
FeI Intermediates in N2O2 Schiff Base Complexes: Effect of Electronic Character of the Ligand and of the Proton Donor on the Reactivity with Carbon Dioxide


Abstract
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Fe Complexes with Tetradentate N2O2 Ligands
3.2. Electrochemical Properties and Spectroscopic Features of FeI Species
- (i)
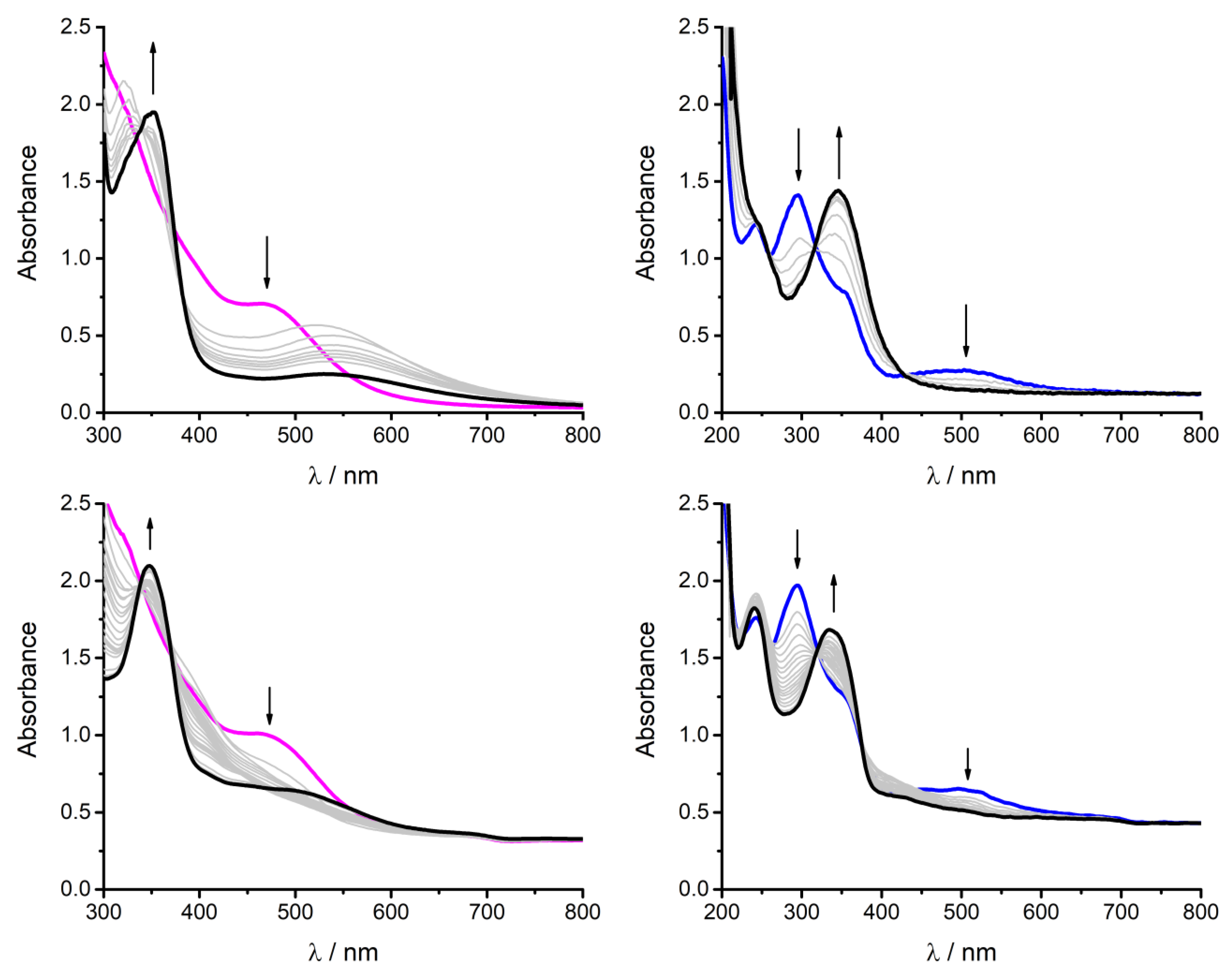
- In the case of the Fe(acacsalen)Cl, two absorption features rise upon generation of FeI at 308 and 379 nm, respectively (see Figure S6); these two MLCT components are expected on the basis of the peculiar character of the asymmetric acacsalen ligand, bearing both the acetylacetoneimine and salicylideneimine pendants. Incidentally, the two absorption maxima observed for FeI(acacsalen) are similar to those observed separately for FeI(acacen) (288 nm) and for FeI(salen) (354 nm).
- (ii)
- The energy of the MLCT band observed for the FeI intermediates correlates linearly with the redox potential of the FeII/I couple; the trend shows that the more negative the potential of the FeII/I couple, the higher the energy of the MLCT band (absorption shifted towards the blue region of the spectrum, Figure S7).
3.3. Reactivity of FeI towards Carbon Dioxide
- (i)
- SEC-UV/Vis analysis in the presence of CO2, where the diagnostic MLCT features of the FeI intermediate are blue-shifted with respect to those observed under dinitrogen (Figure 4 bottom). In particular, the absorption maximum shifts from 354 to 345 nm for Fe(salen) and from 344 nm to 332 nm for Fe(beacen); in the case of Fe(salophen), the MLCT shifted from 405 nm to 385 nm in the presence of CO2 [12]. A spectroelectrochemical analysis in the infrared region (SEC-IR) failed to reveal absorption features ascribable to stretching features of iron-carbonyl intermediates, as was observed in the case of Fe(salophen), ref. [12]. This could be ascribed to an enhanced reactivity of such intermediates, although the interference of platinum working electrode at the negative operative in our setup should be also considered.
- (ii)
- Isolation of FeI species upon constant potential electrolysis under inert atmosphere, followed by the addition of carbon dioxide (in the absence of applied potential), that led to an immediate color change of the solution (see pictures in Figure S8 in Supplementary Information). In the case of Fe(salophen), the reactivity of FeI with CO2 under analogous conditions was associated with a redox process involving the oxidation of FeI to FeIII by CO2, as supported by EPR evidence [12]. Notably, no reduced products of CO2 were detected under these conditions (in particular, carbon monoxide), suggesting that a further reduction of the FeI−CO2 adduct is required in order to close the cycle and release the products (vide infra).
3.4. Effect of Proton Donors and Electrolysis
- (i)
- Carbon monoxide (CO) and hydrogen (H2) were the sole product identified; NMR analysis according to the procedure reported by Nichols et al. [11] did not reveal the production of formate (See Supplementary Information and Figure S14);
- (ii)
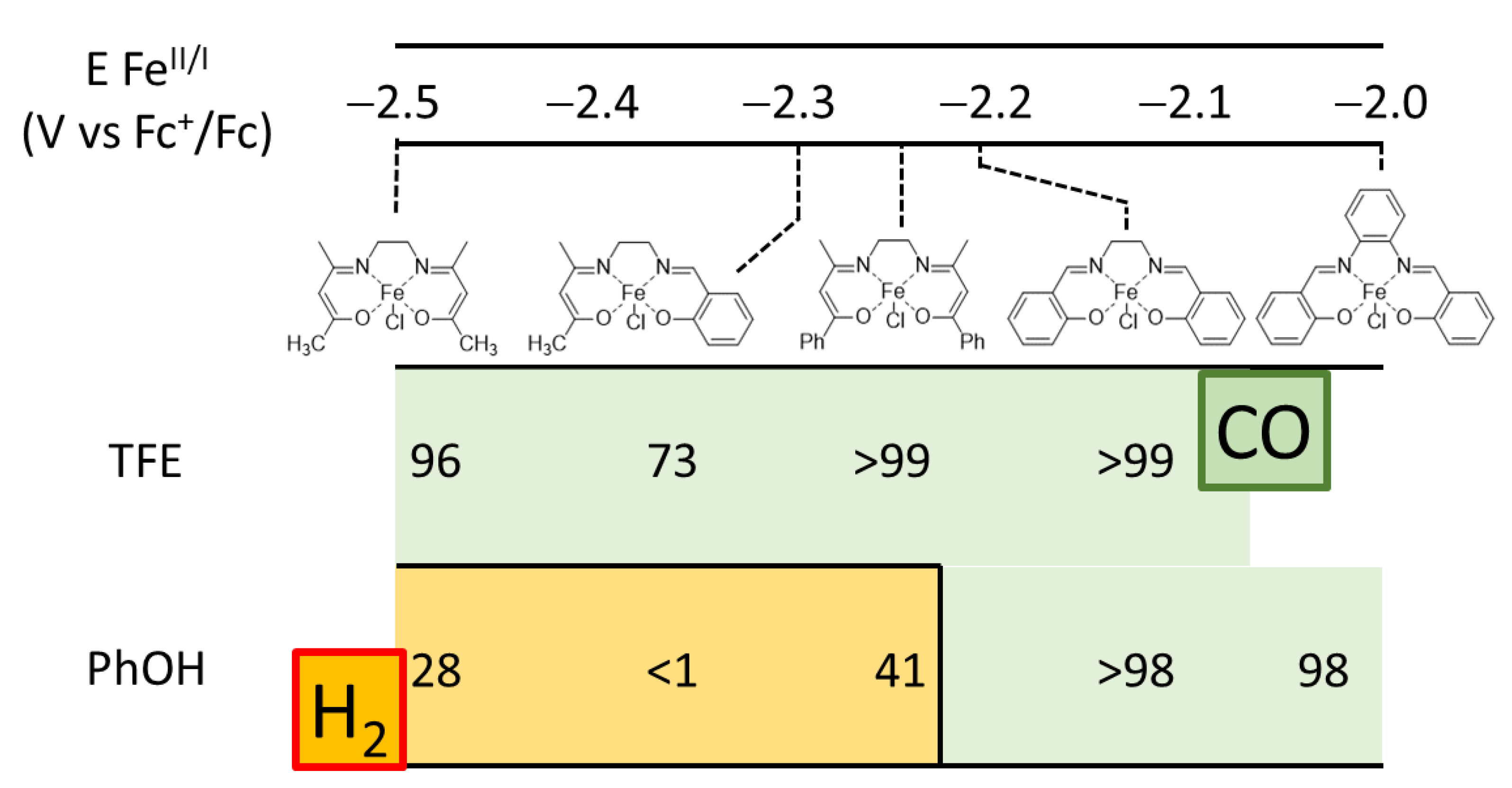
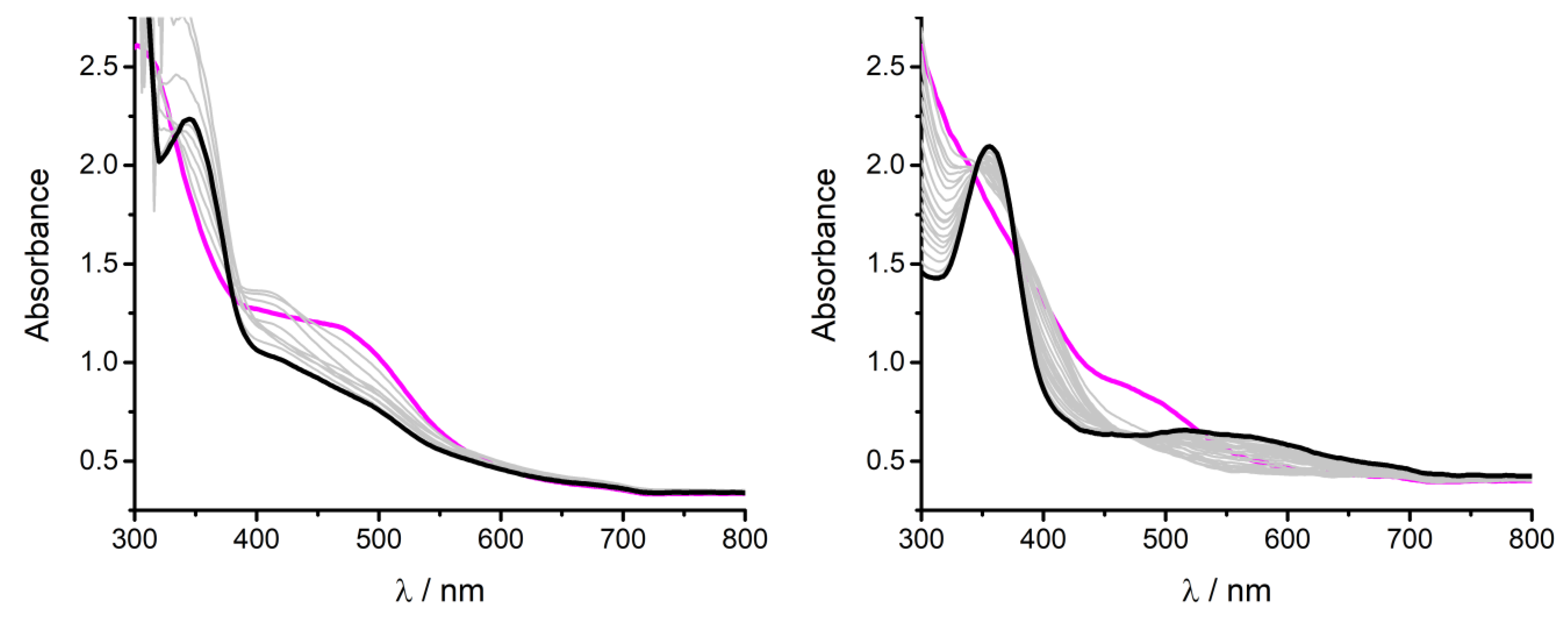
- In all cases, the overall Faradaic yield of the process for CO and H2 production is significantly lower than the ideal value; this is ascribed to both the need for pre-reducing the FeIII to the active FeI state, and the consumption of reducing equivalents by the iron complexes leading to their inactivation and decomposition, as demonstrated by the drop of electrolysis current over time (Figure S15) and by the marked changes in the UV/Vis spectra after electrolysis (Figure S16). Non-quantitative Faradaic yields are not unusual in the electrochemical reduction of CO2 with coordination complexes [5,12,31];
- (iii)
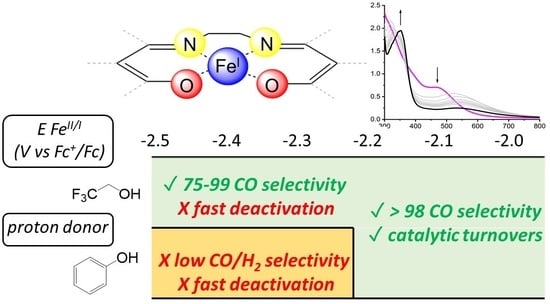
- The use of phenol as the proton donor leads to a marked impact on the selectivity of the process depending on the iron complex. In particular, while high selectivity for CO is observed in the case of Fe(salophen) and Fe(Salen), H2-oriented selectivity is observed for Fe(acacen), Fe(acacsalen), and Fe(beacen), as shown in Table 2 and Figure 6, i.e., for the species where the FeI intermediate is generated at more negative potentials and is thus expected to be more prone to a direct reaction with proton donors.
4. Discussion
5. Conclusions
- (i)
- The FeIII(LN2O2)Cl complexes display two metal-based reductions, involving FeIII/II and FeII/I couples; the potential associated with the FeII/I couple, relevant to reactivity with CO2, correlates with the electronic character of the LN2O2 (in terms of the energy of the highest occupied σ-donating orbital) and with the energy of the metal-to-ligand charge transfer absorption of the FeI intermediate, determined by SEC-UV/Vis.
- (ii)
- The FeI intermediates react with CO2, as proven by CV and SEC-UV/Vis investigation.
- (iii)
- For Fe(salen)Cl, in the presence of phenol or trifluoroethanol proton donors, the process is associated with the selective reduction of CO2 to CO (no H2 and formate are detected along with the electrolysis); in the presence of 0.5 M phenol, key performance indicators are an overpotential of 0.91 V, a catalytic rate constant of 5 × 104 s−1, and a turnover number of 4. CO2 reduction occurs through a homogeneous route, and the transformation of Fe(Salen) into an electrochemically inert species occurs.
- (iv)
- In the case of Fe(acacen)Cl, Fe(beacen(Cl), and Fe(acacsalen)Cl, the production of CO is observed only with TFE proton donor, while phenol leads to the evolution of H2 from the early stage of electrolysis. In all cases, the electrolysis current drops suddenly after three electrons passed per iron center, indicating a higher instability of these species, likely associated with the protonation under cathodic conditions of the ketylacetoneimine pendant of the ligands.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Oliveira, F.T.; Chanda, A.; Banerjee, D.; Shan, X.; Mondal, S.; Que, L.; Bominaar, E.L.; Münck, E.; Collins, T.J. Chemical and spectroscopic evidence for an FeV-oxo complex. Science 2007, 315, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.R.; Groves, J.T. A highly reactive P450 model compound I. J. Am. Chem. Soc. 2009, 131, 9640–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhugun, I.; Lexa, D.; Savéant, J.M. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins: Synergystic effect of weak Brönsted acids. J. Am. Chem. Soc. 1996, 118, 1769–1776. [Google Scholar] [CrossRef]
- Chalkley, M.J.; Drover, M.W.; Peters, J.C. Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes. Chem. Rev. 2020, 120, 5582–5636. [Google Scholar] [CrossRef]
- Cometto, C.; Chen, L.; Lo, P.K.; Guo, Z.; Lau, K.C.; Anxolabéhère-Mallart, E.; Fave, C.; Lau, T.C.; Robert, M. Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs. Co Quaterpyridine Complexes upon Mechanistic Studies. ACS Catal. 2018, 8, 3411–3417. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, S.; Cometto, C.; Anxolabéhère-Mallart, E.; Ng, S.-M.; Ko, C.-C.; Lu, G.; Chen, L.; Robert, M.; Lau, T.-C. Highly Efficient and Selective Photocatalytic CO2 Reduction by Iron and Cobalt Quaterpyridine Complexes. J. Am. Chem. Soc. 2016, 138, 9413–9416. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, S.; Rana, A.; Hematian, S.; Karlin, K.D.; Dey, A. Proton Relay in Iron Porphyrins for Hydrogen Evolution Reaction. Inorg. Chem. 2021. [Google Scholar] [CrossRef]
- Bonetto, R.; Crisanti, F.; Sartorel, A. Carbon Dioxide Reduction Mediated by Iron Catalysts: Mechanism and Intermediates That Guide Selectivity. ACS Omega 2020, 5, 21309–21319. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.M. Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef]
- Pun, S.N.; Chung, W.H.; Lam, K.M.; Guo, P.; Chan, P.H.; Wong, K.Y.; Che, C.M.; Chen, T.Y.; Peng, S.M. Iron(I) complexes of 2,9-bis(2-hydroxyphenyl)-1,10-phenanthroline (H2dophen) as electrocatalysts for carbon dioxide reduction. X-ray crystal structures of [Fe(dophen)Cl]2·2HCON(CH3)2 and [Fe(dophen)(N-MeIm)2]ClO4 (N-MeIm = 1-methylimidazole). J. Chem. Soc. Dalt. Trans. 2002, 575–583. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; MacHan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef]
- Bonetto, R.; Altieri, R.; Tagliapietra, M.; Barbon, A.; Bonchio, M.; Robert, M.; Sartorel, A. Electrochemical Conversion of CO2 to CO by a Competent FeI Intermediate Bearing a Schiff Base Ligand. ChemSusChem 2020, 13, 4111–4120. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Oshio, S.; Kida, S. Synthesis and Magnetic properties of Iron(III) complexes with several quadridentate Schiff bases. Bull. Chem. Soc. Jpn. 1977, 50, 119–122. [Google Scholar] [CrossRef]
- Cisterna, J.; Artigas, V.; Fuentealba, M.; Hamon, P.; Manzur, C.; Hamon, J.R.; Carrillo, D. Pentacoordinated chloro-iron(III) complexes with unsymmetrically substituted N2O2 quadridentate schiff-base ligands: Syntheses, structures, magnetic and redox properties. Inorganics 2018, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Duan, R.L.; Hu, C.Y.; Li, L.L.; Pang, X.; Zhang, W.X.; Chen, X.S. Salen-iron complexes: Synthesis, characterization and their reactivity with lactide. Chinese J. Polym. Sci. 2018, 36, 185–189. [Google Scholar] [CrossRef]
- Cozzolino, M.; Leo, V.; Tedesco, C.; Mazzeo, M.; Lamberti, M. Salen, salan and salalen iron(III) complexes as catalysts for CO2/epoxide reactions and ROP of cyclic esters. Dalt. Trans. 2018, 47, 13229–13238. [Google Scholar] [CrossRef]
- Wang, X.; Pennington, W.T.; Ankers, D.L.; Fanning, J.C. Comparative crystal structure examination of some iron(III) quadridentate schiff base complexes. Polyhedron 1992, 11, 2253–2264. [Google Scholar] [CrossRef]
- Böttcher, A.; Takeuchi, T.; Hardcastle, K.I.; Meade, T.J.; Gray, H.B.; Cwikel, D.; Kapon, M.; Dori, Z. Spectroscopy and Electrochemistry of Cobalt(III) Schiff Base Complexes. Inorg. Chem. 1997, 36, 2498–2504. [Google Scholar] [CrossRef] [Green Version]
- Isse, A.A.; Gennaro, A.; Vianello, E. Electrochemical reduction of Schiff base ligands H2salen and H2salophen. Electrochim. Acta 1997, 42, 2065–2071. [Google Scholar] [CrossRef]
- Chirik, P.J.; Wieghardt, K. Radical ligands confer nobility on base-metal catalysts. Science 2010, 327, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Udugala-Ganehenege, M.Y.; Dissanayake, N.M.; Liu, Y.; Bond, A.M.; Zhang, J. Electrochemistry of nickel(II) and copper(II) N,N′-ethylenebis(acetylacetoniminato) complexes and their electrocatalytic activity for reduction of carbon dioxide and carboxylic acid protons. Transit. Met. Chem. 2014, 39, 819–830. [Google Scholar] [CrossRef]
- Ueda, T.; Inazuma, N.; Komatsu, D.; Yasuzawa, H.; Onda, A.; Guo, S.X.; Bond, A.M. Comparison of chemical interactions with Li+ and catalytic reactivity of electrochemically generated [FeICl(L)]2- and [CoI(L)]- complexes (L = salen or salophen). Dalt. Trans. 2013, 42, 11146–11154. [Google Scholar] [CrossRef] [PubMed]
- Costes, J.P.; Tommasino, J.B.; Carré, B.; Soulet, F.; Fabre, P.L. Electrochemical studies of iron(III) Schiff base complexes-II. Dimeric μ-oxo [FeIII(N2O2)]2O complexes. Polyhedron 1995, 14, 771–780. [Google Scholar] [CrossRef]
- Matosziuk, L.M.; Holbrook, R.J.; Manus, L.M.; Heffern, M.C.; Ratner, M.A.; Meade, T.J. Rational design of [Co(acacen)L2]+ inhibitors of protein function. J. Chem. Soc. Dalt. Trans. 2013, 42, 4002–4012. [Google Scholar] [CrossRef] [PubMed]
- Cini, R.; Cinquantini, A.; Orioli, P.L.; Mealli, C.; Sabat, M. The effect of d electron configuration on a mononuclear transition metal species of a Schiff-base ligand with a N2S2 donor set (sacacen). Can. J. Chem. 1984, 62, 2908–2913. [Google Scholar] [CrossRef]
- Lee, K.J.; McCarthy, B.D.; Dempsey, J.L. On decomposition, degradation, and voltammetric deviation: The electrochemist’s field guide to identifying precatalyst transformation. Chem. Soc. Rev. 2019, 48, 2927–2945. [Google Scholar] [CrossRef]
- Ngo, K.T.; McKinnon, M.; Mahanti, B.; Narayanan, R.; Grills, D.C.; Ertem, M.Z.; Rochford, J. Turning on the Protonation-First Pathway for Electrocatalytic CO2 Reduction by Manganese Bipyridyl Tricarbonyl Complexes. J. Am. Chem. Soc. 2017, 139, 2604–2618. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef]
- Lam, Y.C.; Nielsen, R.J.; Gray, H.B.; Goddard, W.A. A Mn Bipyrimidine Catalyst Predicted to Reduce CO2 at Lower Overpotential. ACS Catal. 2015, 5, 2521–2528. [Google Scholar] [CrossRef] [Green Version]
- Queyriaux, N.; Abel, K.; Fize, J.; Pécaut, J.; Orio, M.; Hammarström, L. From non-innocent to guilty: On the role of redox-active ligands in the electro-assisted reduction of CO2 mediated by a cobalt(II)-polypyridyl complex. Sustain. Energy Fuels 2020, 4, 3668–3676. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A.; Vianello, E.; Floriani, C. Electrochemical reduction of carbon dioxide catalyzed by [CoI(salophen)Li]. J. Mol. Catal. 1991, 70, 197–208. [Google Scholar] [CrossRef]
- Toniolo, D.; