa, respectively. For C series power compounds with three substituents, except for compound C3, the D and P values of all compounds are more than 8.5 km·s−1 and 30 GPa, respectively. The lower energy characteristics of these four compounds are related to their lower density properties. It can be inferred that the order of contribution to the D and P values of these power compounds is –ONO2 > –NO2 > –NHNO2 > –NH2, which is consistent with that of Q and ρ, which is owing to the fact that N in –ONO2 and –NO2 exhibits a higher valence state and has oxidizing properties. This is because both D and P are determined by Q and ρ. Namely, introducing –NO2 is beneficial for significantly increasing the D and P of compounds, while the introduction of –NH2 leads to no obvious increase in the D and P of the compounds. For example, compound A5, with one –ONO2 substituted compound, possesses the highest D (8.88 km·s−1) and P (34.93 GPa) values; compound A1, with one –NO2 substituted, possesses D (8.68 km·s−1) and P (33.25 GPa) values second only to those of A5; compound A3, with one –NHNO2 substituted, possesses D (8.54 km·s−1) and P (31.93 GPa) values in the last place but one; and compound A8, with one –NH2 substituted, possesses the lowest D (7.91 km·s−1) and P (26.40 GPa) values. It is noted that compounds B1, C1, C4, and C6 possess higher D values than that of CL-20 (9.4 km·s−1), and C4 possesses a similar P value to that of CL-20 (42 GPa), indicating their outstanding detonation performances. Moreover, it was also found that the substituent located on the N atom connecting two tetrazole rings is more advantageous for improving the D and P values of the compound than it is when located on the tetrazole ring, which agrees well with that of ρ. For example, compounds A4, A6, and A7 containing one substituent located on the N atom connecting two tetrazole rings have higher D and P values than those of their corresponding compounds A3, A5, and A8 containing one substituent located on the tetrazole ring, respectively.
In addition, OB is also an important parameter that measures oxygen deficiency or excess oxygen in the molecule when compounds explode to form carbon and hydrogen oxides. Generally, the higher the OB, the better the energy characteristics. As can be seen from Figure 6g, all these power compounds show negative OB values except for the positive OB value (11.11%) of compound C4. It is worth noting that compound C4, with a trinitro substituted, exhibits the best detonation performance, which can be attributed to it having the highest ρ and OB values. On the whole, most these power compounds show relatively higher OB values, except for compounds A7, A8, B2, and C3, which is consistent with the D and P values. Due to the determined elemental composition of the compound, the OB value is independent of the position of the substituent. However, the OB value is related to the type and quantity of substituents. It was found that introducing the groups containing an O atom (–ONO2 and –NO2) is beneficial for increasing the OB values of compounds, while the introduction of the groups containing an H atom (–NHNO2 and –NH2) reduces the OB values of the compounds. For example, compound A5, with one –ONO2 substituted, possesses the highest OB value (−14.95%); compound A1, with one –NO2 substituted, possesses an OB value (−24.23%) second only to that of A5; compound A3, with one –NHNO2 substituted possesses an OB value (−26.28%) in the last place but one; and compound A8, with one –NH2 substituted, possesses the lowest OB value (−57.10%).
In short, compounds B1, B7, B8, C1, C4, and C6 have higher ρ, ΔfH298K(s), Q, D, P, and OB values than those of RDX and can be selected as candidates for high-energy-density compounds.
3.4. Safety Evaluation
Safety performance is another important feature of power molecular systems. Impact sensitivity (h50, cm) is an accurate way to evaluate the safety performance of these power compounds. Generally, the higher the h50 value, the less sensitive the molecule is. Table 2 lists the h50 values of these power compounds and of several common energetic compounds for comparison. As can be seen in Figure 6f, it is clear that the type, number, and position of the substituents have a significant effect on the h50 of the compounds. Generally, the more energetic groups there are, the worse the safety performance of the compound is. On the whole, all these power compounds exhibit relatively high h50 values, although the h50 of compound C4 is slightly lower than that of CL-20, which indicates that our approach to the safety design of energetic compounds is effective. Specifically, the h50 values of A series power compounds with one substituent are in the range of 14.35–43.69 cm, the h50 values of B series power compounds with two substituents are in the range of 15.96–46.40 cm, and the h50 values of C series power compounds with three substituents are in the range of 9.04–49.74 cm. However, the effect of the type of substituents on the h50 of these power compounds is different from that of effect of the position of the substituents on the h50 of these power compounds. It was found that the order of contribution to h50 of the compounds with the substituent located on the N atom connecting two tetrazole rings is –NH2 > –NO2 > –ONO2 > –NHNO2, while that of the compounds with the substituent located on the tetrazole ring is –ONO2 > –NH2 > –NHNO2 > –NO2. Namely, introducing mono-substituents –NO2 and –NH2 onto the N atom connecting two tetrazole rings and composite substituents –NHNO2 and –ONO2 onto the tetrazole ring will increase the h50 value of the compound, thereby improving its safety performance. For example, compound A2 a possesses higher h50 value than A1 and A7 possesses a higher h50 value than A8, while compound A4 possesses a higher h50 value than A3 and A6 possesses a higher h50 value than A5. In short, the variety of safety performance for these power compounds is the result of the combined effect of the type and position of substituents.
