
Visible Light-Driven Hydrogen Evolution Catalysis by Heteroleptic Ni(II) Complexes with Chelating Nitrogen Ligands: Probing Ligand Substituent Position and Photosensitizer Effects


Abstract
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Instruments
2.3. Synthesis
2.3.1. Synthesis of Complex 1
2.3.2. Synthesis of Complex 2
2.3.3. Synthesis of Complex 3
2.3.4. Synthesis of TGA-Coated CdTe QDs
2.4. Cyclic Voltammetry
2.5. Photocatalysis
3. Results and Discussion
3.1. Synthesis and Characterization
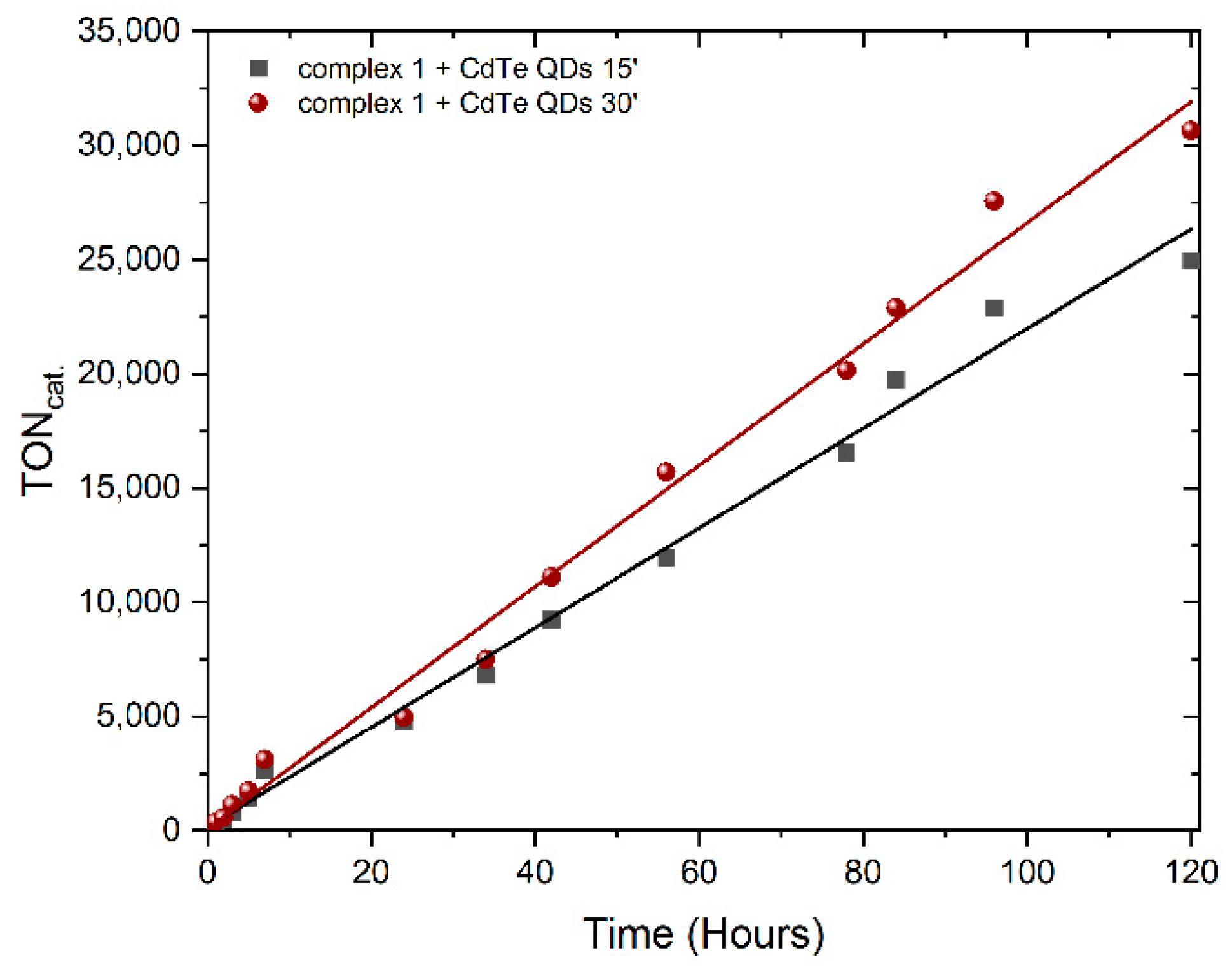
3.2. Photocatalytic Hydrogen Production
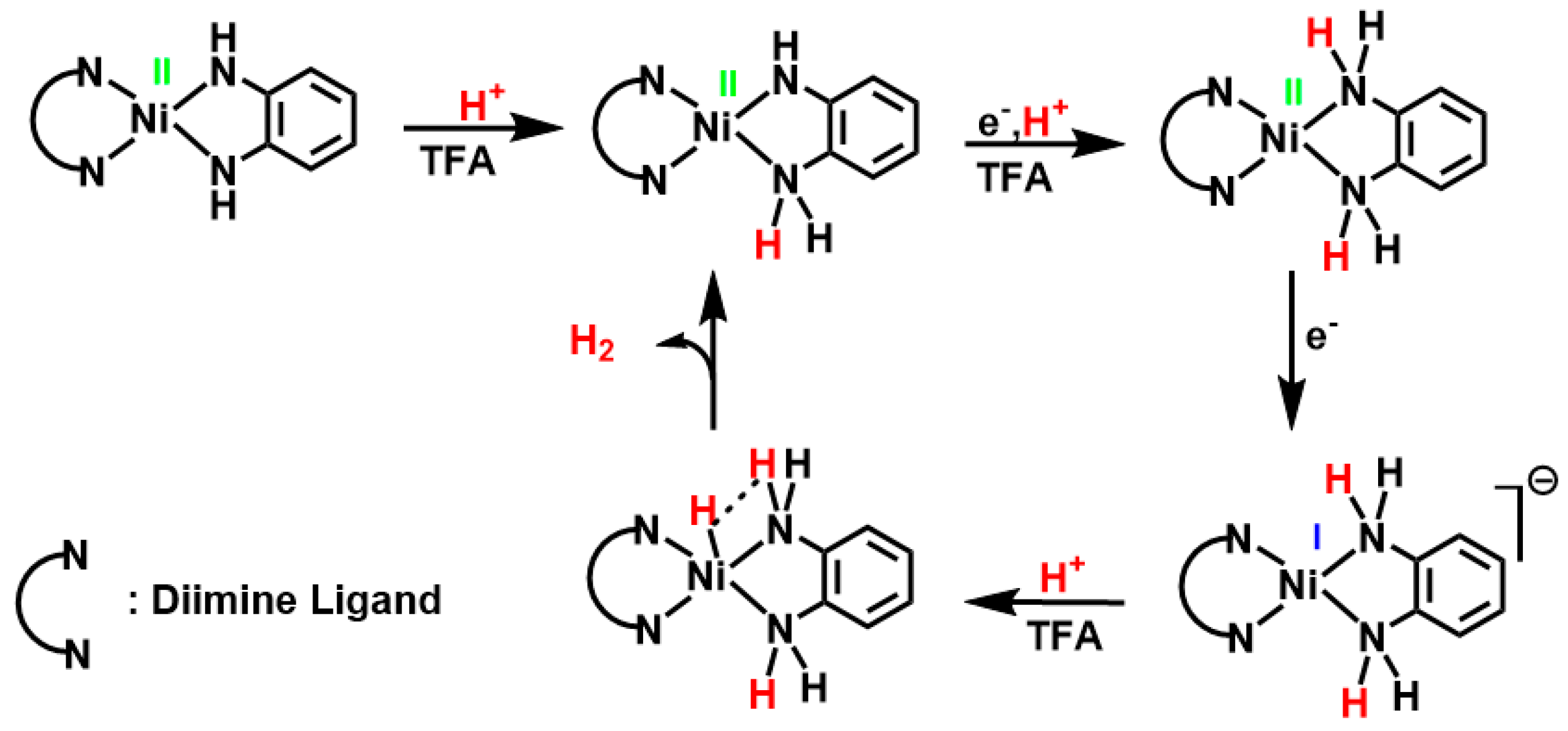
3.3. Electrocatalytic Hydrogen Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ibn Shamsah, S.M. Earth-Abundant Electrocatalysts for Water Splitting: Current and Future Directions. Catalysts 2021, 11, 429. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, L.; Zhao, P.; Lee, L.Y.S.; Wong, K.-Y. Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles. Chem. Rev. 2020, 120, 851–918. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lai, W.; Xu, B. A Mini Review on Doped Nickel-Based Electrocatalysts for Hydrogen Evolution Reaction. Energies 2020, 13, 4651. [Google Scholar] [CrossRef]
- Ferriday, T.B.; Middleton, P.H.; Kolhe, M.L. Review of the Hydrogen Evolution Reaction—A Basic Approach. Energies 2021, 14, 8535. [Google Scholar] [CrossRef]
- Benghanem, M.; Mellit, A.; Almohamadi, H.; Haddad, S.; Chettibi, N.; Alanazi, A.M.; Dasalla, D.; Alzahrani, A. Hydrogen Production Methods Based on Solar and Wind Energy: A Review. Energies 2023, 16, 757. [Google Scholar] [CrossRef]
- Gray, H.B. Powering the planet with solar fuel. Nat. Chem. 2009, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Bockris, J.O.; Conway, B.E. Studies in hydrogen overpotential. The effect of catalytic poisons at platinized platinum and nickel. Trans. Faraday Soc. 1949, 45, 989–999. [Google Scholar] [CrossRef]
- Thoi, V.S.; Sun, Y.; Long, J.R.; Chang, C.J. Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev. 2013, 42, 2388–2400. [Google Scholar] [CrossRef]
