1. Introduction
The use of plastic products in the world is rising every day. Because they are flexible, durable, lightweight and cheap in cost, plastic materials have become an inseparable part of our lives, as they are frequently utilized in homes, industry and agriculture to make a wide variety of products related to daily human life. Plastics are polymeric materials derived from natural resources [
1]. Excessive usage of these polymers is connected with creating enormous amounts of waste, representing a great threat to the environment. They are disposed of using various techniques, including burning, land-filling, reusing and conversion into value-added products. However, most of these techniques are inexpensive; they create pollution with several environmental consequences. Among these approaches, conversion into value-added products is now being explored as a result of its many scientifically proven benefits [
2]. Plastic waste management has become a global issue due to its non-biodegradable nature. Plastic trash is classified as either pre-consumer or post-consumer waste. Plastic scraps created by industries during product manufacture are included in pre-consumer plastic trash. Because they are clean and similar, these plastics are typically easier to recycle. Consumers generate post-consumer plastic wastes after using products. Because they are difficult to collect, dirty after use and heterogeneous, these plastics are more challenging to recycle. Approximately, only 9 percent of the 400 million tons of plastic generated each year in the world is recycled, and 12 percent is burned. Landfills, rivers and seas are becoming dangerously polluted with plastic trash [
1,
3]. If burned, they emit carbon dioxide and potentially hazardous gases, which accounted for 5.9 million cubic meters of carbon dioxide in 2015 [
3]. As a result, recycling and plastic waste management are becoming increasingly important. The waste plastic polymer can be recycled into original monomers or other useful compounds such as fuel, since plastic and petroleum have the same basic hydrocarbon components. Waste plastic fuel has similar properties to diesel fuel, and it has been concluded that it can be used instead of diesel. The calorific value of fuel generated from plastic waste is about 9829.3515 kcal/kg, which is almost equal to the calorific value of diesel. It is also possible to produce many other petroleum derivatives from waste plastics such as gasoline, kerosene, heavy fuel oil (HFO), light fuel oil (LFO) and furnace oil, thus reducing the high demand for fossil fuel products [
4].
Pyrolysis is the thermal breakdown of big molecules in an inert environment with the absence of oxygen at high operating conditions such as temperature and pressure [
1,
4]. In general, plastics are long-chain molecules of hydrocarbon and include several kinds such as high-density polyethylene (HDPE), polypropylene (PP), polystyrene (PS), polyethylene terephthalate (PET), low-density polyethylene (LDPE), polyvinyl chloride (PVC) and other less famous plastics, which have been used less as polymers. Among these groups, a higher amount of PP is produced in comparison with other types of plastic products that generate large amounts of waste each year [
3]. The GC/MS instrument has been used to successfully characterize the thermal degradation of PP waste plastic to generate fuel oil. The heavy oil generated had a dark color and an unpleasant odor. According to the GC/MS study, heavy fuel has a carbon chain range of C
4 to C
35, similar to fuel-grade oil. The fuel oil produced from PP could be an alternative to renewable energy and be used in industry and transportation [
3]. Vijayakumar et al. [
5] reviewed a summary of different types of plastics and the potential of pyrolysis for fuel production. The pyrolysis process is approved as a potential method to generate energy from plastic waste. The pyrolysis results of PP polymer in a batch reactor were achieved with a temperature of 380 °C. The ratio of oil obtained was 80.1 wt%, while the gas and solid ratios were 6.6 and 13.3 wt%, respectively. Pyrolysis of PP was performed by Samosa et al. [
6] at a temperature of 350 °C, and the product was a liquid oil with a density of 0.8204 g/cm
3. GC/MS analysis confirmed that the compounds obtained from the thermal cracking had carbon chains between C
7 and C
27, which was an indication that this compound is a mixture of kerosene (C
8 to C
19)–diesel (C
7 to C
27) fractions. Hakeem et al. [
7] studied the pyrolysis process that converts polypropylene plastic waste into usable liquid fuel at an optimal temperature of 450 °C and a catalyst-to-plastic ratio of 1 to 3. Kaolin was shown to be an efficient, low-cost catalyst for converting PP to gasoline/diesel grade fuel. The kaolin was more selective for liquid pyrolysis products than solid and gaseous pyrolysis products. The oil obtained from the kaolin-catalyzed pyrolysis reaction is higher than the oil obtained from the uncatalyzed reaction. Moreover, thermal pyrolytic liquid fuel requires further development to be commercially viable and suitable for use as fuel. When the catalyst was used, the FTIR findings revealed that the oil included hydrocarbons with different functional groups due to the secondary breaking of the oil into lighter products. In fact, the feedstock plastic-type, cracking temperature, heating rate, operation pressure, reactor type, residence time, catalyst feed ratio and catalyst type are the key parameters impacting the plastic pyrolysis process and pyrolysis product molecular dispersion. Temperature is one of the main significant operational variables since it controls the polymer materials’ cracking response. By raising the temperature, not all polymer materials can be fractured. The Van der Waals force is the force between molecules that keeps them together and prevents them from collapsing. When the vibration of molecules is high enough, they evaporate from the liquid surface [
8]. However, if the energy produced by the Van der Waals force along the polymer chains is higher than the enthalpy of the C-C bond in the chain, the carbon chain will be broken [
9]. This is why when a high molecular weight polymer is heated, it decomposes rather than boils [
8,
9,
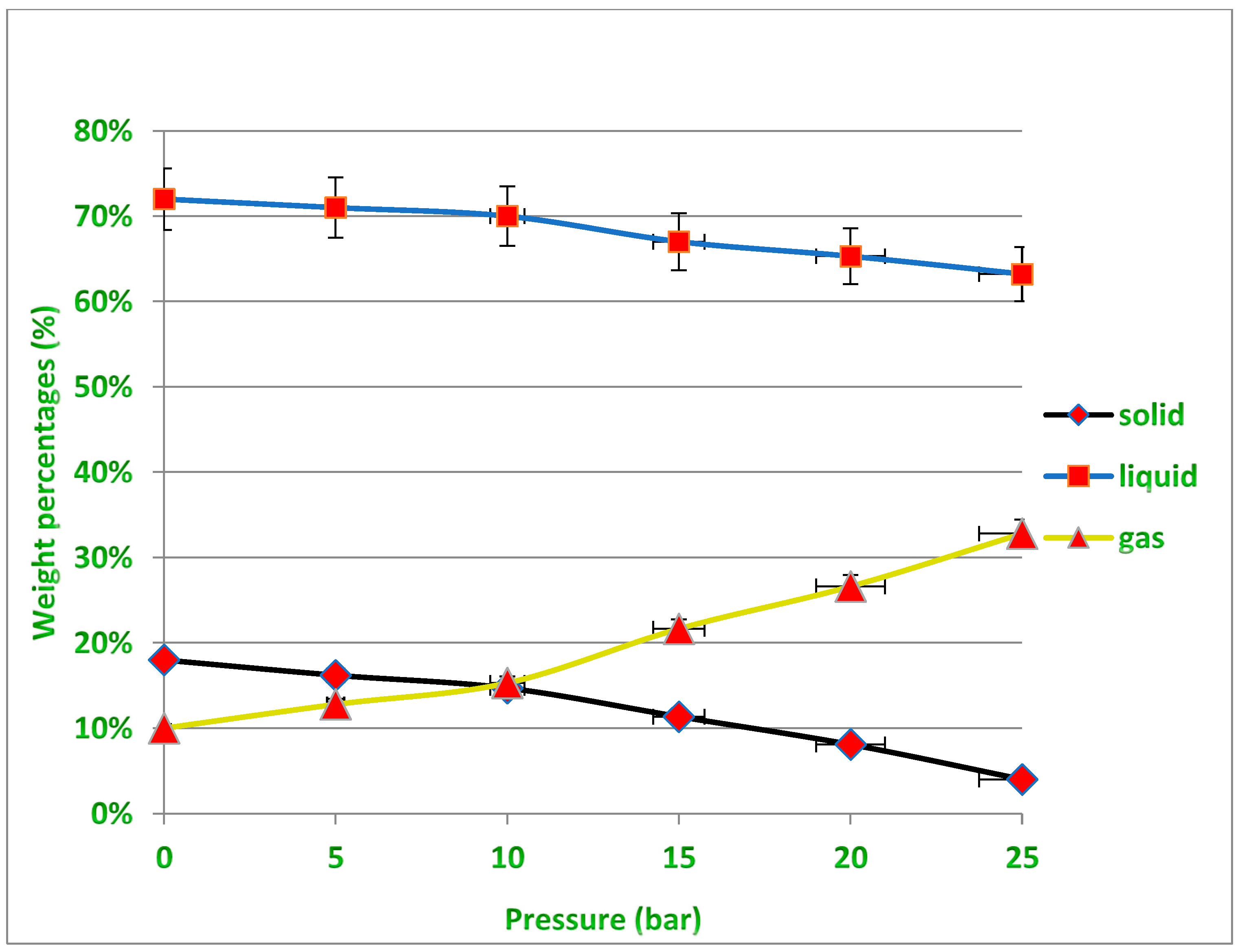
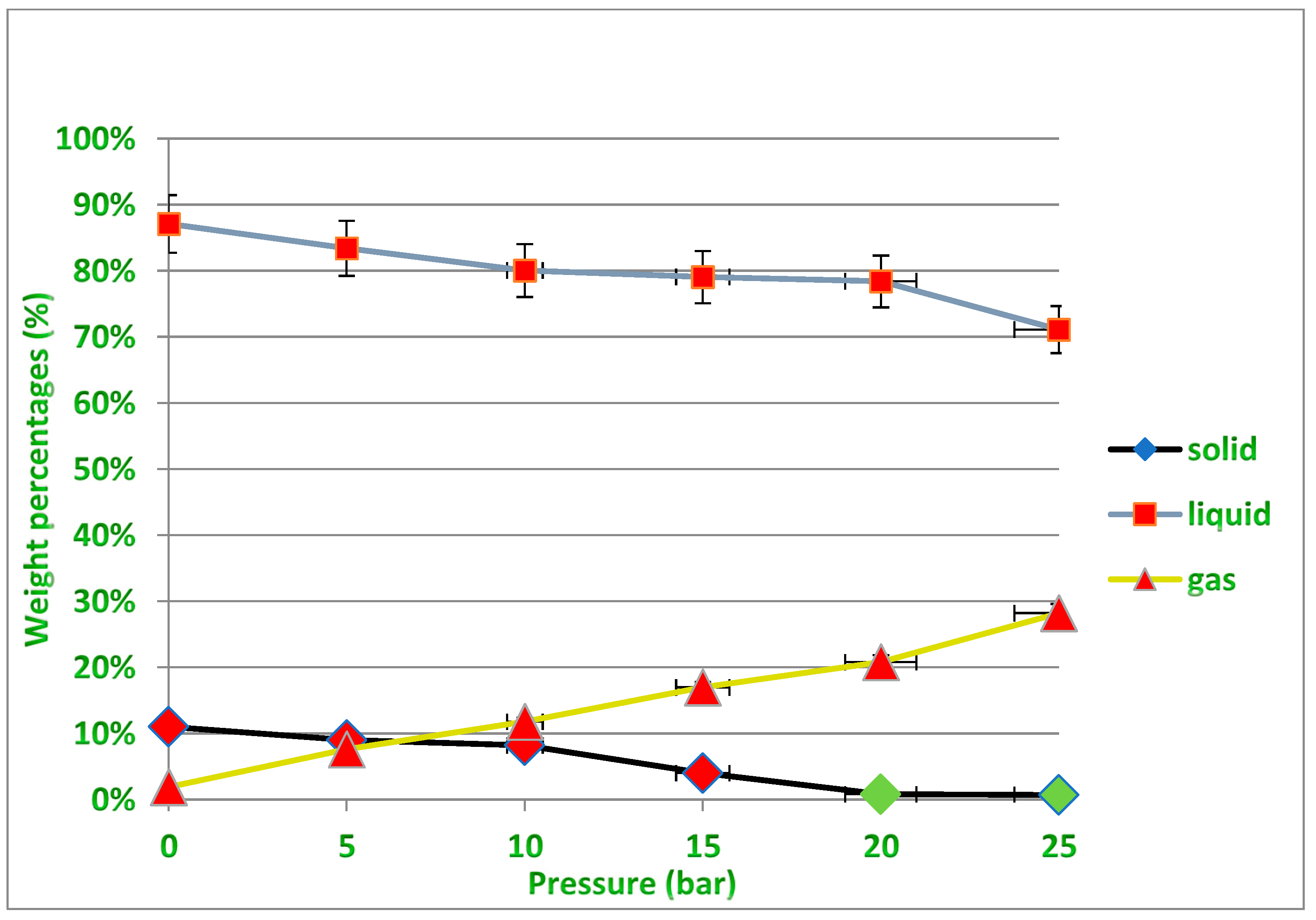
10]. The second parameter that researchers focused on is pressure; both the pyrolysis process and the amount of hydrocarbons and their fractions in pyrolysis products are affected by operating pressure. Because the boiling points of pyrolysis products grow with increasing pressure, heavier hydrocarbons are further pyrolyzed rather than evaporated at a given operating temperature in a pressurized atmosphere. In practice, additional energy is necessary for subsequent hydrocarbon cracking in pressured pyrolysis. High pressure may increase the product gases and lower the yield of liquid products and solid residue. As a result, the average molecular weight of the gas, liquid and solid products also decreases with pressure [
10,
11,
12,
13]. In order to optimize plastic pyrolysis reactions and modify the distribution of pyrolysis products, catalysts are widely used in research and industrial pyrolysis processes, which is the third and most important parameter that has attracted the attention of researchers in this field. The thermal decomposition of waste plastics can be accelerated with the use of appropriate catalysts. Zeolite, alumina, silica alumina, the FCC catalyst, the reforming catalyst and other catalysts are commonly employed in this process [
4,
14,
15,
16,
17].
Table 1 summarizes some of the most common catalysts used in polymer cracking reactions.
In catalytical pyrolysis, lower operating conditions will be needed for the thermal reaction [
10]. If the cracking process is achieved by a reaction with hydrogen instead of an inert gas, it is known as hydrocracking [
21]. Hydrocracking of polymer waste typically involves complex reactions that occur on the surfaces of special metal-loaded catalysts, including the reactions of hydrogenation and dehydrogenation inside a stirred batch autoclave at moderate temperatures and pressures, typically 150–450 °C and 20–100 bar, and obtained products containing high-quality fuel [
21]. The application of cracking reactions in the presence of hydrogen reduces unwanted solid residues and increases the selectivity of single-ring aromatics compared to inert pyrolysis. In other words, hydrogen is more beneficial to pyrolysis in terms of lowering coke yield and increasing hydrocarbon yield, and this fact was concluded by Yuan Xue et al. [
22]. An alumina catalyst was preferred because it is classically used in the petroleum refineries for cracking reactions in various parts of the world [
21,
23]. Therefore, it is possible to take the spent catalyst from these refineries and use it in polymer waste hydrocracking reactions after regenerating. In addition, the high acidity properties and the moderate price of alumina catalysts makes it possible to use them without affecting the economic viability of the process compared to other expensive catalysts such as zeolites [
24,
25]. Moreover, it was shown that adding metals such as molybdenum, platinum and nickel to a catalyst can increase the acidity of the catalyst, and more cracking of large molecules with better liquid quality would be achieved due to the saturation of unsaturated hydrocarbon, and produce a more stable range of gasoline–diesel products [
21].
The aim of this work was to compare the quantity and quality of the reaction products from non-catalytic thermal- and catalytic hydrocracking processes. However, the focus was on making the hydrocracking process economical in terms of its practical application by separating the deactivated catalyst (i.e., spent catalyst) from the solid residues at the end of the plastic waste hydrocracking reactions. This was then further utilized after regeneration (i.e., reactivation) and its properties tested through its catalytic behavior and reaction products because the cost of using a fresh catalyst in each reaction makes recycling the waste into useful products an expensive and uneconomical procedure. Thus, the reaction conditions that give the best yield (i.e., maximizing the amount of useful products, such as oils and gases, while at the same time minimizing environmentally harmful solid waste as much as possible) are taken into account in this research. Since reducing the percentage of solid residues inside the reactor facilitates the complete separation of the spent catalyst after the end of the reaction, it can then be used again after reactivation in subsequent reactions. In addition, the data of the produced fuel from the conversion of waste polypropylene were compared experimentally and theoretically, through mathematical modeling, to determine the efficiency of the catalysts, calculating the activation energies of the reactions that took place on their surface, and comparing them with the experimental results.
4. Kinetic Modeling
Building the kinetic model requires conducting a series of practical experiments at different temperatures. Therefore, in this study, for the purpose of building the lump kinetic model, six catalytic hydrocracking experiments were conducted on two types of catalysts. Studying the behavior of each catalyst needs three experiments at different temperatures (i.e., 400, 425 and 450 °C) to evaluate the model parameters.
Table 8 shows these results.
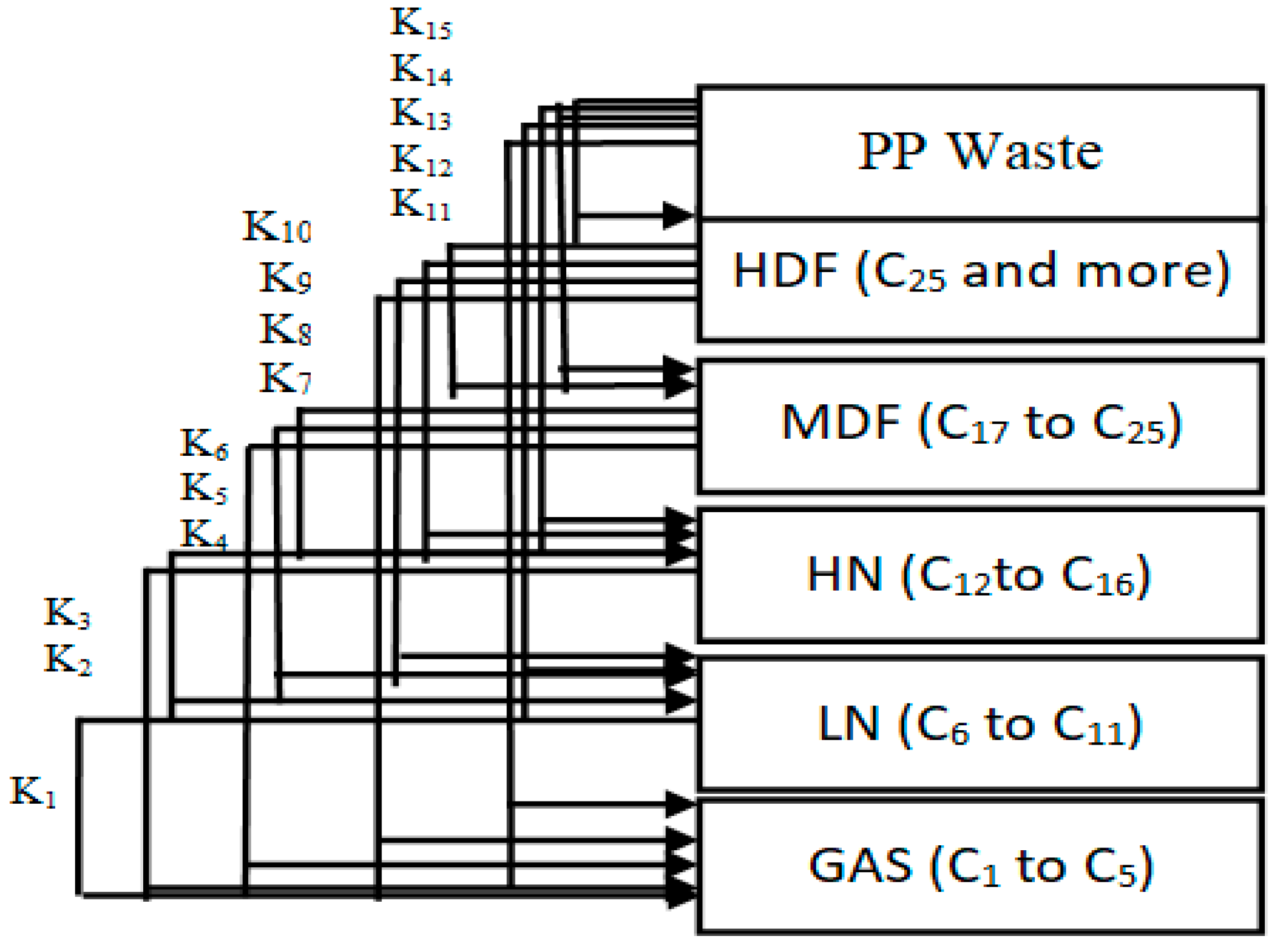
The products of the hydrocracking process were divided into five lumps, depending on the GC-MS results. The division was based on the number of carbon atoms in the produced hydrocarbons. The five lumps were gas (GAS), light distillates including light naphtha (LN) heavy naphtha (HN), middle distillate fuel (MDF), and finally, heavy distillate fuel (HDF). It is clear from
Table 8 that the tendency to produce lower lumps such as gas, light naphtha and heavy naphtha increases as the temperature rises due to further hydrocracking of heavy hydrocarbons into lighter weights. On the other hand, a lower temperature causes middle distillate fuel and heavy distillate fuel to increase. The reaction pathways for the hydrocracking of plastic wastes proposed by Ramdoss and Tarrer assume five lumps and seven reactions [
52,
53]. The researchers studied the kinetic model of hydrocracking by assuming the dependence of the reaction rate constant on the Arrhenius equation. Pre-exponential factor values and activation energies for each reaction were determined by solving differential equations and regression of the data simultaneously. This method proposed by Ramdoss and Tarrer for the investigation of the kinetic model has been widely used, especially in the study of crude oil hydrocracking residue [
54,
55,
56,
57]. All the kinetic models suggested by these studies use the lumps model with a different number of lumps for each study. The increase in the number of lumps gives more accurate results for calculating model parameters. Generally, this process is very similar to the hydrocracking of waste plastics. Thus, the same Ramdoss and Tarrer methodology is utilized, considering the modification on the use of six lumps and fifteen reactions, as shown in
Figure 14.
Reactions 11 to 15 are called primary catalytic reactions because they are the first step of the decomposition of PP into other lumps (i.e., the slowest step in a reaction mechanism, which is known as the rate-determining step), and the other reactions (i.e., from reaction 1 to 10) are called secondary catalytic reactions because they follow the primary catalytic reaction. For each lump in
Figure 14, a mole balance was made, and six-mole balances were solved using MATLAB software. The MATLAB program optimizes the mole balances equations and the experimental results in
Table 8 depending on the genetic algorithm that is designed to fit the Arrhenius equation.
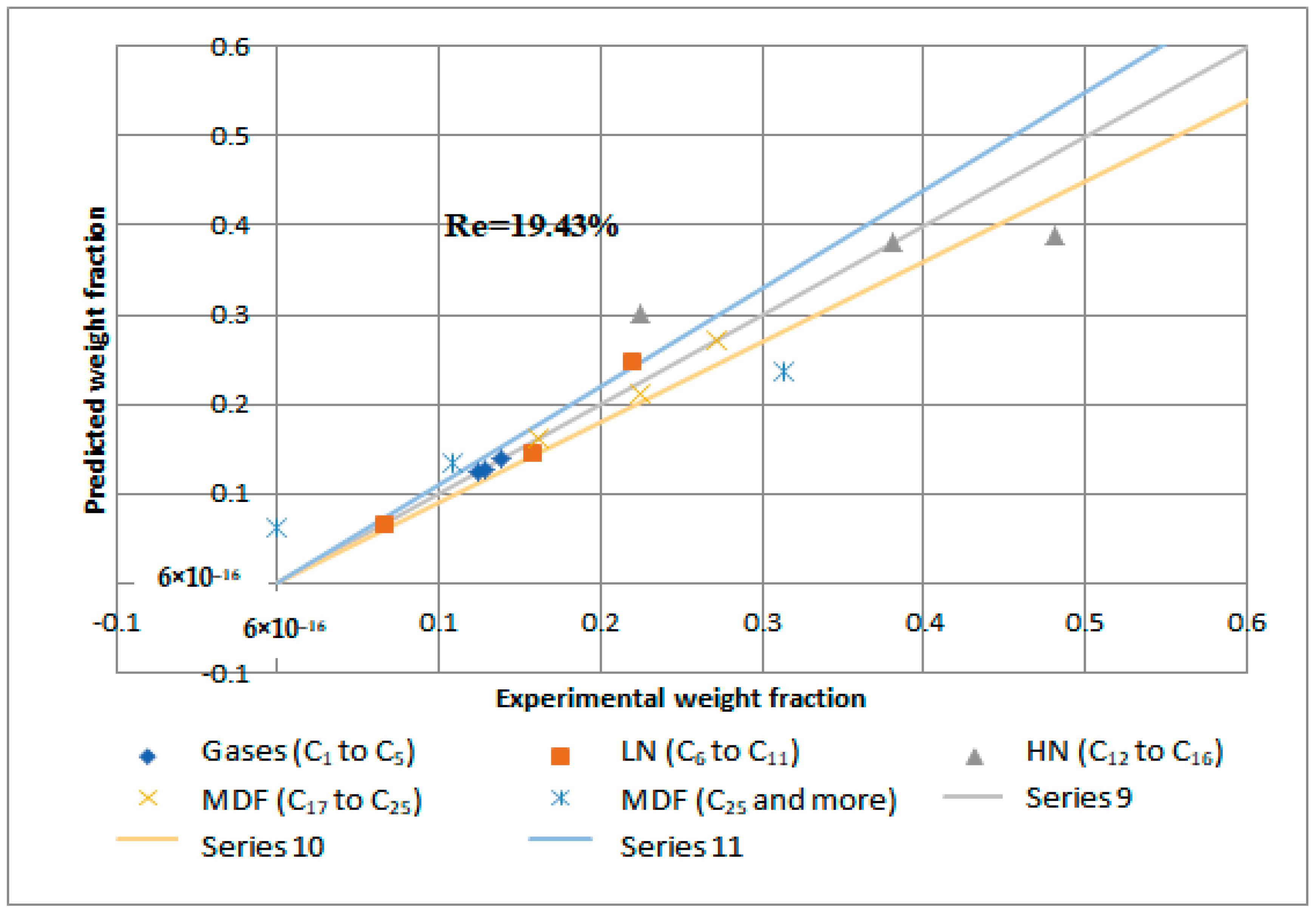
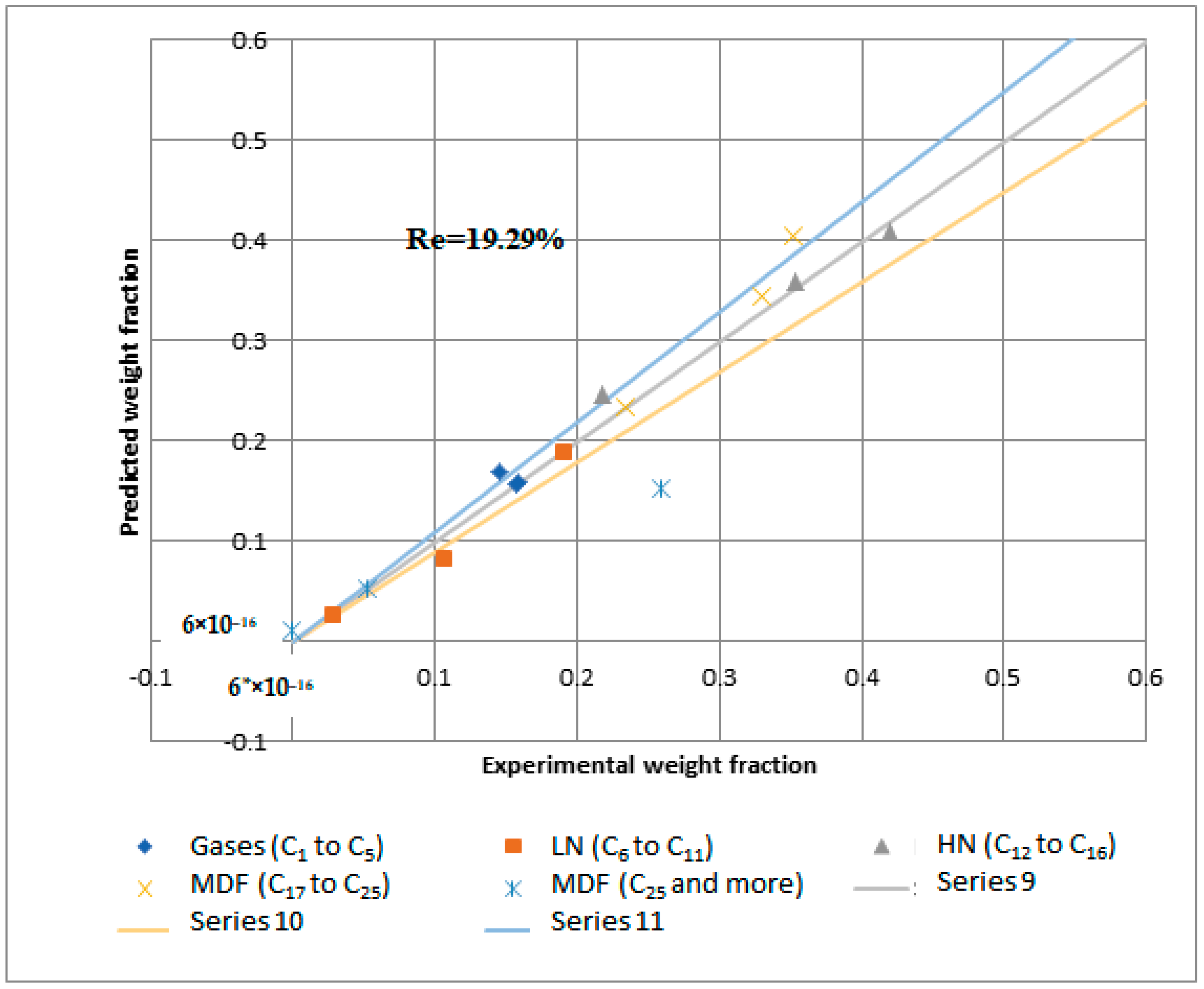
Figure 15 and
Figure 16 demonstrate that the regression of the kinetic model is acceptable. However, it had a relative error value of 19.43 and 19.29% for the NiMo/Al
2O
3 and Pt/Al
2O
3 catalysts, respectively.
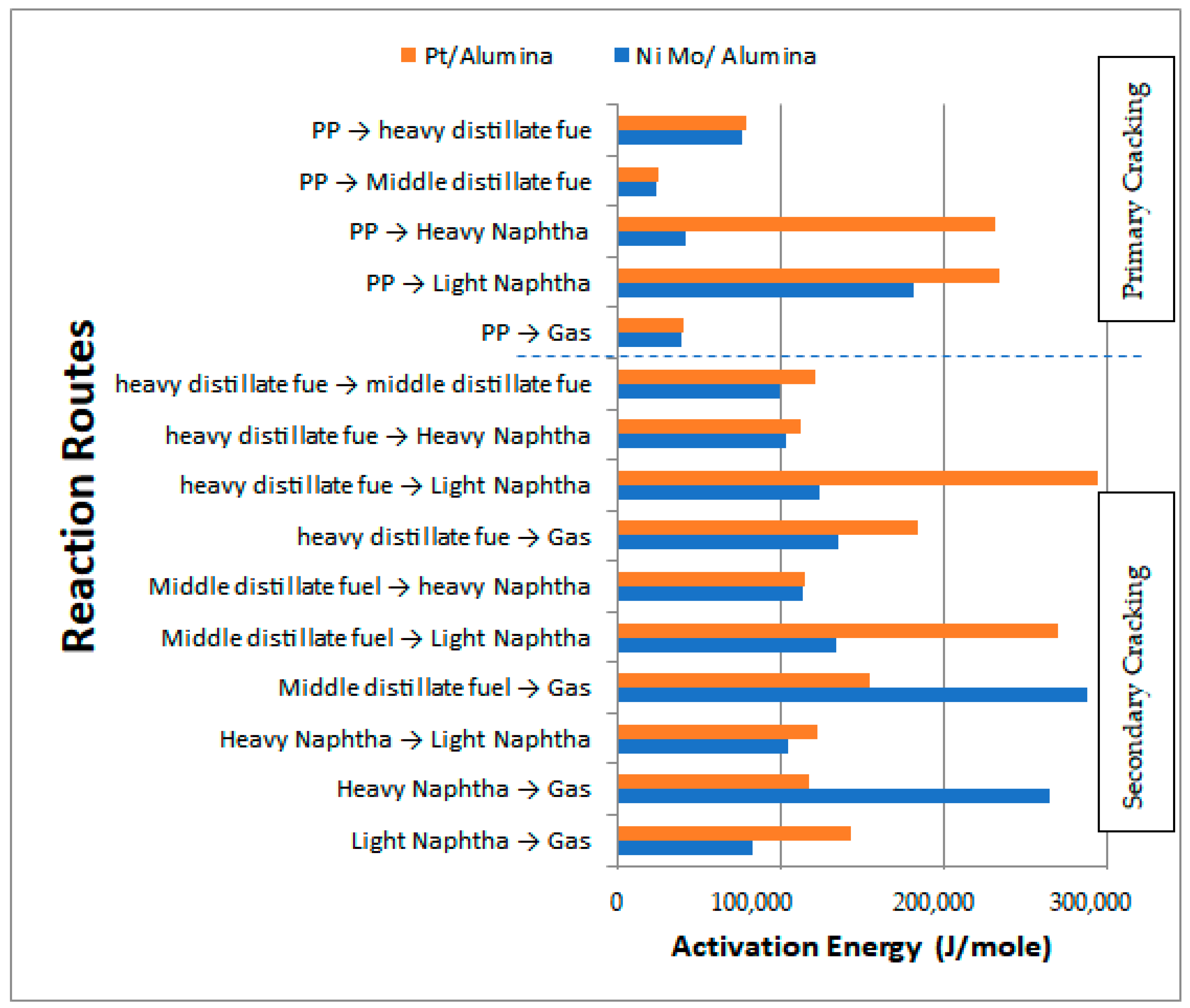
Table 9 shows the activation energies and reaction rate constants for all reactions at a reaction temperature of 450 °C over the two types of catalysts used, while
Figure 17 illustrates the obtained activation energies for the primary and secondary catalytic reactions. In general, the values of the activation energies over the Pt/Al
2O
3 catalyst showed a larger value than those on the NiMo/Al
2O
3 catalyst. In particular, the primary reactions demonstrated high values for the activation energies of HN and LN of 232,017.95 and 234,521.65 J/mol, respectively, with the values for MDF and HDF being 25,332.65 and 78,823.90 J/mol, respectively. This result demonstrates that the decomposition of the large PP molecules on the surface of the Pt/Al
2O
3 catalyst into primary hydrocarbon lumps is more difficult than the case of the large PP molecules on the surface of the NiMo/Al
2O
3 catalyst, and thus more energy is required to break the large PP molecules into smaller ones.
In summary, all primary catalytic reactions show greater activation energy values on the Pt/Al
2O
3 catalyst except for the production of the gas lump, where its value decreased to 39,245.980 J/mol compared to 39,844.25 J/mol on the NiMo/Al
2O
3 catalyst. This result is consistent with the experimental results previously reported in
Table 4 where less liquid and a higher amount of gas were obtained on the Pt/Al
2O
3 catalyst as compared to the products obtained on the NiMo/Al
2O
3 catalyst since the lower the activation energy value on the surface of the catalyst, the amount of production of a particular product increases due to the increase in the reaction rate. On the other hand, the activation energies of the secondary catalytic reactions also give a higher value over Pt/Al
2O
3 than that over NiMo/Al
2O
3 in seven out of ten reactions (i.e., that is, except for reactions 2 to 4).
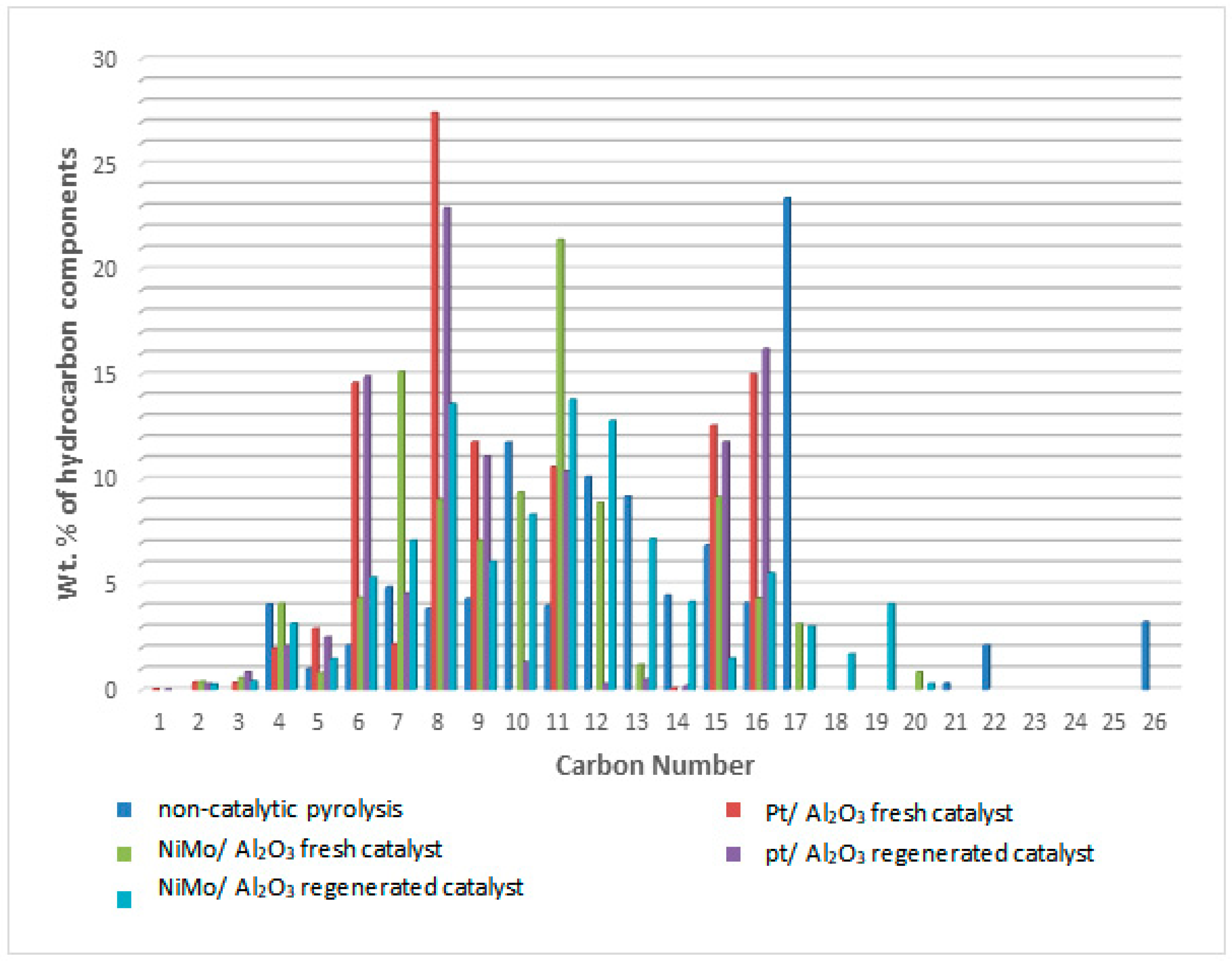
For the purpose of comparing the result of the kinetic model with the results of the experimental work, five product lumps were selected to increase the accuracy of the parameters determined in the kinetic model and compared with the data of three fuel fractions in the experimental work. For the comparison between the kinetic model and the experimental results, the lump of LN in the model is consistent with the data for the gasoline fraction, while the HN, MDF and HDF lumps are consistent with the diesel data; this division of components certainly depends on the number of carbon atoms in the hydrocarbons produced.
Therefore, the apparent activation energy was calculated for the gas, gasoline and diesel ranges to build a clear view of this comparison. The apparent activation energy of the parallel reaction was calculated using Equation (2) [
53], and
Table 10 demonstrates the apparent activation energy for the liquid and gas on one side, and for the components of the liquid fraction (i.e., gasoline and diesel) on the other side.
where
Eapp is the apparent activation energy and
K is the reaction rate constant.
Firstly, the apparent activation energy value was calculated from the results of the kinetic modeling for the gas product depending on the reactions that produce the gas only (i.e., reactions 1, 2, 4, 7 and 11), as shown in
Table 10 and
Figure 17. The calculated apparent activation energy shows a lower value in the case of using the Pt/Al
2O
3 catalyst, which is in the order of 39,252.03 J/mol, while the corresponding apparent activation energy obtained using the NiMo/Al
2O
3 catalyst is in the range of 39,845.02 J/mol, and this result matches the experimental work data in
Table 4, which showed a lower weight ratio of the gas produced if the NiMo/Al
2O
3 catalyst is employed. Secondly, the liquid products include the hydrocarbons produced within the range of LN, HN, MDF and HDF (i.e., reactions 3, 5, 6, 8, 9 and 10). The apparent activation energy value of the liquid components was calculated based on Equation (2) as well. It is noted that in the case of a liquid, the behavior is opposite to that of a gas. The apparent activation energy was 69,260.76 and 66,301.42 J/mol for the Pt/Al
2O
3 and NiMo/Al
2O
3 catalysts, respectively. These results are also consistent with the data of the experimental work, where the liquid production increased over the NiMo/Al
2O
3 catalyst in comparison with the liquid produced over the Pt/Al
2O
3 catalyst. On the other hand, the gasoline and diesel fractions were investigated to ensure that the results of the kinetic model matched the data of the experimental work. The LN lumps’ production reactions that correspond to the gasoline production reactions demonstrated a large value of activation energies when the NiMo/Al
2O
3 catalyst was used. This means that more energy is required to produce the LN range fractions on this catalyst. Moreover, in
Table 10, the apparent activation energy for gasoline production (i.e., reactions 3, 5, 8 and 12) on the catalyst surface of NiMo/Al
2O
3 is in the order of 108,234.89 J/mol, and then this value decreases to 103,404.02 J/mole when the Pt/Al
2O
3 catalyst is used. This result is consistent with the experimental results documented in
Table 5 where they were 34.45 and 49.85 wt% for the NiMo/Al
2O
3 and Pt/Al
2O
3 catalysts, respectively. Gasoline is produced in larger quantities on the surface of the Pt/Al
2O
3 catalyst. The production of the diesel fraction includes the reactions 6, 9, 10, 13, 14 and 15. The value of the apparent activation energy was recorded as 66,301.29 J/mole using the NiMo/Al
2O
3 catalyst. In contrast, the value of 69,260.46 J/mole was determined using the Pt/Al
2O
3 catalyst, and here there is also a logical agreement in the obtained results with the results of the experimental work. Diesel is produced in larger quantities on the surface of the NiMo/Al
2O
3 catalyst. Finally, it is possible to conclude that the results of the kinetic model conclusively agree with the results of the experimental work for the catalytic hydrocracking of PP waste plastic. Thus, the suggestive pathways of the reaction mechanism in this work have confirmed and correctly explained the reactions that occurred throughout the process.
5. Conclusions
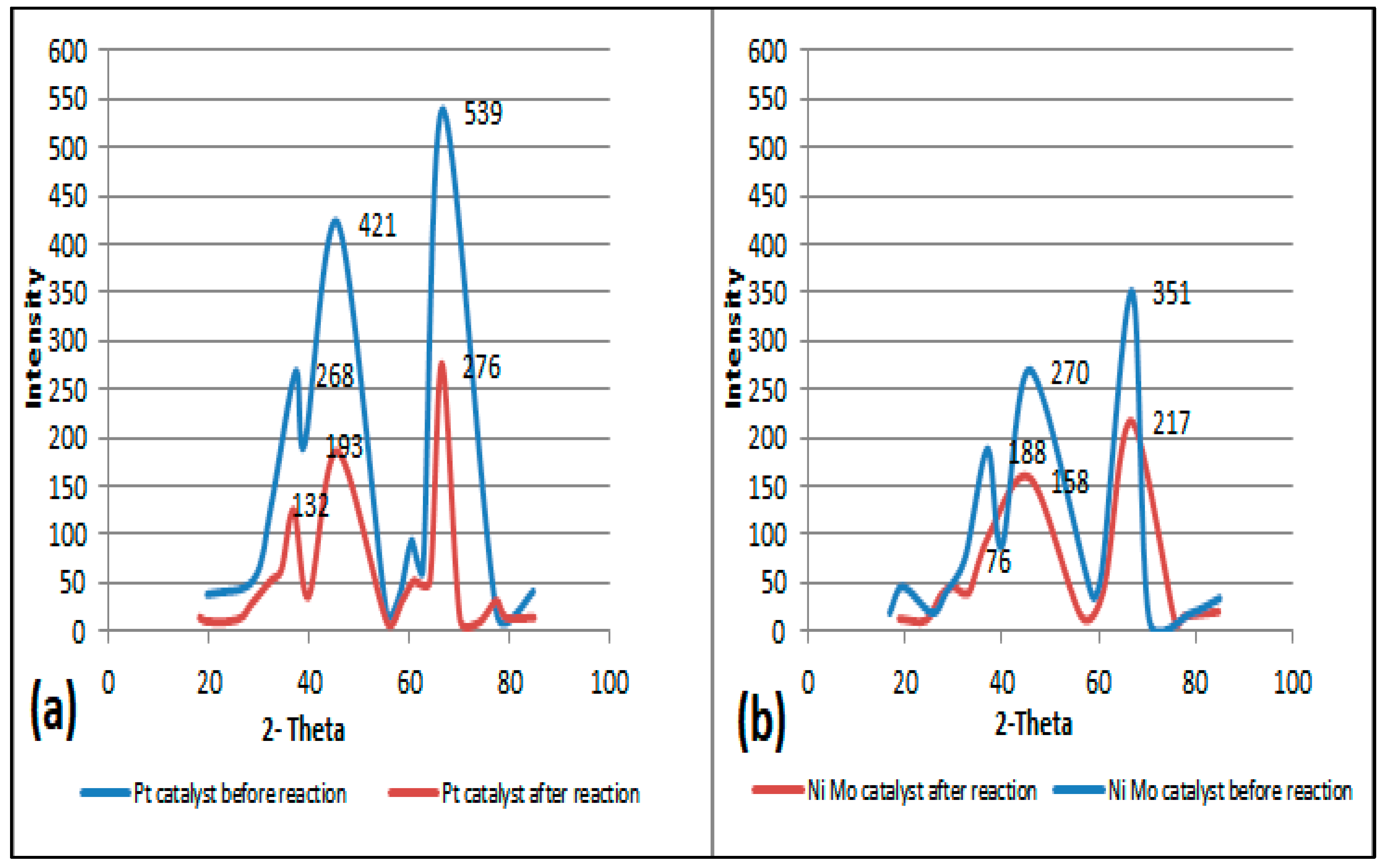
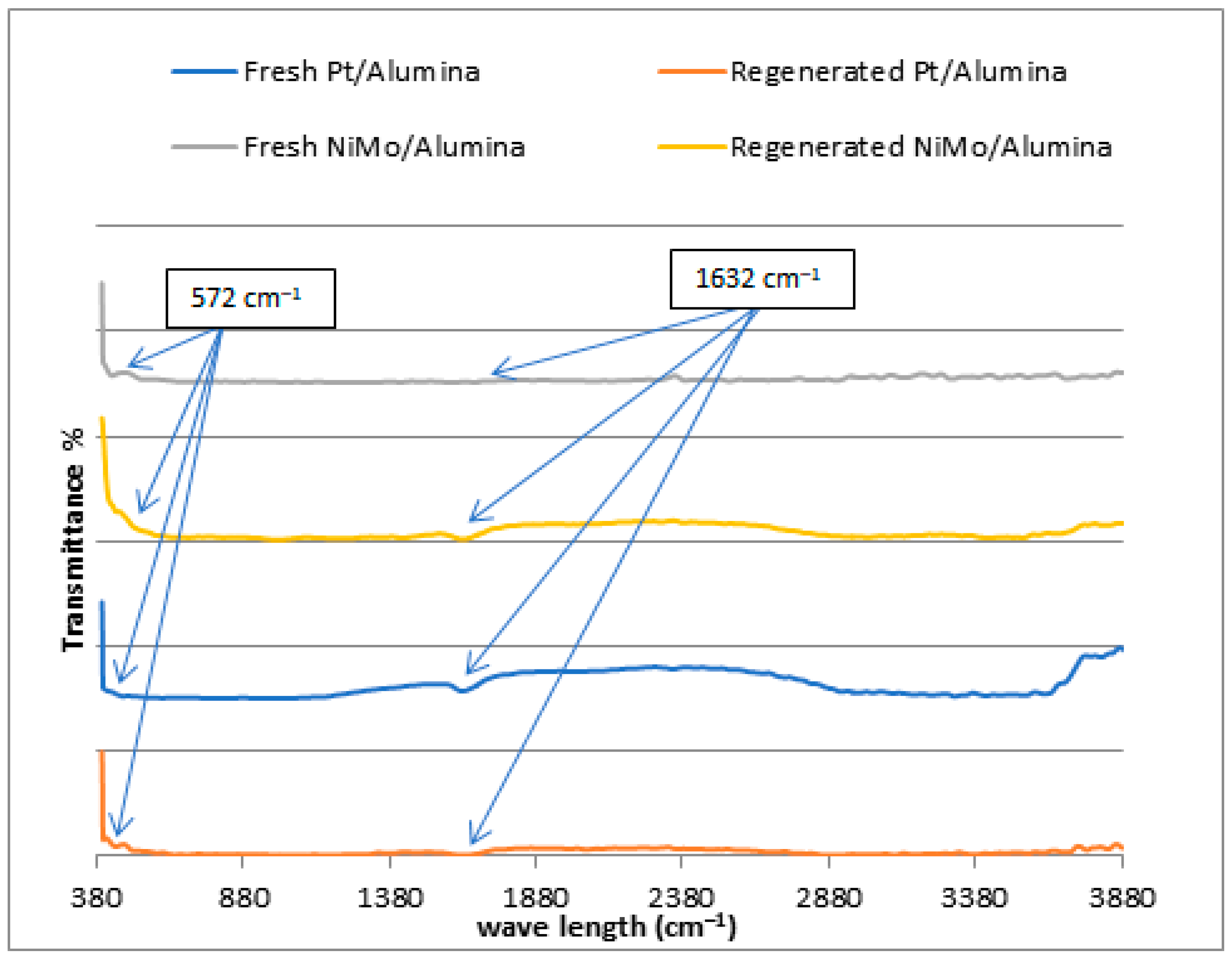
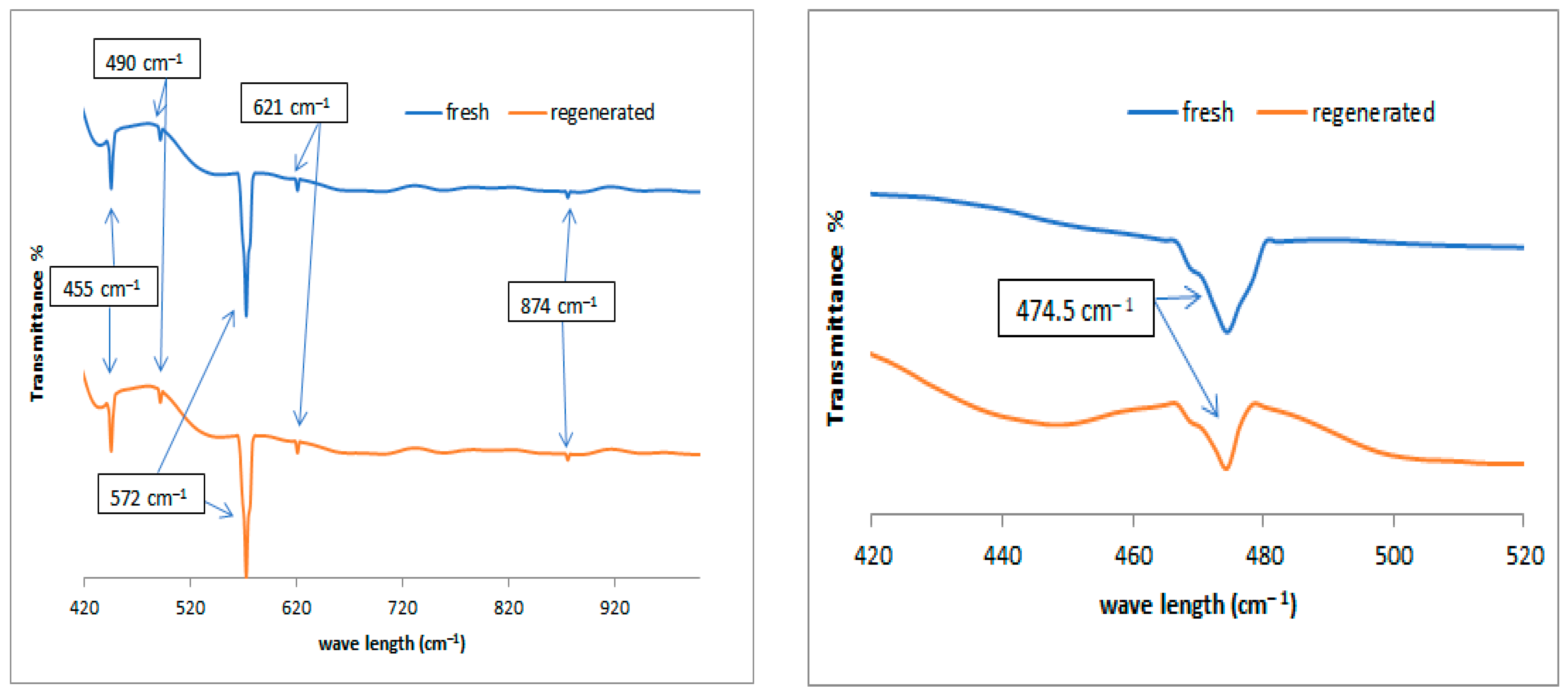
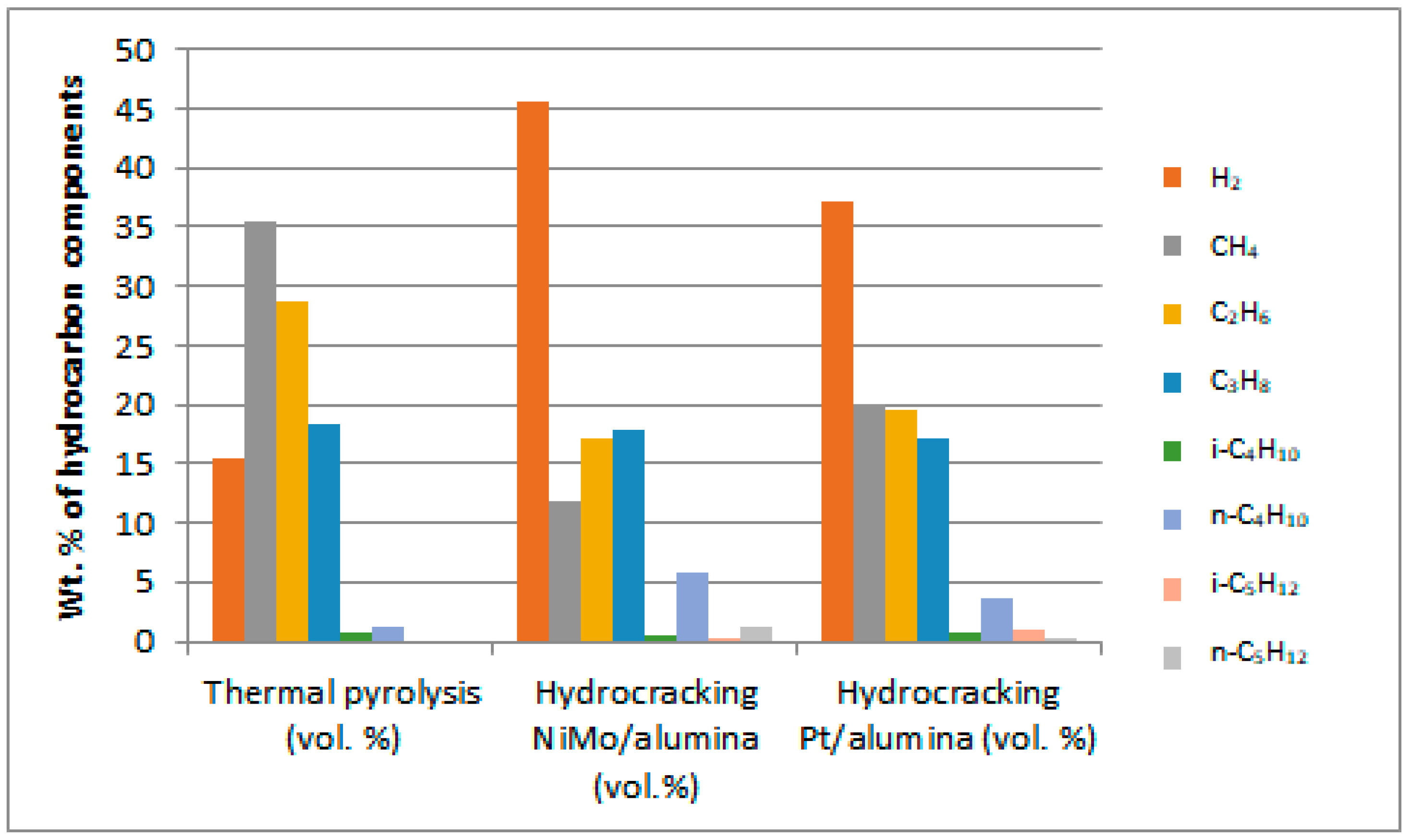
Non-catalytic thermal pyrolysis and catalytic hydrocracking using an alumina catalyst after metal loading via the wetness impregnation method gives an effective catalyst for converting waste plastics into valuable liquid and gaseous fuels. A temperature of 450 °C and a pressure of 20 bar in thermal and hydrocracking were seen as the best conditions for minimizing the solid residue to ensure that the used catalyst could easily be extracted from the reaction residues at the end of the reaction. Burning the spent catalyst at 700 °C proved successful in regenerating it to give excellent catalytic behavior while being used in the reaction again. The properties of the fresh and reactivated catalyst were investigated using TGA, XRD, SEM, EDX, BET and FTIR analyses. This was to ensure that severe reactivation conditions did not affect the structure or catalytic activity of the catalysts. GC-MS analysis showed that the fresh NiMo/Al2O3 catalyst gives the best liquid hydrocarbons yield of 86 wt% compared with non-catalytic pyrolysis and the fresh Pt/Al2O3 catalyst, which recorded 78.4 and 84 wt%, respectively. After using the regenerated catalyst, the proportion of the liquid produced was slightly decreased to 83.3 and 82.9 wt% over the NiMo/Al2O3 and Pt/Al2O3 catalysts, respectively. The obtained solids were 0.22, 0.4 and 0.8 wt% and the gases were found to be 13.78, 15.6 and 20.8 wt% over the NiMo/Al2O3 and Pt/Al2O3 fresh catalysts and non-catalytic pyrolysis, respectively. It was then observed that the ratios of the produced gas increased to reach 16.02 and 16.1 wt% when using the regenerated NiMo/Al2O3 and Pt/Al2O3 catalysts, respectively. In addition, the results showed that the hydrocracking over the Pt/Al2O3 catalysts gave the highest weight of products of the gasoline fractions (C6–C12), as the percentage of the produced gasoline range increased from 15.94 wt% in non-catalytic pyrolysis to 34.45 and 49.85 wt% over the NiMo/Al2O3 and Pt/Al2O3 fresh catalysts, respectively. When a regenerated catalyst was used instead of a fresh one, the gasoline yield decreased slightly to 48.17 and 31.39 wt% over the Pt/Al2O3 and NiMo/Al2O3 catalysts, respectively. Overall, the non-catalytic pyrolysis process led to an increase in the weight percentage within the diesel fuel (C13–C28) to 80.83 wt% compared to the content of the produced gasoline fuel. Similarly, in the case of using catalytic hydrocracking with the presence of either fresh or regenerated catalysts, taking into account a reduction in the proportion of diesel fuel produced compared to the non-catalytic cracking process, the gas in range of (C1–C5) showed that the most significant proportion of the resulting gases were combustible gases (i.e., methane, ethane, propane, butane and pentane) with a large proportion of hydrogen gas as well. Moreover, the availability of a greater proportion of oxygen gas in non-catalytic pyrolysis reactions led to the emergence of a greater proportion of carbon dioxide gas in its products compared to the gaseous products in the hydrocracking process. Furthermore, the results of the viscosity of the produced liquid show that the viscosity follows the trend: Pt/Al2O3 < NiMo/Al2O3 < non-catalytic thermal cracking, whereas the lowest pour point and flash point possessed by the liquid products were found on the surface of the fresh and regenerated Pt/Al2O3 catalysts. A decrease in the pour point and flash point values indicates the enhancement in the flowability of the crude oil and an increase in the volatile hydrocarbon components of ignition when exposed to a spark. Finally, in this work, the same methodology as that used by Ramdoss and Tarrer was applied, with the usage modified to six lumps and fifteen reactions. All primary reactions showed greater activation energy on the Pt/Al2O3 catalyst except for the production of the gas lump where its value decreased to 39,245.980 J/mol compared to 39,844.25 J/mol on the NiMo/Al2O3 catalyst. Moreover, the activation energies of the secondary reactions gave a higher value over Pt/Al2O3 than that over NiMo/Al2O3 in seven out of ten reactions. The apparent activation energies for gasoline production were 108,234.89 and 103,404.02 J/mole on the catalyst surface of the NiMo/Al2O3 and Pt/Al2O3 catalysts, respectively, while the value of the apparent activation energies for diesel fraction production were 66,301.29 and 69,260.46 J/mole on the catalyst surface of the NiMo/Al2O3 and Pt/Al2O3 catalysts, respectively.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}