Overall, taking both energy characteristics and safety performance into consideration compared with CL-20, it can be concluded from the above discussion that the power molecular systems B1(N-(1-nitro-1H-tetrazol-5-yl)-N-(1H-tetrazol-5-yl)nitramide), B7(N’-(1-nitro-1H-tetrazol-5-yl)-N’-(1H-tetrazol-5-yl)nitric hydrazide), B8(N-(1-(nitroamino)-1H-tetrazol-5-yl)-N-(1H-tetrazol-5-yl)nitramide), C1(5,5′-(hydrazine-1,1-diyl)bis(1-nitro-1H-tetrazole)), C4(N,N-bis(1-nitro-1H-tetrazol-5-yl)nitramide), and C6(N-(1-amino-1H-tetrazol-5-yl)-N-(1-nitro-1H-tetrazol-5-yl)nitramide) can be regarded as candidates for high-energy-density compounds.
4. Conclusions
With the increasing emphasis on environmental protection and the demand for sustainable development, innovation in energy storage technology and its solutions for energetic materials has become increasingly important. Therefore, series of tetrazole-based ultra-high-energy-density high-nitrogen heterocyclic power compounds were designed and their energy characteristics and safety performances were evaluated theoretically in this work. The results indicate that the type, number, and position of substituents have a significant effect on the comprehensive performance of these compounds.
The studies on energy gaps between HOMO and LUMO indicates that less energetic substituents increase the ΔE value of the compound. The discussions on energy characteristics reveal that the more energetic groups there are, the better the detonation performance of the compound is, and introducing –NO2 is beneficial for significantly increasing the D and P of the compounds, while the introduction of –NH2 leads to no obvious increase in the D and P of the compounds. The analysis on safety performance suggests that introducing mono-substituents –NO2 and –NH2 onto the N atom connecting two tetrazole rings and composite substituents –NHNO2 and –ONO2 onto the tetrazole ring increases the h50 value of the compound, thereby improving its safety performance. Overall, taking both energy characteristics and safety performance into consideration, the power molecular systems B1(N-(1-nitro-1H-tetrazol-5-yl)-N-(1H-tetrazol-5-yl)nitramide), B7(N’-(1-nitro-1H-tetrazol-5-yl)-N’-(1H-tetrazol-5-yl)nitric hydrazide), B8(N-(1-(nitroamino)-1H-tetrazol-5-yl)-N-(1H-tetrazol-5-yl)nitramide), C1(5,5′-(hydrazine-1,1-diyl)bis(1-nitro-1H-tetrazole)), C4(N,N-bis(1-nitro-1H-tetrazol-5-yl)nitramide), and C6(N-(1-amino-1H-tetrazol-5-yl)-N-(1-nitro-1H-tetrazol-5-yl)nitramide) can be regarded as candidates for high-energy-density compounds.
Our study provides new solutions for the innovation and progress of energy storage technologies in the energetic materials field.
Author Contributions
Funding acquisition, Q.Y.; supervision, Q.Y.; writing—original draft, Y.L.; writing—review and editing, Q.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (52371131).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shi, C.; Hamann, T.; Takeuchi, S.; Alexander, G.V.; Nolan, A.M.; Limpert, M.; Fu, Z.; O’Neill, J.; Godbey, G.; Dura, J.A.; et al. 3D Asymmetric Bilayer Garnet-hybridized High-energy-density Lithium-sulfur Batteries. ACS Appl. Mater. Interfaces 2023, 15, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Aghmadi, A.; Mohammed, O.A. Energy Storage Systems: Technologies and High-Power Applications. Batteries 2024, 10, 10040141. [Google Scholar] [CrossRef]
- Ali, Z.M.; Calasan, M.; Aleem, S.H.E.A.; Jurado, F.; Gandoman, F.H. Applications of Energy Storage Systems in Enhancing Energy Management and Access in Microgrids: A Review. Energies 2023, 16, 5930. [Google Scholar] [CrossRef]
- Aghmadi, A.; Hussein, H.; Polara, K.H.; Mohammed, O.A. Comprehensive Review of Architecture, Communication, and Cybersecurity in Networked Microgrid Systems. Inventions 2023, 8, 84. [Google Scholar] [CrossRef]
- Kandari, R.; Neeraj, N.; Micallef, A. Review on Recent Strategies for Integrating Energy Storage Systems in Microgrids. Energies 2022, 16, 317. [Google Scholar] [CrossRef]
- Aghmadi, A.; Ali, O.; Mohammed, O.A. Enhancing DC Microgrid Stability under Pulsed Load Conditions through Hybrid Energy Storage Control Strategy. In Proceedings of the 2023 IEEE Industry Applications Society Annual Meeting (IAS), Nashville, TN, USA, 29 October–2 November 2023; pp. 1–6. [Google Scholar] [CrossRef]
- Faisal, M.; Hannan, M.A.; Ker, P.J.; Hussain, A.; Mansor, M.B.; Blaabjerg, F. Review of Energy Storage System Technologies in Microgrid Applications: Issues and Challenges. IEEE Access 2018, 6, 35143–35164. [Google Scholar] [CrossRef]
- Gao, H.; Shreeve, J.M. Azole-based Energetic Salts. Chem. Rev. 2011, 111, 7377–7436. [Google Scholar] [CrossRef]
- Zhang, Q.; Shreeve, J.M. Growing Catenated Nitrogen Atom Chains. Angew. Chem. Int. Ed. 2013, 52, 8792–9794. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Qi, C.; Zhao, X.; Zhang, J.; Zhang, S.; Pang, S. 3D Energetic Metal-Organic Frameworks: Synthesis and Properties of High Energy Materials. Angew. Chem. Int. Ed. 2013, 52, 14031–14035. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Petermayer, C.; Piercey, D.G.; Jo, S. 1,3-Bis(nitroimido)-1,2,3-triazolate Anion, the N-Nitroimide Moiety, and the Strategy of Alternating Positive and Negative Charges in the Design of Energetic Materials. J. Am. Chem. Soc. 2012, 134, 20827–20836. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, J.; Mitchell, L.A.; Parrish, D.A.; Shreeve, J.N.M. Taming of 3,4-Di(nitramino)furazan. J. Am. Chem. Soc. 2015, 137, 15984–15987. [Google Scholar] [CrossRef] [PubMed]
- Chavez, D.E.; Parrish, D.A.; Mitchell, L.; Imler, G.H. Azido and Tetrazolo 1,2,4,5-Tetrazine N-Oxides. Angew. Chem. Int. Ed. 2017, 56, 3575–3578. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Martin, F.A.; Stierstorfer, J. C2N14: An Energetic and Highly Sensitive Binary Azidotetrazole. Angew. Chem. Int. Ed. 2011, 50, 4227–4229. [Google Scholar] [CrossRef]
- Stierstorfer, J.; Klapötke, T.M.; Hammerl, A.; Chapman, R.D. 5-Azido-1H-tetrazole-Improved Synthesis, Crystal Structure and Sensitivity Data. Z. Anorg. Allg Chem. 2008, 634, 1051–1057. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Piercey, D.G. 1,1′-Azobis(tetrazole): A Highly Energetic Nitrogen-Rich Compound with a N10 Chain. Inorg. Chem. 2011, 50, 2732–2734. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Mukundan, T.; Muthurajan, H.; Sikder, A.K.; Gandhe, B.R.; Rao, A.S. Environmentally Compatible Next Generation Green Energetic Materials (GEMs). J. Hazard. Mater. 2009, 161, 589–607. [Google Scholar] [CrossRef]
- Liu, H.; Wang, F.; Wang, G.X.; Gong, X.D. Theoretical Studies on 2-(5-Amino-3-nitro-1,2,4-triazolyl)-3,5-dinitropyridine (PRAN) and Its Derivatives. J. Phys. Org. Chem. 2013, 26, 30–36. [Google Scholar] [CrossRef]
- Zhao, G.; Yin, P.; Kumar, D.; Imler, G.H.; Parrish, D.A.; Shreeve, J.N.M. Bis-(3-nitro-1-(trinitromethyl)-1H-1,2,4-triazol-5-yl)methanone: An Applicable and Very Dense Green Oxidizer. J. Am. Chem. Soc. 2019, 141, 19581–19584. [Google Scholar] [CrossRef]
- Yang, H.; Huang, L.; Xu, M.; Tang, Y.; Wang, B.; Cheng, G. Strategy for Extending the Nitrogen Chain: The Bis-(1,2,3-triazole) Formation Reaction from Tosylhydrazones and N-Amino Azole. J. Org. Chem. 2019, 84, 10629–10634. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, G.; Li, J.; Zhang, Z.; Lu, H.; Liao, L.; Fan, G.; Nie, F. Pyrazol-triazole energetic hybrid with high thermal stability and decreased sensitivity: Facile synthesis, characterization and promising performance. Chem. Eng. J. 2020, 379, 31734–31736. [Google Scholar] [CrossRef]
- Benz, M.; Klapötke, T.M.; Stierstorfer, J. Combining Performance with Thermal Stability: Synthesis and Characterization of 5-(3,5-Dinitro-1H-pyrazol-4-yl)-1H-tetrazole and its Energetic Derivatives. Z. Anorg. Allg Chem. 2020, 646, 1380–1388. [Google Scholar] [CrossRef]
- Gettings, M.L.; Thoenen, M.T.; Byrd, E.F.; Sabatini, J.J.; Zeller, M.; Piercey, D.G. Tetrazole Azasydnone (C2N7O2H) And Its Salts: High-Performing Zwitterionic Energetic Materials Containing A Unique Explosophore. Chemistry 2020, 26, 14530–14535. [Google Scholar] [CrossRef]
- Shlomovich, A.; Pechersky, T.; Cohen, A.; Yan, Q.L.; Kosa, M.; Petrutik, N.; Tal, N.; Aizikovich, A.; Gozin, M. Energetic isomers of 1,2,4,5-tetrazine-bis-1,2,4- triazoles with low toxicity. Dalton Tran. 2017, 46, 5994–6002. [Google Scholar] [CrossRef]
- Fei, T.; Du, Y.; Chen, P.; He, C.; Pang, S. N-Fluoro functionalization of heterocyclic azoles: A new strategy towards insensitive high energy density materials. New J. Chem. 2018, 42, 16244–16257. [Google Scholar] [CrossRef]
- Johnson, E.C.; Sabatini, J.J.; Chavez, D.E.; Wells, L.A.; Banning, J.E.; Sausa, R.C.; Byrd, E.F.; Orlicki, J.A. Bis-(Nitroxymethylisoxazolyl) Furoxan: A Promising Standalone Melt-Castable Explosive. Chempluschem 2020, 85, 237–239. [Google Scholar] [CrossRef]
- Yu, Q.; Yang, H.; Imler, G.H.; Parrish, D.A.; Cheng, G.; Shreeve, J.N.M. Derivatives of 3,6-Bis(3-aminofurazan-4-ylamino)-1,2,4,5- tetrazine: Excellent Energetic Properties with Lower Sensitivities. ACS Appl. Mater. Interfaces 2020, 12, 31522–31531. [Google Scholar] [CrossRef]
- Thottempudi, V.; Gao, H.X.; Shreeve, J.M. Trinitromethyl-Substituted 5-Nitro- or 3-Azo-1,2,4-triazoles: Synthesis, Characterization, and Energetic Properties. J. Am. Chem. Soc. 2011, 133, 6464–6471. [Google Scholar] [CrossRef]
- Hahre, W.J.; Radom, L.; Schleyer, P.V.R.; Pole, J.A. Ab Initio Molecular Orbital Theory; Wiley-Interscience: New York, NY, USA, 1986. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 09; Gaussian Inc.: Wallingford, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Joo, Y.H.; Shreeve, J.M. High-density Energetic Mono-or Bis (oxy) -5-nitroiminotetrazoles. Angew. Chem. Int. Ed. 2010, 49, 7320–7323. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Jacobs, S.J. A Simple Method for Calculating Detonation Properties of C-H-N-O Explosives. J. Chem. Phys. 1968, 48, 23–35. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Some Perspectives on Estimating Detonation Properties of C, H, N, O Compounds. Cent. Eur. J. Energ. Mat. 2011, 8, 209–220. [Google Scholar] [CrossRef]
- Pospíšil, M.; Vavra, P.; Concha, M.C.; Murray, J.S.; Politzer, P. A possible crystal volume factor in the impact sensitivities of some energetic compounds. J. Mol. Model. 2010, 16, 895–901. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).