3.1. Effects Related to Seawater Composition
According to the experimental plan, in order to define the chemical physical characteristics of water inside sediments samples, after the leaching treatment with ultrapure water, measurements were performed on the obtained water solutions. All the results were calculated by considering averages of humidity % values for each carrot (30.89% for the GC01 and 31.64% for the GC02 series) [
39] obtained by the previous characterization of the sentiments. The identification and quantification of all analytes were carried out using specific standard mixtures of elements or anions.
The measured pH and conductivity values (mS/cm) of the obtained eluates samples are reported in the
Table 3.
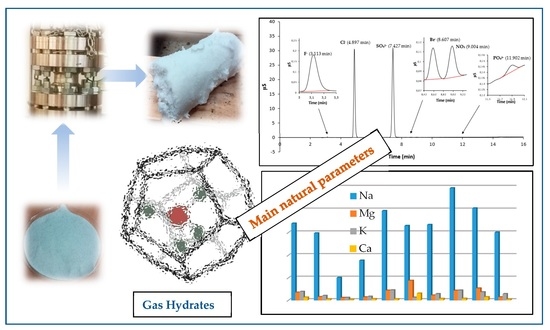
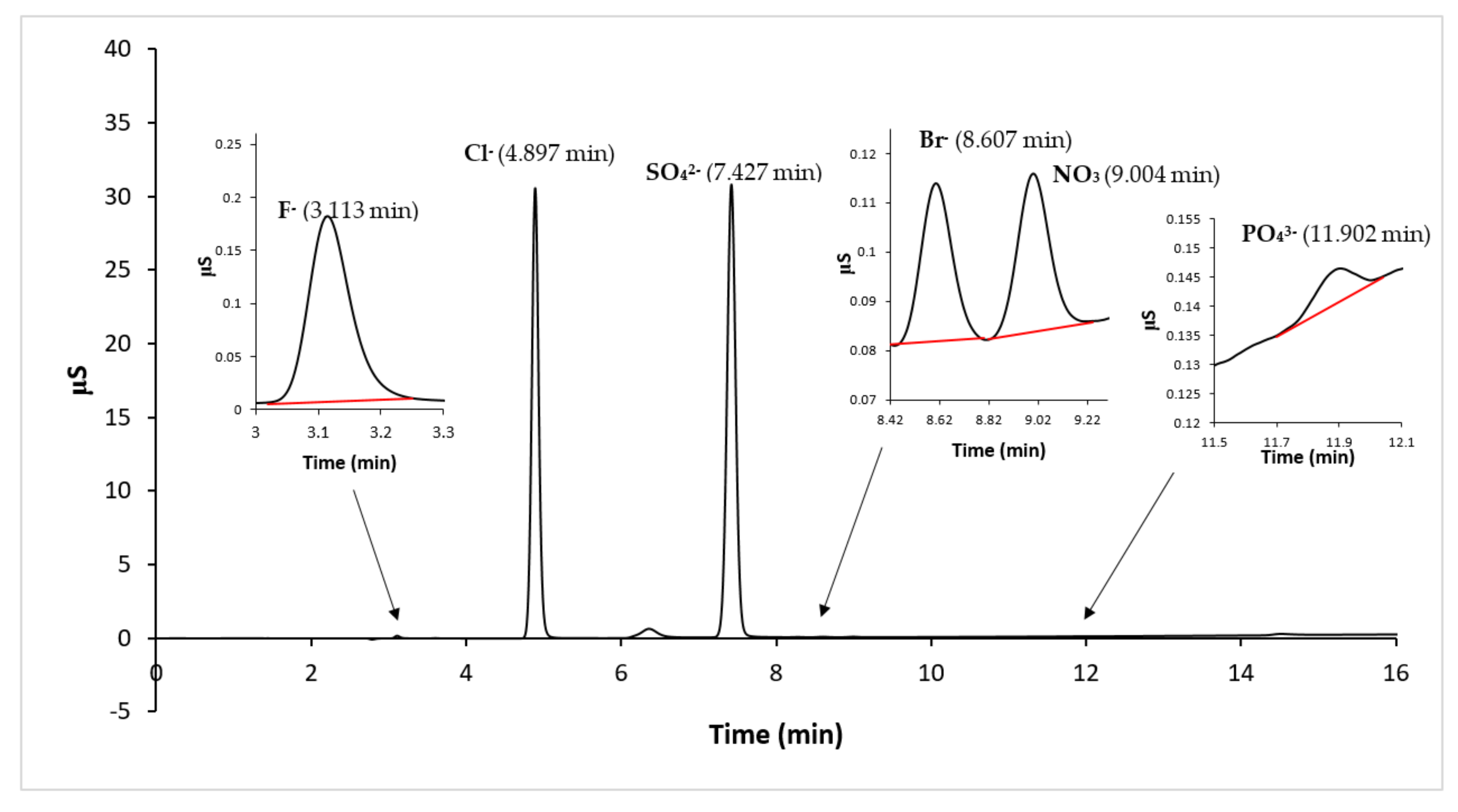
The anionic composition of the sample solutions, related to the water eluate samples obtained by the leaching test, were determined by IC analysis, using KOH as eluent, with the gradient method and conductimetric detection. A typical obtained chromatogram is reported in
Figure 6, where it is possible to observe that the major anionic compounds are chlorides and sulfates.
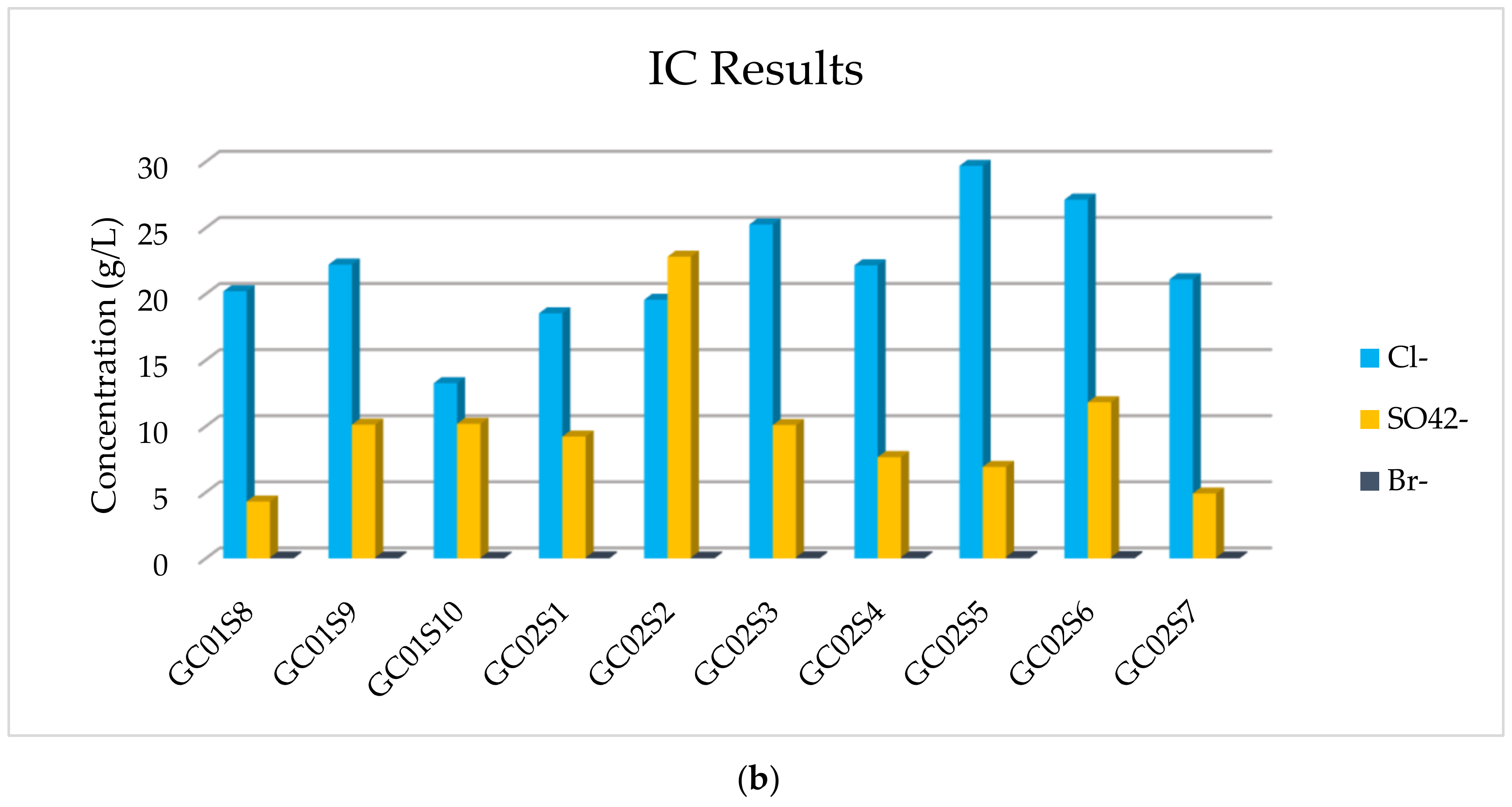
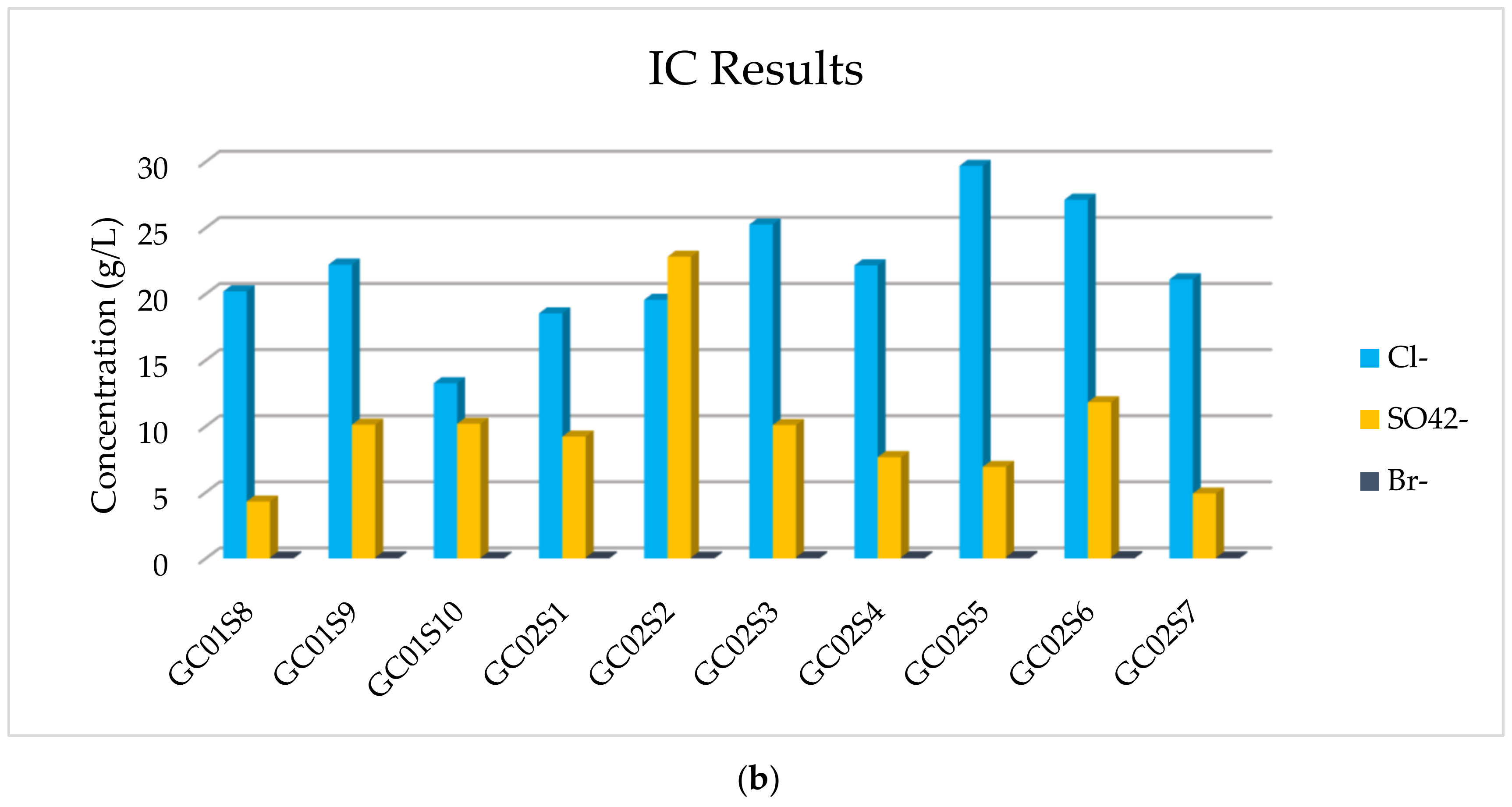
The results of the IC analysis related to the anions’ concentration of all samples are listed in
Table 4, in which it is possible to observe that the major anions are Cl
− and SO
42− and a very low concentration of Br
− is detected. The more abundant anion is Cl
−, but appreciable, and in one sediment comparable, is SO
42−.
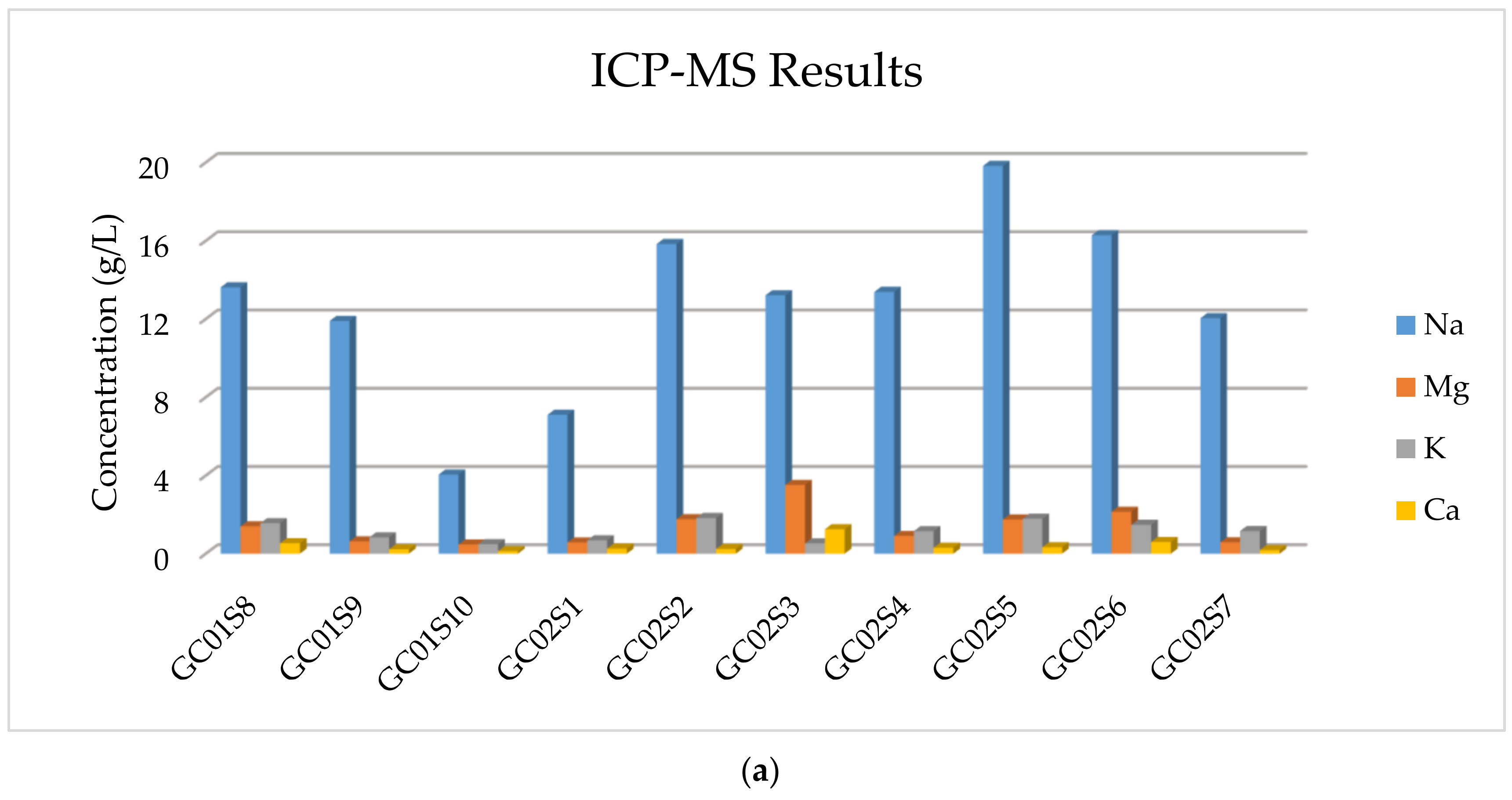
The results of the multi-element ICP-MS analysis are reported in
Table 5, showing that the more abundant element is Na and those in smaller quantities, but not negligible, are Ca, K, and Mg.
In addition, in
Figure 7, the concentration of majority elements and anionic compounds is graphically expressed.
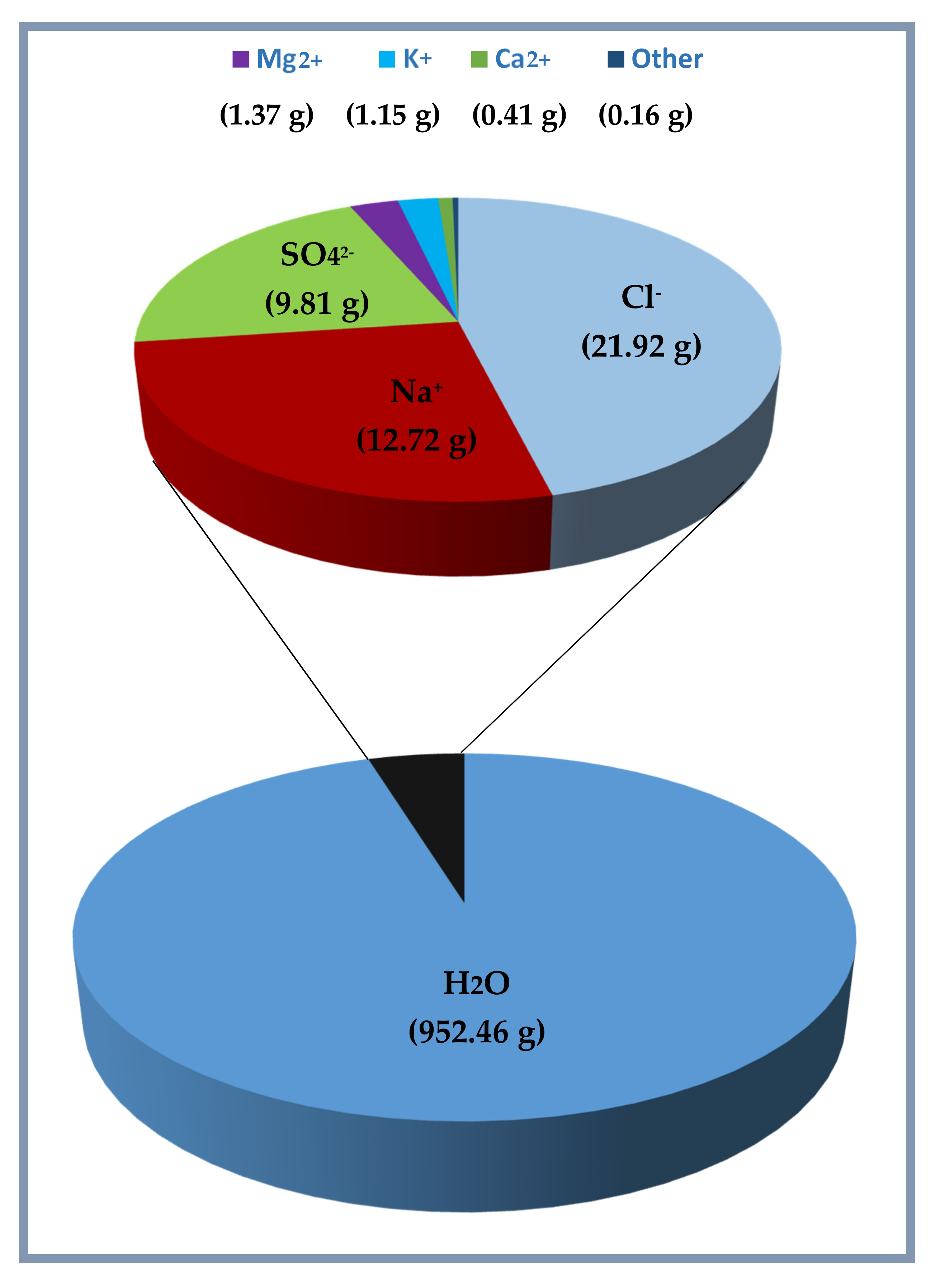
By its chemical composition, the water contained in the analyzed sediment samples represents, therefore, a solution consisting of about 95.2% of water, where dissolved salts are about 4.7%. In
Figure 8, the results reported in
Table 4 and
Table 5 are summarized in relation to chemical composition of 1 kg of water contained in the sediment samples. The obtained results demonstrate that the salinity obtained by the mean of different sediment samples is 47.54‰, and it can be converted as a function of the chlorinity Cl (‰) (mass of halides in a given mass of sea water) that, for the results reported here, is defined by the relationship S (‰) = 2.18 CI (‰).
The obtained results are in agreement with those related to other studies about the composition of the major components of sea water. The oscillation referred to single ion concentration is not largely relevant, and demonstrated that the contained major cations and anions are constant and that they contribute to physical chemical properties, permitting to treat ocean waters as an electrolyte solution. However, the obtained mean of salinity is higher when compared with that of marine water, which is about 35‰; this is because of the fact that, in sediments, the dissolution/precipitation processes, which can happen in different manners at different depths, can change the relative composition of dissolved ions. In addition, it was reported that the cations (Na
+, Mg
2+, K
+, Sr
2+) concentration is independent of the depth, with the exception of Ca
2+, which increases with the depth as a result of the dissolution of CaCO
3, like in deep ocean waters, where the high pressure can affect the solubility [
40].
3.2. Effects Related to Sand Composition and Its Grainsize
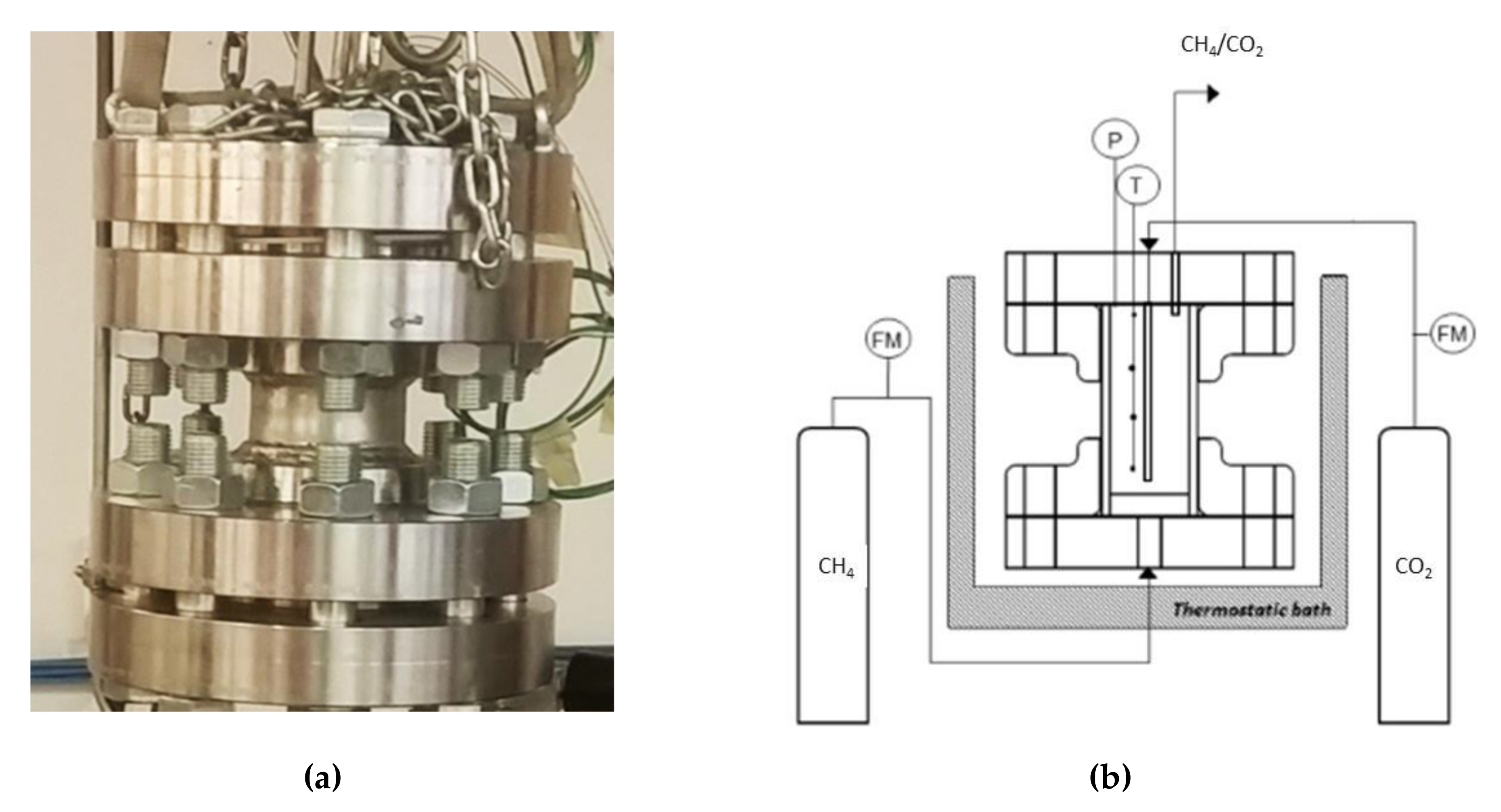
As explained before, two types of experiments were made. In both cases, pure methane hydrate formation was performed, and the difference stayed in the type of sand used. All tests were described by showing the pressure and temperature trend over time as well as the pressure trend as a function of temperature.
Table 6 and
Table 7 contain the most significant parameters related to tests carried out in pure quartz sand, while
Table 8 and
Table 9 are referred to tests made with sand TS.
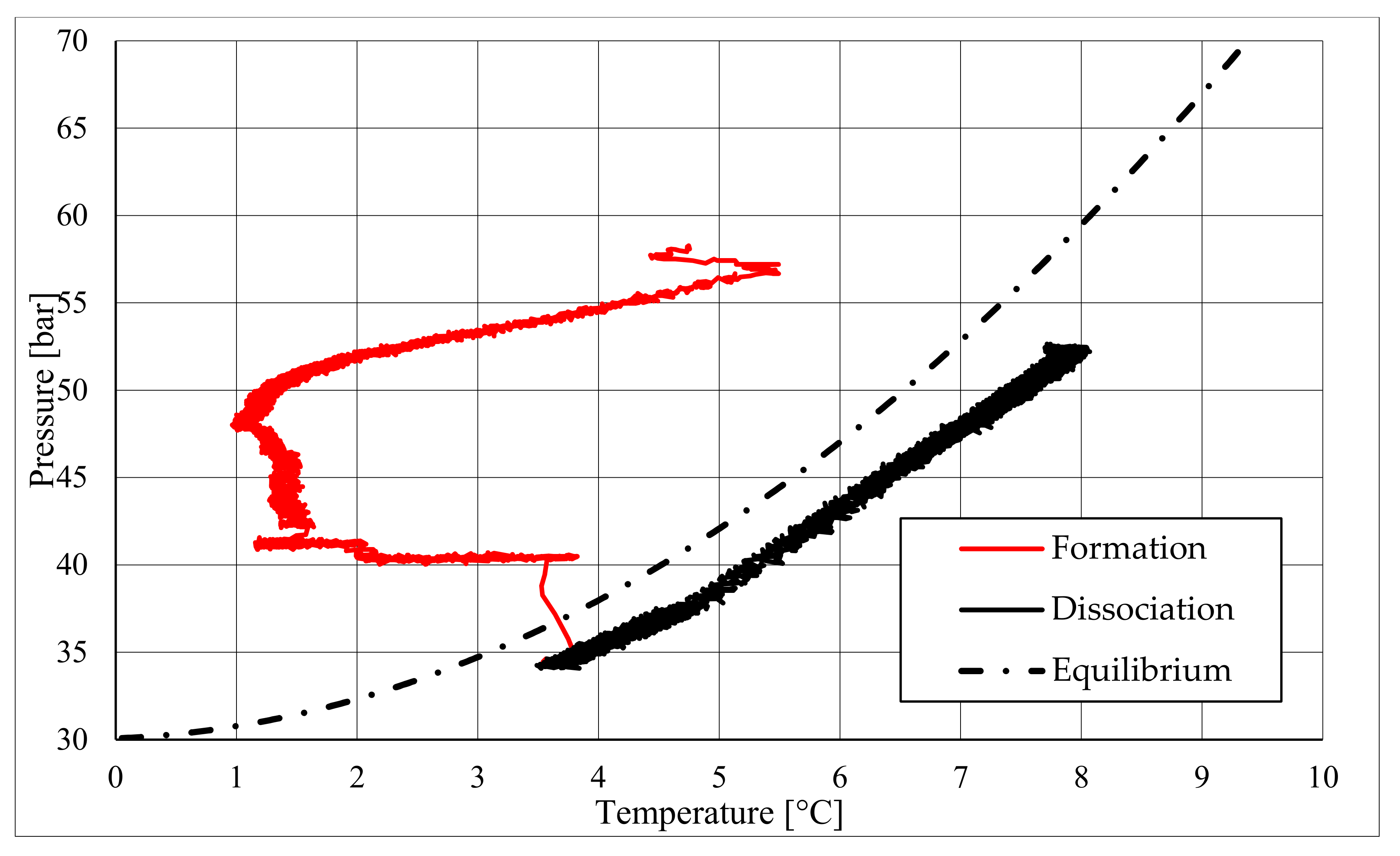
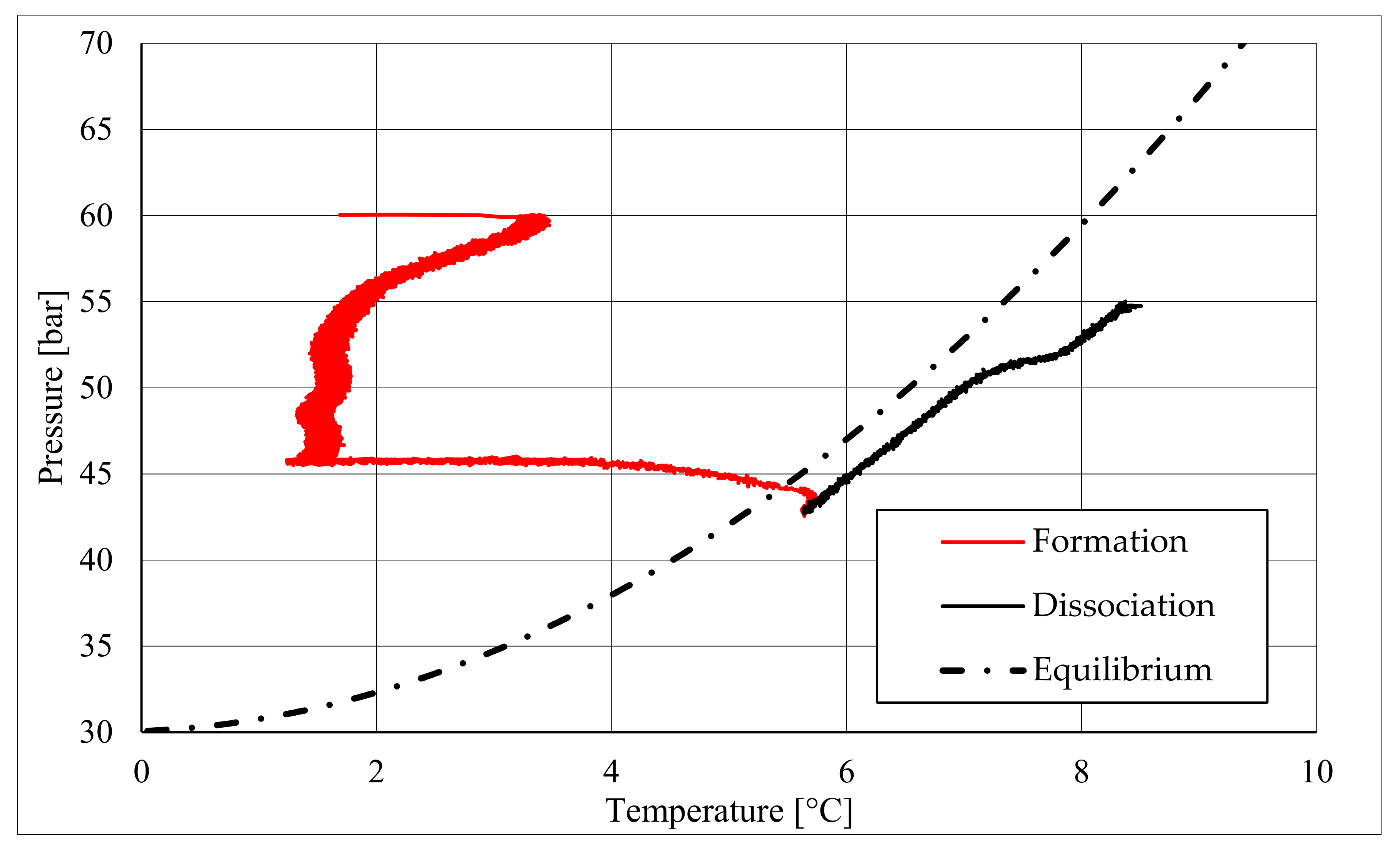
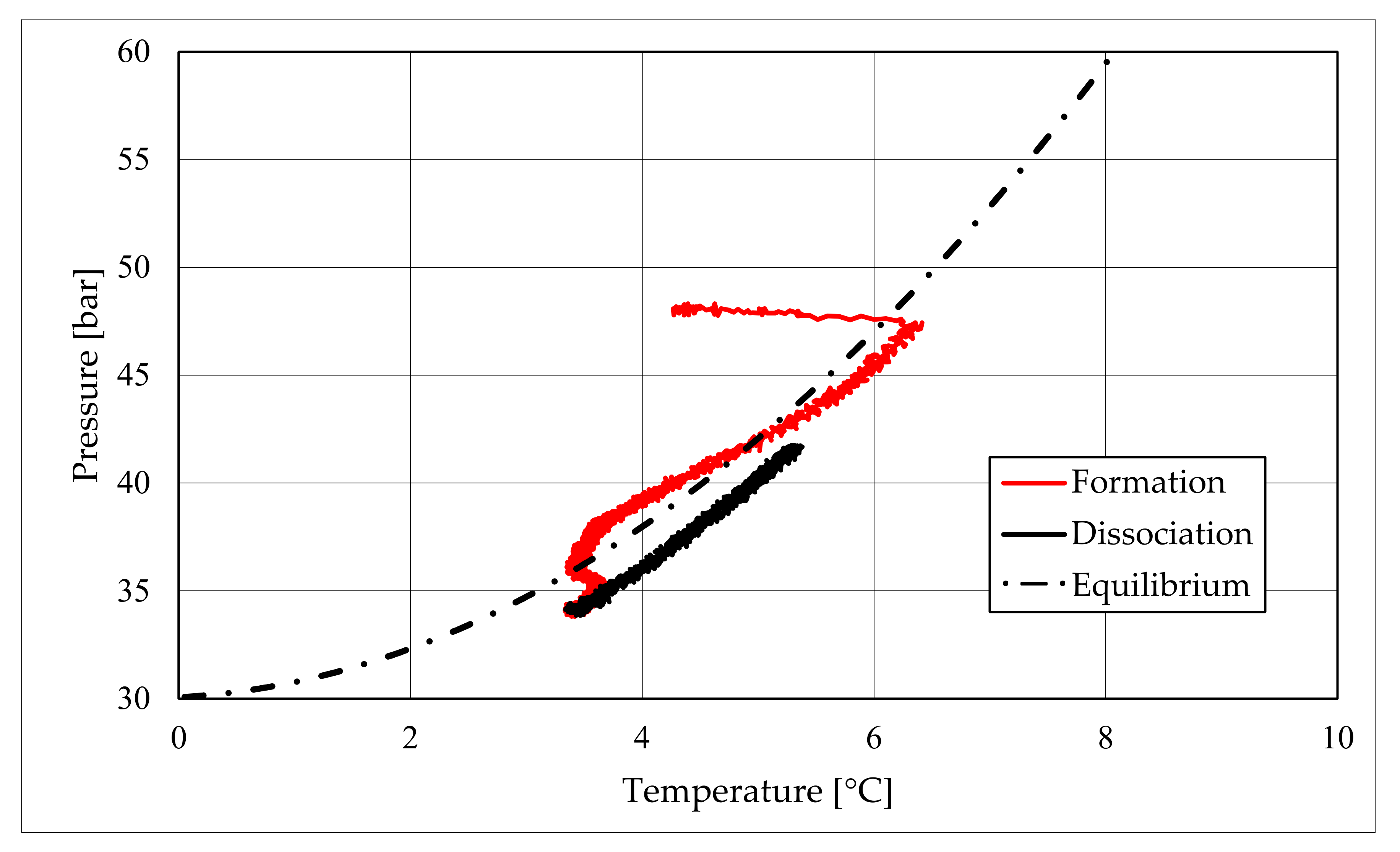
The parameters described in those tables reveal substantial differences between these two types of test. First of all, pressure required to trigger hydrate formation was considerably higher in tests made in sand TS. In this latter case, a pressure about 58.27–60.05 bar was needed, while in the presence of sand PQ, pressure ranged from 47.56 to 48.18 bar. Considering that the same quantity of liquid water was inserted inside the reactor in both cases, such a difference in pressure led to higher values of parameter CH4g and [CH4g] in Tests 3 and 4. This difference is visible in all points. Concerning time, hydrate formation was completed in 1617–1878 min in pure quartz sand (see PT3, absolute values); conversely, 2157–4642 min was necessary for sand TS. This means that the natural silica-based sand tested in this work acted as a kinetic inhibitor for hydrate formation, thus requiring more time to reach results similar to those produced in sand PQ. This difference in time required was observed mainly in PT3* and PT3, while PT2 did not provide meaningful information in this sense, because even the corresponding time lapse measured is extremely limited. Concerning kinetic, a small difference was also noticed when analyzing parameter dCH4/dt. Considering its absolute value, in Tests 1 and 2, this parameter increased with time. In particular, the pressure reduction due to hydrate formation did not cause a process deceleration; on the contrary, the reaction seemed to proceed faster in the last phase of hydrate formation, during which pressure obviously assumed the lowest values. In Test 1, dCH4/dt started from −4.91 × 10−4 mol/min and reached −9.78 × 10−4 mol/min in PT3, even if in PT3*, it slightly decreased compared with its previous value registered in PT2. In the same way, in Test 2, it was equal to −2.71 × 10−4 mol/min in PT 2, −3.15 × 10−4 mol/min in PT3*, and −3.39 × 10−4 mol/min in PT3. Conversely, in Tests 3 and 4, it constantly decreased with the ongoing process; in Test 3, it moved from −1.85 × 10−3 to −1.11 × 10−4 mol/min, and in Test 4, it moved from −2.59 × 10−4 to −4.05 × 10−5 mol/min. Finally, a slightly higher number of formed hydrae was measured in the presence of TS. Here, 0.297–0.366 moles of hydrate were observed, rather than 0.21–0.217 moles measured in Tests 1 and 2. This means that, even if this second type of sand clearly acted as a kinetic inhibitor for hydrate formation, it did not prevent reaching similar or even better results than those in sand PQ in terms of hydrate formation. This last aspect was also highlighted with parameter GU%, which proved how the gas uptake was higher in Tests 3 and 4.
Starting from an initial sand pores’ saturation with water equal to 55%,
Table 10 shows how that parameter varied due to hydrate formation.
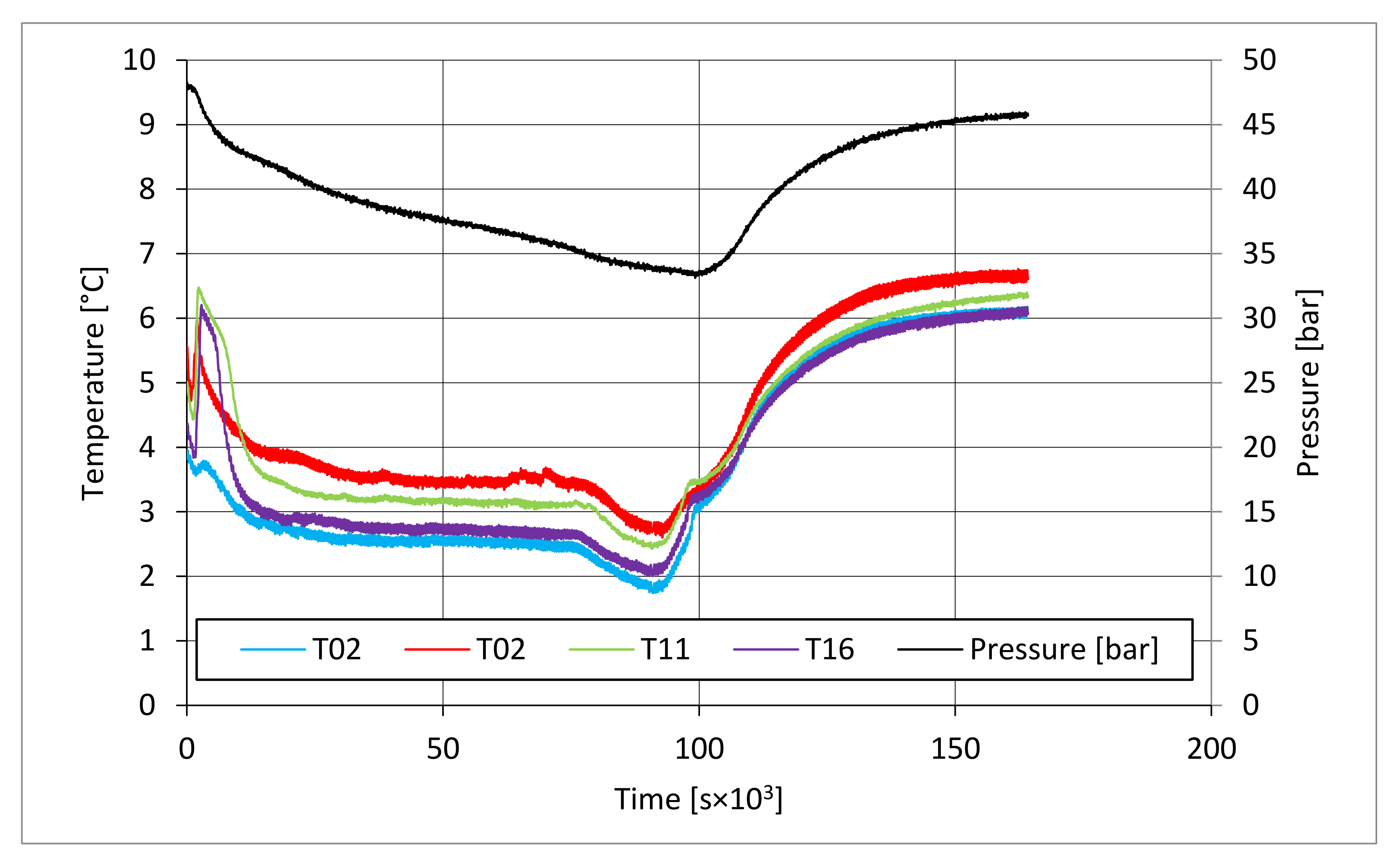
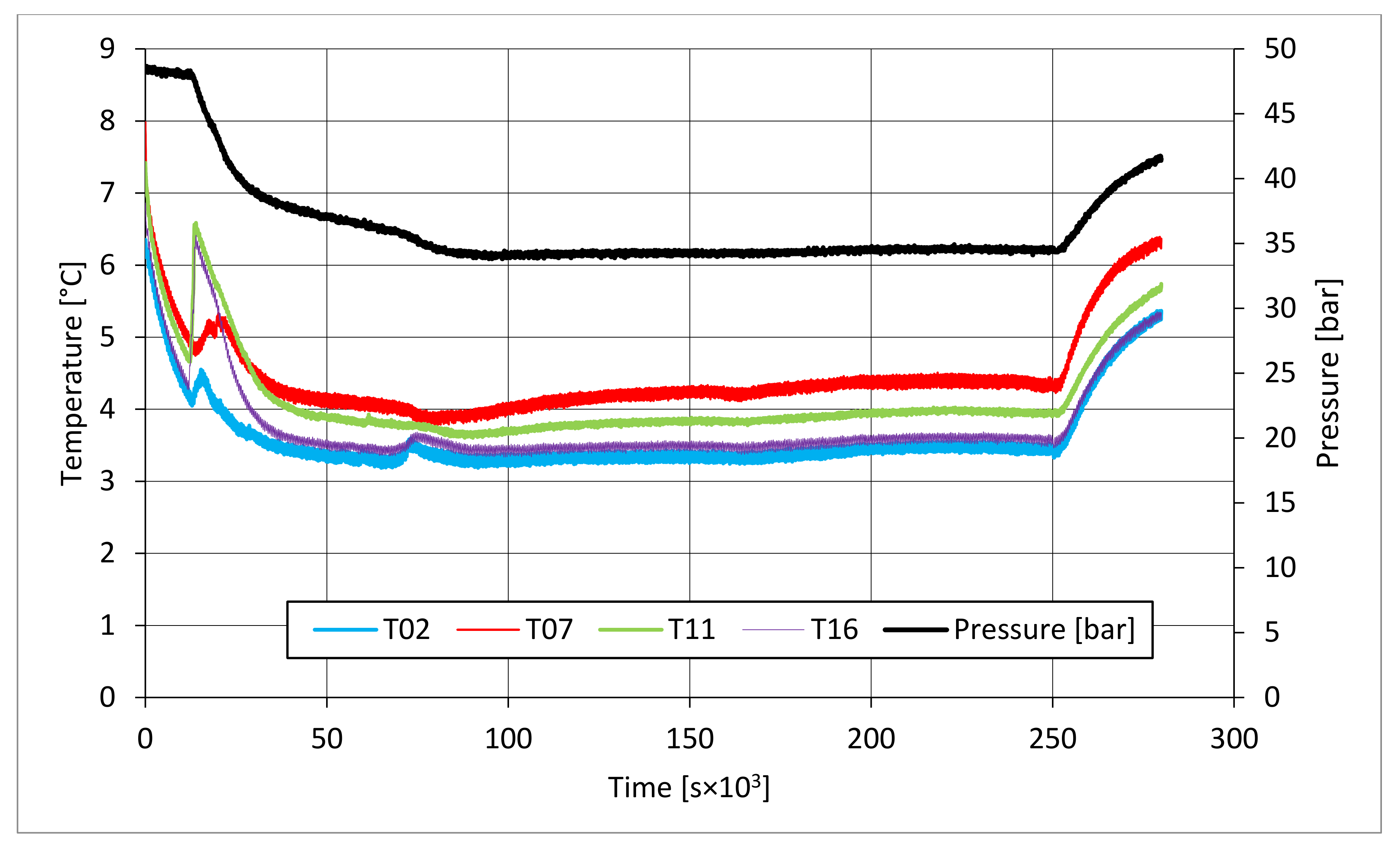
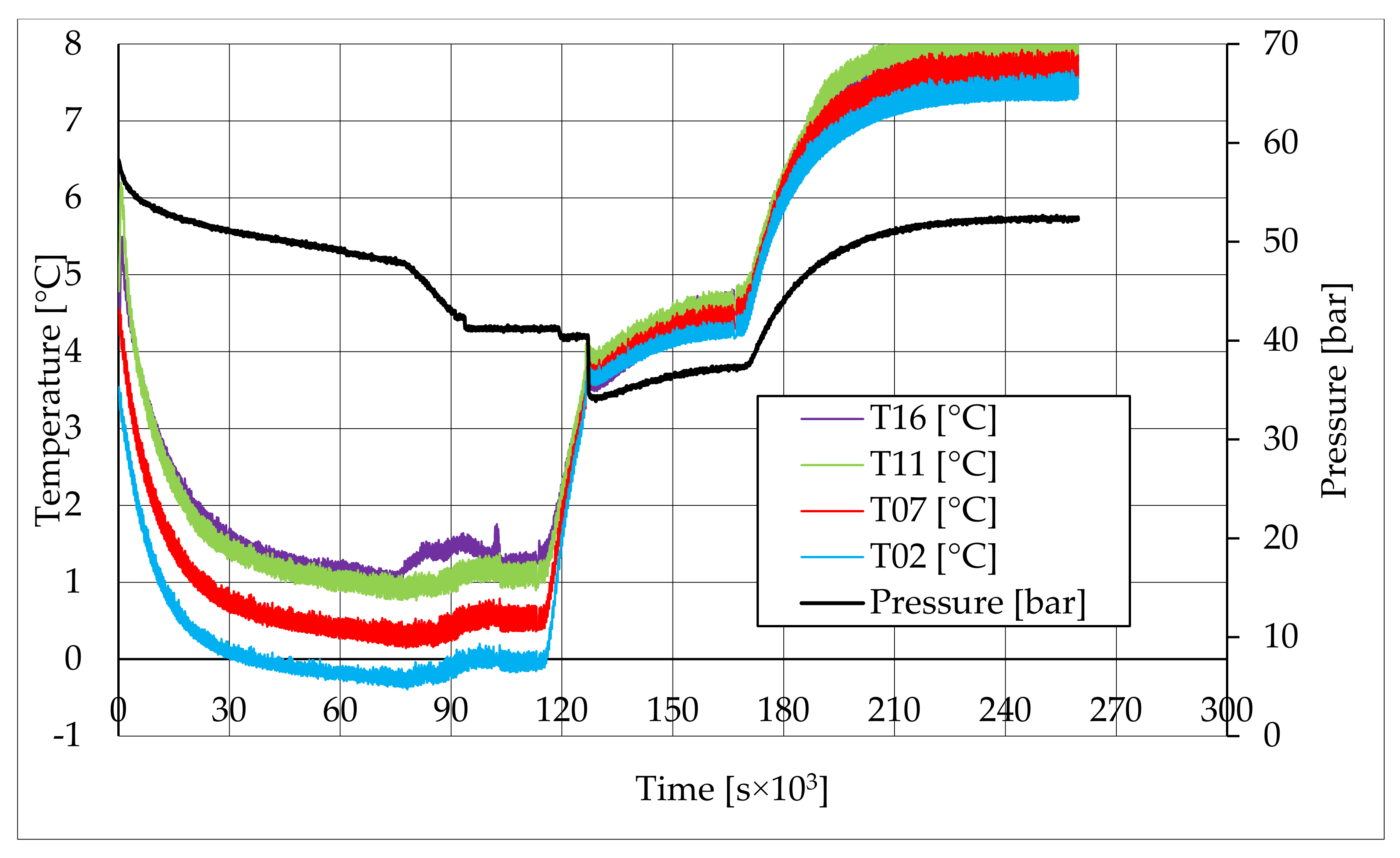
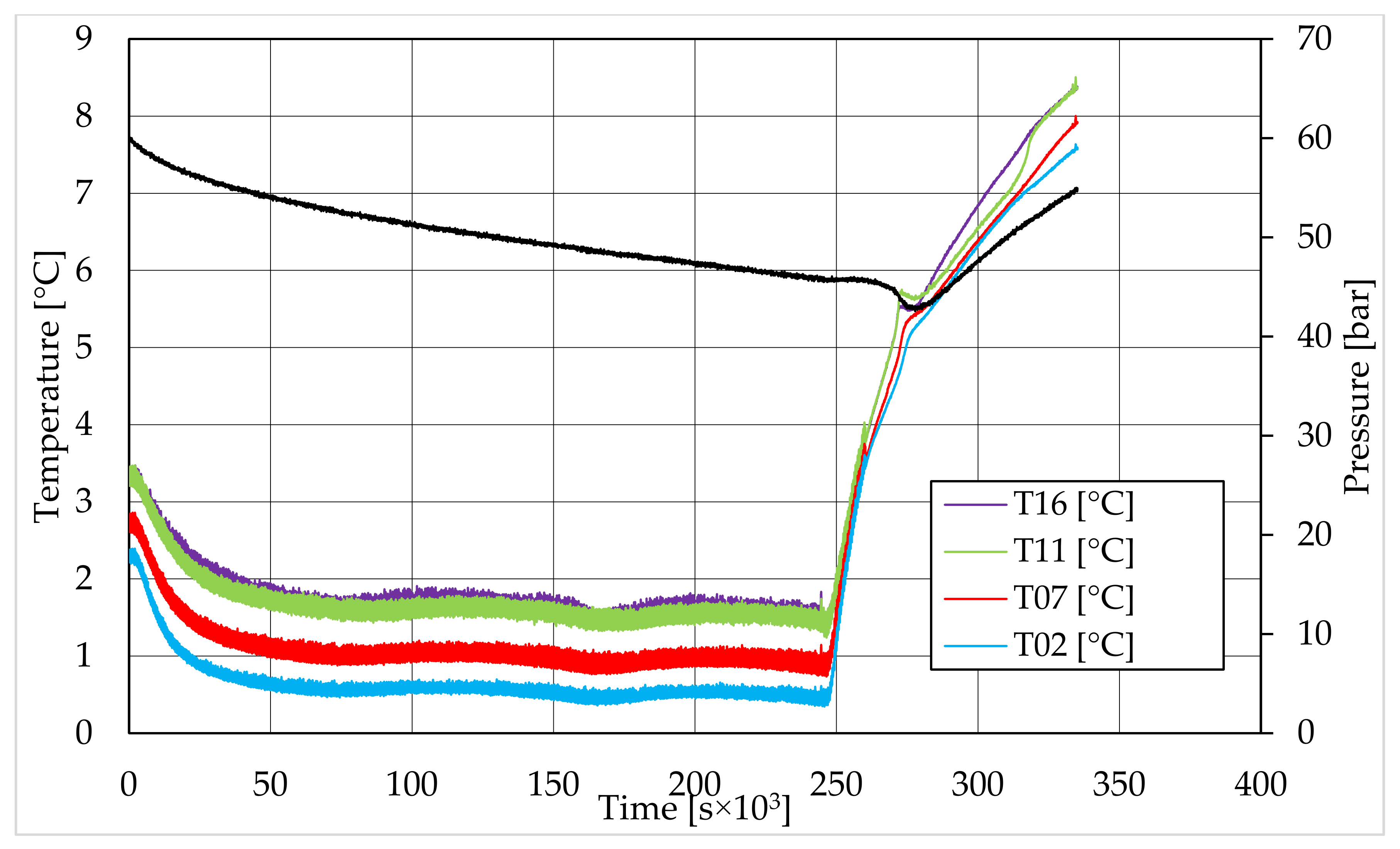
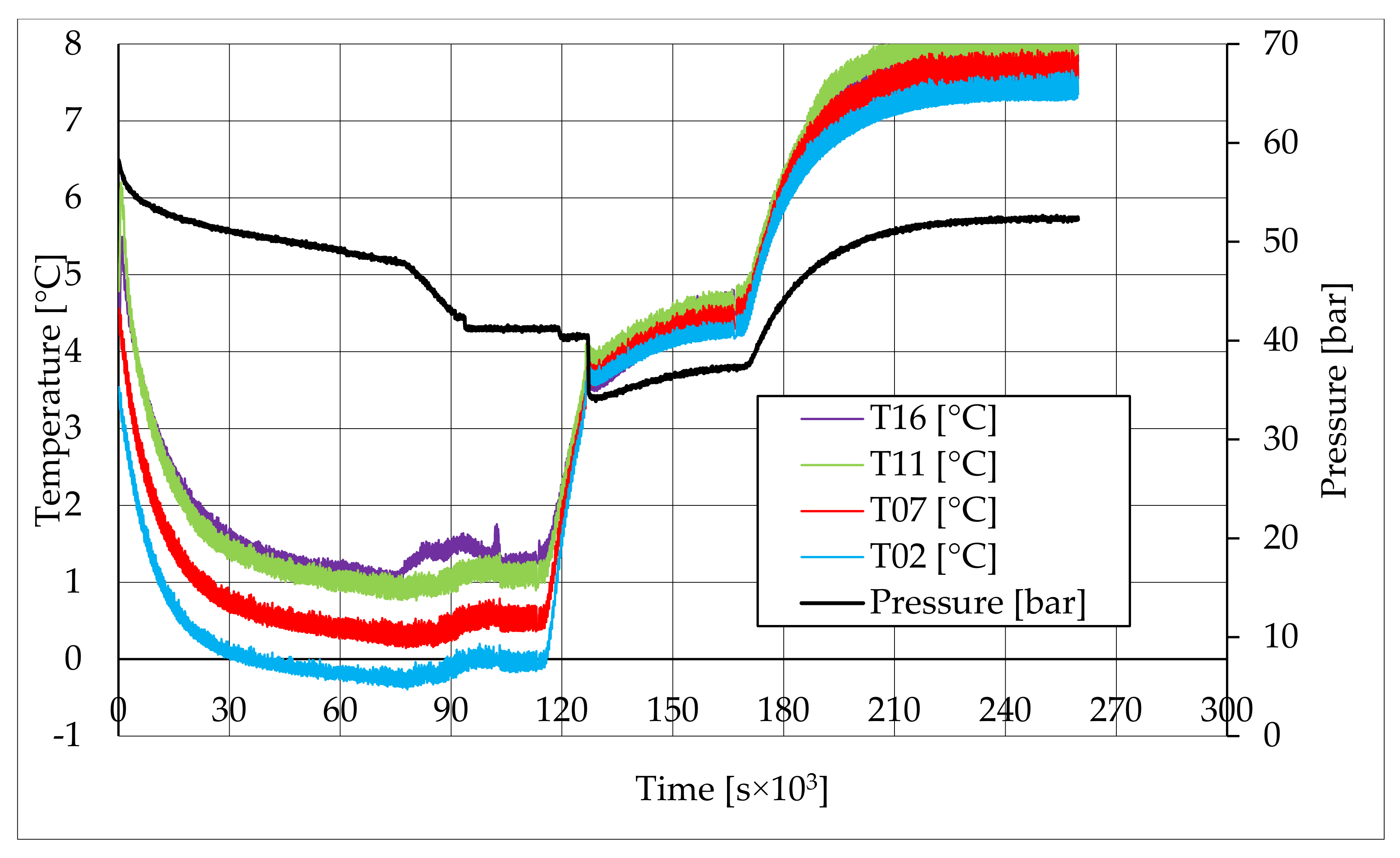
In all tests, the temperature trend over time revealed the exothermic nature of hydrate formation; in all cases, an initial temperature peak was observed, then temperature decreased till the whole system again established the thermal balance. In Test 1 and Test 2, this temperature peak did not occur immediately, or as soon as pressure was kept to its maximum value, it showed a slight delay. This mainly depended on the stochastic nature of hydrate, which may not occur immediately when thermodynamic conditions are feasible for its formation. Such a delay was useful to well highlight the triggering of hydrate formation. Once temperature quickly increased, pressure started decreasing significantly. This aspect occurred in all experiments; however, the slight delay present in Test 1 and Test 2 allowed to clearly identify the difference between the pressure trend when only a sporadic hydrate formation occurred and when the reaction concerned the whole reactor. The temperature trend over time also proved how methane hydrate formation mainly concerned the lower region of the internal volume. In contrast to the others, thermocouple T02 did not measure any relevant temperature variation. It is the only device positioned above the water–sand mixture, where only gaseous methane is present immediately before beginning the experiment. Its trend proved that, in this region, hydrate formation did not occur or, more in general, it was negligible, as already proved in previous works [
41]. For instance, in Test 2, thermocouple T02 registered a small temperature peak; however, it was an order of magnitude lower than those measured by other thermocouples. Thermocouple T07 is positioned immediately below the free surface of the water–sand mixture, and registered a consistent temperature variation, proving that massive hydrate formation occurred in the corresponding region. In particular, it was noticed during Test 1 that the most abundant hydrate formation was observed in correspondence of T11 and T16, or in the lower portion of the internal volume. Both thermocouples registered the maximum temperature values during all tests. Even if they are positioned at different depths inside the reactor, at 11 and 16 cm, respectively, from the upper surface, both of them are situated in a similar environment; they are immersed in the water–sand mixture and are far from the water free surface. This is the reason their trend was found to be extremely similar in all experiments. A moderate difference was observed in Test 3 (see
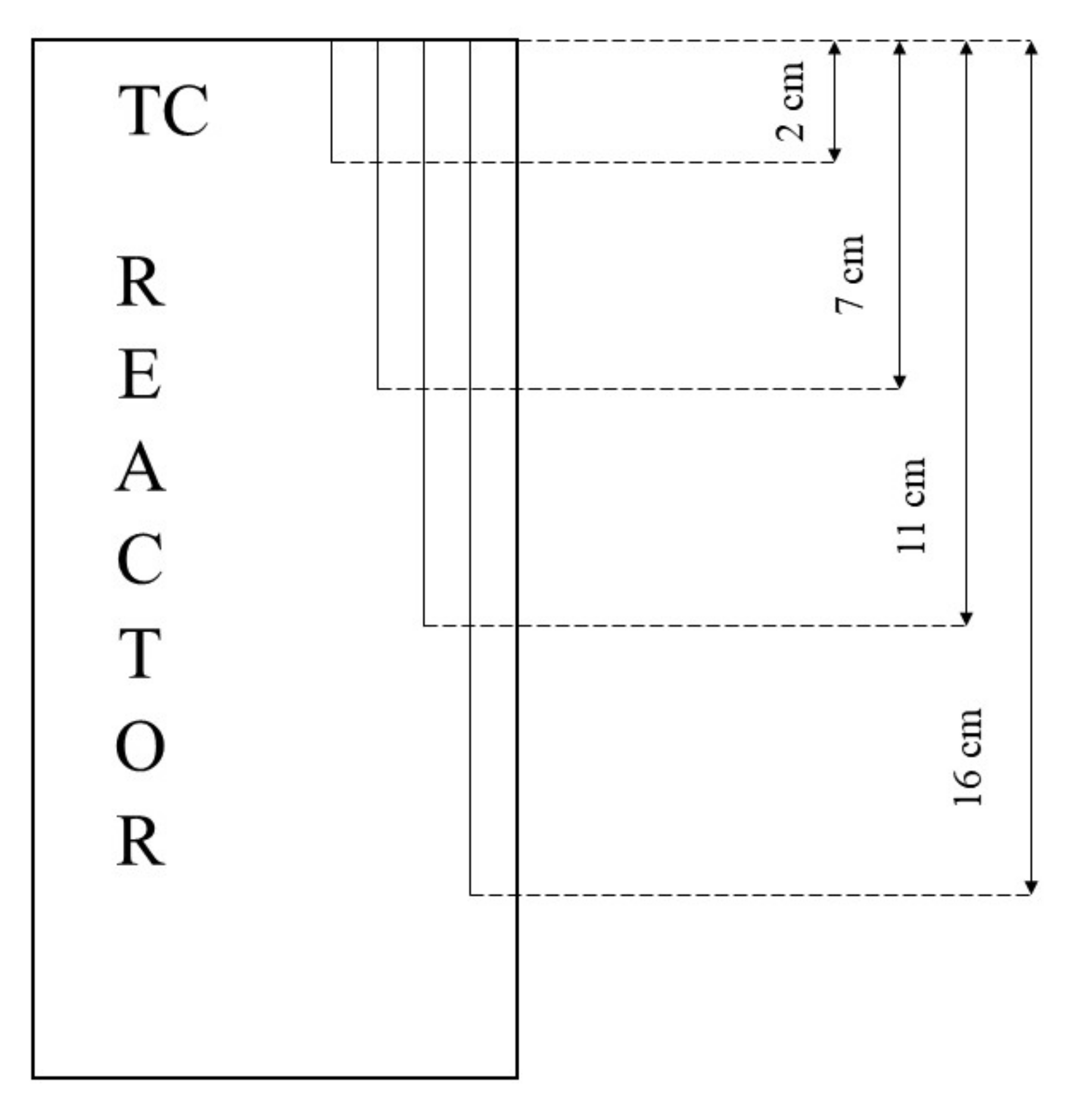
Figure 13), where T16 measured a secondary temperature peak, while, in correspondence with T11, any variation occurred. Such a temperature increase denoted the formation of further hydrate nuclei in the lower region; at the same time, the pressure trend also varied and, in particular, it started decreasing more intensively for the whole duration of the temperature peak.
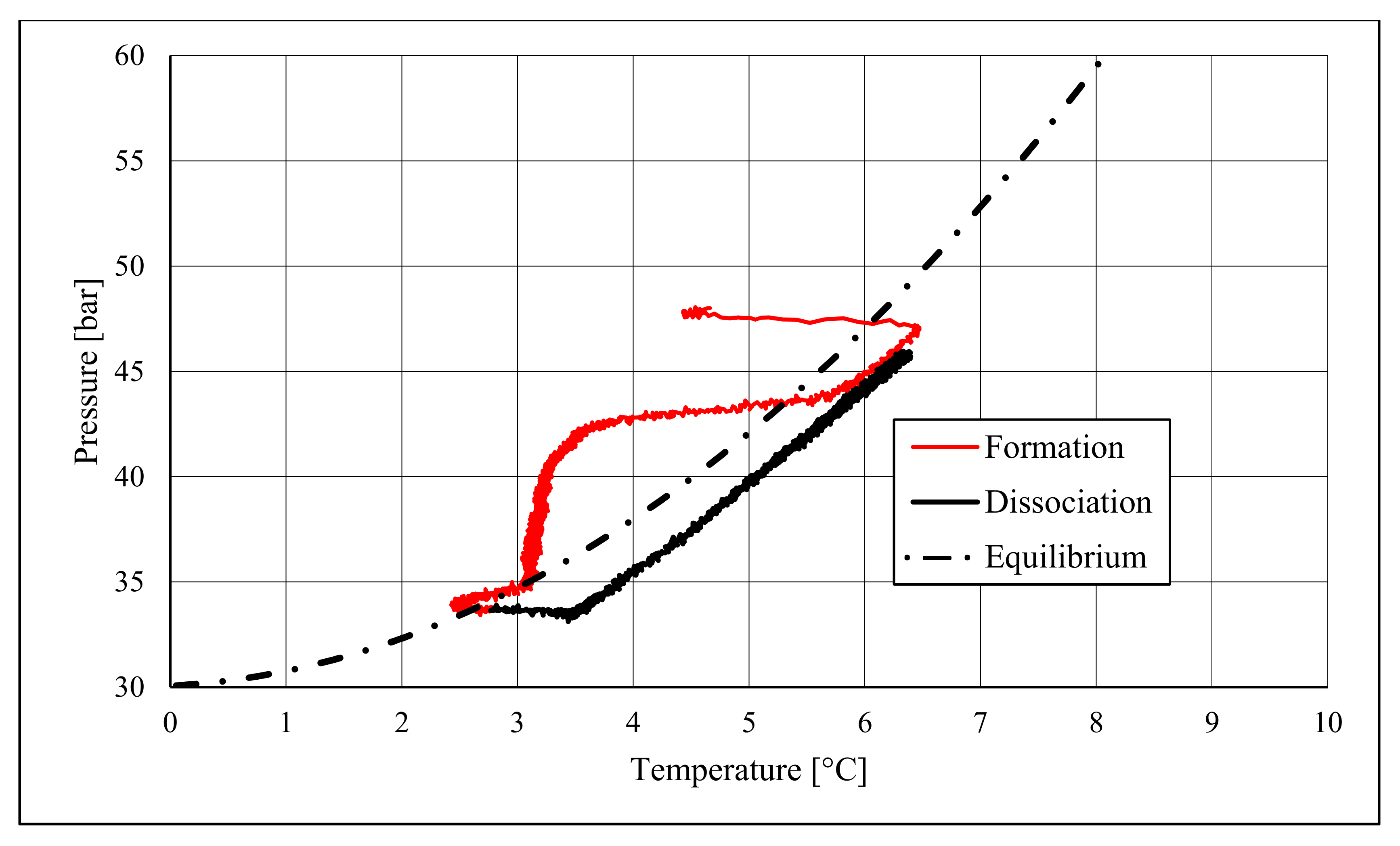
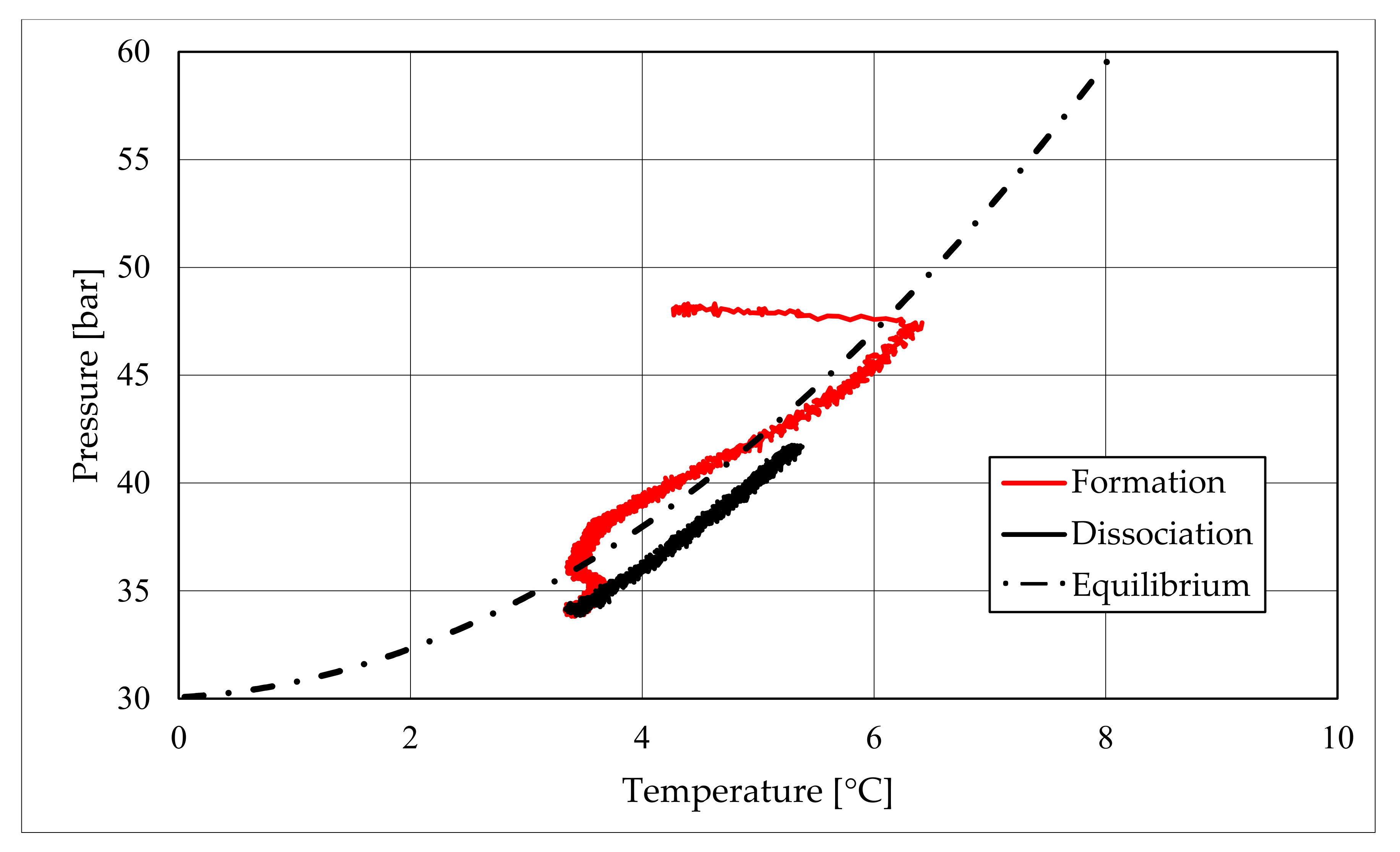
The right side of those diagrams describe the partial hydrate dissociation phase. In Test 1 and Test 2, where pure quartz sand was present inside the reactor, the dissociation was massive and occurred as soon as temperature started increasing, proving that, when hydrate formation finished, the internal thermodynamic conditions precisely corresponded to the equilibrium values. This latter aspect is well visible in
Figure 10, or the pressure–temperature diagram related to the same test. Here, it is possible to see that the start point of hydrate dissociation is completely superimposed on the equilibrium curve. On the contrary, when sand TS was used, the pressure increased due to hydrate dissociation had a remarkable delay compared with the temperature increase, proving in this latter case that, when the hydrate formation finished, the internal thermodynamic conditions were considerably above the equilibrium curve for methane hydrate, thus favoring hydrate stability for a prolonged period of time. About dissociation temperature, the two different types of experiments showed significantly different behaviors. In the first group of experiments, temperature increased constantly and slowed down only at the end of the dissociation phase, when the internal temperature was close to the external temperature, fixed with the chiller. Conversely, in tests performed with the natural sand TS, temperatures assumed two different velocities during dissociation. In the first part, it was extremely elevated and, in the corresponding diagrams, it seems to assume an almost vertical diagram. Then, it continued increasing, but following a lower gradient, close to that observed in Test 1 and Test 2. This aspect is mainly associated to hydrate dissociation, which is an endothermic process, and thus lowers the temperature in the surrounding environment. In Test 1 and Test 2, hydrate dissociation occurred immediately and temperature rose slowly compared with the second group of experiments. In this latter case, because of the initial pressure and temperature values present inside the reactor, hydrate formation did not occur immediately and temperature increased drastically, till hydrate dissociation started and the temperature trend become completely similar to that observed in Test 1 and Test 2.
Rather than experimental values, in all diagrams describing the pressure–temperature trend during the experiments, a black dotted line was inserted to represent methane hydrate equilibrium. This line was drawn with data present elsewhere in the literature [
42,
43,
44,
45,
46,
47,
48,
49,
50]. While the tables well indicated the effect of TS sand on the process kinetic, in those diagrams, the thermodynamics of concern are clearly shown. In the presence of sand PQ, methane hydrate formation occurred at conditions close to equilibrium values. In Test 1, during the last part of the hydrate formation phase, the two curves were completely overlapped. Conversely, with sand TS, the P–T formation values were extremely far from the black dotted line. In this latter situation, hydrate required higher pressures (the reaction started at 58–60 bar, instead of 47–48 bar) and/or lower temperature values. However, the pressure drop present in
Figure 7 and in
Figure 8 proves that massive hydrate formation occurred. While the hydrate formation phase was completely different as a function of the type of porous medium involved in the process, the hydrate dissociation phase was extremely similar. In both cases, the experimental hydrate dissociation denoted behavior equal to the ideal trend extrapolated from the literature, with the only difference that experimental values are slightly shifted at the right of the equilibrium curve. This slight deviation is associated with the presence of sand, whose grains are able to hinder and decelerate the dissociation process [
51].
During hydrate formation, in tests made with sand PQ, a “hump” is clearly visible. In Tests 3 and 4, it is also present; however, the greater distance from the equilibrium of the whole curve made it less observable. Such a temporary deviation from the current trend was usually observed and reported in the literature; moreover, it might be useful to develop a more in-depth understanding of the process. In the present work, that specific point was called PT3* and, as for other points, all kinetic and thermodynamic parameters were evaluated and reported in the respective tables.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}