- McKone, J.R.; Marinescu, S.C.; Brunschwig, B.S.; Winkler, J.R.; Gray, H.B. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 2014, 5, 865–878. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, C.; Liu, S.; Wang, J.-L.; Lin, W. A Biomimetic Copper Water Oxidation Catalyst with Low Overpotential. J. Am. Chem. Soc. 2014, 136, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Sutra, P.; Igau, A. Emerging Earth-abundant (Fe, Co, Ni, Cu) molecular complexes for solar fuel catalysis. Curr. Opin. Green Sustain. Chem. 2018, 10, 60–67. [Google Scholar] [CrossRef]
- Hu, C.; Lv, C.; Liu, S.; Shi, Y.; Song, J.; Zhang, Z.; Cai, J.; Watanabe, A. Nickel Phosphide Electrocatalysts for Hydrogen Evolution Reaction. Catalysts 2020, 10, 188. [Google Scholar] [CrossRef]
- Drosou, M.; Kamatsos, F.; Mitsopoulou, C.A. Recent advances in the mechanisms of the hydrogen evolution reaction by non-innocent sulfur-coordinating metal complexes. Inorg. Chem. Front. 2020, 7, 37–71. [Google Scholar] [CrossRef]
- Mitsopoulou, C.A. Identifying of charge-transfer transitions and reactive centers in M (diimine)(dithiolate) complexes by DFT techniques. Coord. Chem. Rev. 2010, 254, 1448–1456. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Koutsouri, E.; Mitsopoulou, C.A. A perspective on solar energy conversion and water photosplitting by dithiolene complexes. Coord. Chem. Rev. 2012, 256, 2424–2434. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Field, M.J.; Papatriantafyllopoulou, C.; Fize, J.; Artero, V.; Mitsopoulou, C.A. Experimental and Theoretical Insight into Electrocatalytic Hydrogen Evolution with Nickel Bis(aryldithiolene) Complexes as Catalysts. Inorg. Chem. 2016, 55, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Koutsouri, E.; Mitsopoulou, C.A. Photocatalytic Hydrogen Evolution by tris-dithiolene tungsten complexes. Open Chem. 2016, 14, 393–403. [Google Scholar] [CrossRef]
- Kamatsos, F.; Bethanis, K.; Mitsopoulou, C.A. Synthesis of Novel Heteroleptic Oxothiolate Ni(II) Complexes and Evaluation of Their Catalytic Activity for Hydrogen Evolution. Catalysts 2021, 11, 401. [Google Scholar] [CrossRef]
- Lelj, F.; Rosa, A.; Ricciardi, G.; Casarin, M.; Cristinziano, P.; Morelli, G. On the spectroscopic behaviour of o-bis(o-phenylendiimido) nickel. Chem. Phys. Lett. 1989, 160, 39–42. [Google Scholar] [CrossRef]
- Bachler, V.; Olbrich, G.; Neese, F.; Wieghardt, K. Theoretical Evidence for the Singlet Diradical Character of Square Planar Nickel Complexes Containing Two o-Semiquinonato Type Ligands. Inorg. Chem. 2002, 41, 4179–4193. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Dou, J.; Zhang, J.; Zhang, S. A simple and economical one-pot method to synthesize high-quality water soluble CdTe QDs. J. Mater. Chem. 2012, 22, 14573. [Google Scholar] [CrossRef]
- Kamatsos, F.; Drosou, M.; Mitsopoulou, C.A. Heteroleptic thiolate diamine nickel complexes: Noble-free-metal catalysts in electrocatalytic and light-driven hydrogen evolution reaction. Int. J. Hydrogen Energy 2021, 46, 19705–19716. [Google Scholar] [CrossRef]
- Himmel, H.-J.; Manceron, L. Ni(N2)4 revisited: An analysis of the Ni–N2 bonding properties of this benchmark system on the basis of UV/Vis, IR and Raman spectroscopy. Dalton Trans. 2005, 15, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.M.; Versiane, O.; Felcman, J.; Soto, C.A.T. FT-IR vibrational spectrum and DFT:B3LYP/6-31G structure and vibrational analysis of guanidinoaceticserinenickel(II) complex: [Ni(GAA)(Ser)]. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 67, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Halli, M.B.; Patil, V.B. Synthesis, spectroscopic, thermal, and biological activities of mixed ligand complexes containing E-N′-(3,4,5-trimethoxybenzylidene)benzofuran-2-carbohydrazide and 2-aminothiophenol. J. Coord. Chem. 2011, 64, 3740–3750. [Google Scholar] [CrossRef]
- Ghiasi, Z.; Amani, V.; Mirzaei, P.; Safari, N.; Abedi, A. Trichloridothallium(III) Complexes with Bipyridine Derivatives: From Structure to Luminescence Properties. Aust. J. Chem. 2013, 66, 676. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectroscopy; Elsevier: Amsterdam, The Netherlands, 1968. [Google Scholar]
- Patra, A.K.; Mukherjee, R. Bivalent, trivalent, and tetravalent nickel complexes with a common tridentate deprotonated pyridine bis-amide ligand. Molecular structures of nickel (II) and nickel (IV) and redox activity. Inorg. Chem. 1999, 38, 1388–1393. [Google Scholar] [CrossRef]
- Kaasjager, V.E.; Puglisi, L.; Bouwman, E.; Driessen, W.L.; Reedijk, J. Synthesis, characterization and crystal structures of nickel complexes with dissymmetric tetradentate ligands containing a mixed-donor sphere. Inorg. Chim. Acta 2000, 310, 183–190. [Google Scholar] [CrossRef]
- Herebian, D.; Bothe, E.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Experimental Evidence for the Noninnocence of o-Aminothiophenolates: Coordination Chemistry of o-Iminothionebenzosemiquinonate(1-) π-Radicals with Ni(II), Pd(II), Pt(II). J. Am. Chem. Soc. 2001, 123, 10012–10023. [Google Scholar] [CrossRef]
- Das, A.; Han, Z.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. Nickel Complexes for Robust Light-Driven and Electrocatalytic Hydrogen Production from Water. ACS Catal. 2015, 5, 1397–1406. [Google Scholar] [CrossRef]
- Goss, C.A.; Abruna, H.D. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorg. Chem. 1985, 24, 4263–4267. [Google Scholar] [CrossRef]
- Saravani, H.; Rezvani, A.R.; Mansouri, G.; Rad, A.R.S.; Khavasi, H.R.; Hadadzadeh, H. Crystal structure, magnetic and electrochemical properties of five-coordinate copper (II) complexes with 1,10-phenanthroline-5,6-dione. Inorg. Chim. Acta 2007, 360, 2829–2834. [Google Scholar] [CrossRef]
- Hadadzadeh, H.; Mansouri, G.; Rezvani, A.R.; Khavasi, H.R. A Novel 1:1 Co-Crystal of Bis(1,10-Phenanthroline)(1,10-Phenanthroline-5,6-Dione)Nickel(II) Hexafluorophosphate and Tris(1,10-Phenanthroline)Nickel(II) Hexafluorophosphate Complexes, [Ni(phen)2(phen-dione)] [Ni(phen)3] (PF6)4. J. Chem. Crystallogr. 2012, 42, 486–493. [Google Scholar] [CrossRef]
- Drosou, M.; Kamatsos, F.; Ioannidis, G.; Zarkadoulas, A.; Mitsopoulou, C.A.; Papatriantafyllopoulou, C.; Tzeli, D. Reactivity and Mechanism of Photo- and Electrocatalytic Hydrogen Evolution by a Diimine Copper(I) Complex. Catalysts 2020, 10, 1302. [Google Scholar] [CrossRef]
- Fourmond, V.; Canaguier, S.; Golly, B.; Field, M.J.; Fontecave, M.; Artero, V. A nickel–manganese catalyst as a biomimic of the active site of NiFe hydrogenases: A combined electrocatalytical and DFT mechanistic study. Energy Environ. Sci. 2011, 4, 2417–2427. [Google Scholar] [CrossRef]
- Elgrishi, N.; McCarthy, B.D.; Rountree, E.S.; Dempsey, J.L. Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes. ACS Catal. 2016, 6, 3644–3659. [Google Scholar] [CrossRef]
- Fang, M.; Engelhard, M.H.; Zhu, Z.; Helm, M.L.; Roberts, J.A.S. Electrodeposition from Acidic Solutions of Nickel Bis(benzenedithiolate) Produces a Hydrogen-Evolving Ni–S Film on Glassy Carbon. ACS Catal. 2014, 4, 90–98. [Google Scholar] [CrossRef]
- Rountree, E.S.; McCarthy, B.D.; Eisenhart, T.T.; Dempsey, J.L. Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 9983–10002. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts. ChemElectroChem 2014, 1, 1226–1236. [Google Scholar] [CrossRef]
- Cavell, A.C.; Hartley, C.L.; Liu, D.; Tribble, C.S.; McNamara, W.R. Sulfinato Iron(III) Complex for Electrocatalytic Proton Reduction. Inorg. Chem. 2015, 54, 3325–3330. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, W.T. Molecular catalysts of Co, Ni, Fe, and Mo for hydrogen generation in artificial photosynthetic systems. Coord. Chem. Rev. 2018, 373, 295–316. [Google Scholar] [CrossRef]
- Zhao, P.-H.; Li, J.-R.; Gu, X.-L.; Jing, X.-B.; Liu, X.-F. Diiron and trinuclear NiFe2 dithiolate complexes chelating by PCNCP ligands: Synthetic models of [FeFe]- and [NiFe]-hydrogenases. J. Inorg. Biochem. 2020, 210, 111126. [Google Scholar] [CrossRef] [PubMed]
- Appel, A.M.; Helm, M.L. Determining the Overpotential for a Molecular Electrocatalyst. ACS Catal. 2014, 4, 630–633. [Google Scholar] [CrossRef]
- Fourmond, V.; Jacques, P.-A.; Fontecave, M.; Artero, V. H2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation. Inorg. Chem. 2010, 49, 10338–10347. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mingot, M.; Porcher, J.-P.; Todorova, T.K.; Fogeron, T.; Mellot-Draznieks, C.; Li, Y.; Fontecave, M. Bioinspired Tungsten Dithiolene Catalysts for Hydrogen Evolution: A Combined Electrochemical, Photochemical, and Computational Study. J. Phys. Chem. B 2015, 119, 13524–13533. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.Z.; Cronin, S.P.; Mashuta, M.S.; Buchanan, R.M.; Grapperhaus, C.A. Metal-Assisted Ligand-Centered Electrocatalytic Hydrogen Evolution upon Reduction of a Bis(thiosemicarbazonato)Cu(II) Complex. Inorg. Chem. 2017, 56, 11254–11265. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, B.D.; Martin, D.J.; Rountree, E.S.; Ullman, A.C.; Dempsey, J.L. Electrochemical Reduction of Brønsted Acids by Glassy Carbon in Acetonitrile—Implications for Electrocatalytic Hydrogen Evolution. Inorg. Chem. 2014, 53, 8350–8361. [Google Scholar] [CrossRef] [PubMed]
- Zarkadoulas, A.; Koutsouri, E.; Kefalidi, C.; Mitsopoulou, C.A. Rhenium complexes in homogeneous hydrogen evolution. Coord. Chem. Rev. 2015, 304, 55–72. [Google Scholar] [CrossRef]
- Solis, B.H.; Hammes-Schiffer, S. Proton-Coupled Electron Transfer in Molecular Electrocatalysis: Theoretical Methods and Design Principles. Inorg. Chem. 2014, 53, 6427–6443. [Google Scholar] [CrossRef]
- Greene, B.L.; Wu, C.-H.; McTernan, P.M.; Adams MW, W.; Dyer, R.B. Proton-Coupled Electron Transfer Dynamics in the Catalytic Mechanism of a [NiFe]-Hydrogenase. J. Am. Chem. Soc. 2015, 137, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, G.; Cai, X. Study on deactivation and reaction mechanism of Co thiolate complexes in photocatalytic hydrogen production system. Int. J. Energy Res. 2018, 42, 977–984. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [PubMed]
- Schröder, D.; Shaik, S.; Schwarz, H. Two-State Reactivity as a New Concept in Organometallic Chemistry. Acc. Chem. Res. 2000, 33, 139–145. [Google Scholar] [CrossRef]
- Artero, V.; Saveant, J.-M. Toward the rational benchmarking of homogeneous H2-evolving catalysts. Energy Environ. Sci. 2014, 7, 3808–3814. [Google Scholar] [CrossRef]
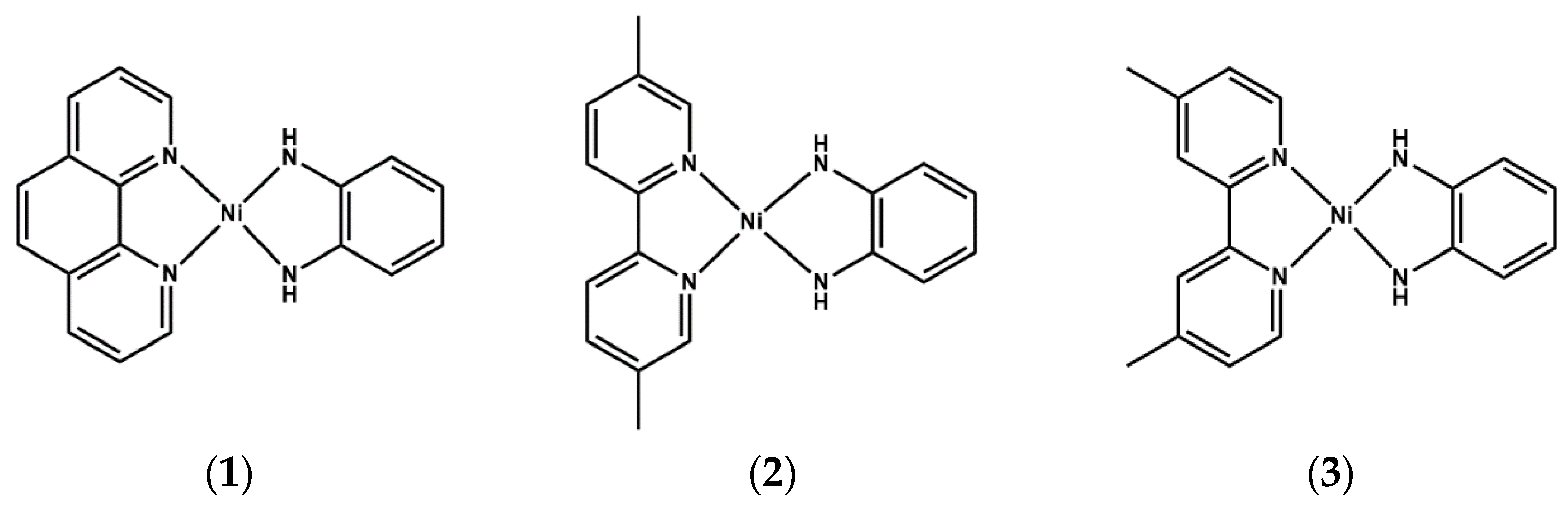




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Complex]. | Solvent | λmax (ε *) | λmax (ε *) | λmax (ε *) | λmax (ε *) |
|---|---|---|---|---|---|
| 1 (5 × 10−5 M) | DMF | 274 (80,000) | 295 (43,400) | 634 (1080) | 785 (8200) |
| 2 (5 × 10−5 M) | DMF | 274 (5600) | 298 (800) | 635 (12,400) | 787 (23,000) |
| 3 (5 × 10−5 M) | DMF | 275 (3800) | 295 (480) | 643 (25,800) | 784 (40,600) |
| Complex | Ediimine0/−1 V,(ip,a/ip,c) | Ediamine0/−1 V,(ip,a/ip,c) | E0NiII/I V,(ip,a/ip,c) | |
|---|---|---|---|---|
| 1 | Ep,a = −0.45 * Ep,c = −0.30 | Ep,a = −0.95 Ep,c = −0.89 | Ep,a = −1.38 *** Ep,c = −1.32 | Ep,a = −2.12 * Ep,c = −1.96 |
| 2 | - | - | Ep,a = −1.74 ** | Ep,a = −2.12 ** |
| 3 | - | Ep,a = −0.94 *** Ep,c = −0.92 | Ep,a = −1.38 *** Ep,c = −1.32 | Ep,a = −2.04 Ep,c = −1.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kourmousi, M.; Kamatsos, F.; Mitsopoulou, C.A. Visible Light-Driven Hydrogen Evolution Catalysis by Heteroleptic Ni(II) Complexes with Chelating Nitrogen Ligands: Probing Ligand Substituent Position and Photosensitizer Effects. Energies 2024, 17, 2777. https://doi.org/10.3390/en17112777
Kourmousi M, Kamatsos F, Mitsopoulou CA. Visible Light-Driven Hydrogen Evolution Catalysis by Heteroleptic Ni(II) Complexes with Chelating Nitrogen Ligands: Probing Ligand Substituent Position and Photosensitizer Effects. Energies. 2024; 17(11):2777. https://doi.org/10.3390/en17112777
Chicago/Turabian StyleKourmousi, Maria, Fotios Kamatsos, and Christiana A. Mitsopoulou. 2024. "Visible Light-Driven Hydrogen Evolution Catalysis by Heteroleptic Ni(II) Complexes with Chelating Nitrogen Ligands: Probing Ligand Substituent Position and Photosensitizer Effects" Energies 17, no. 11: 2777. https://doi.org/10.3390/en17112777
APA StyleKourmousi, M., Kamatsos, F., & Mitsopoulou, C. A. (2024). Visible Light-Driven Hydrogen Evolution Catalysis by Heteroleptic Ni(II) Complexes with Chelating Nitrogen Ligands: Probing Ligand Substituent Position and Photosensitizer Effects. Energies, 17(11), 2777. https://doi.org/10.3390/en17112777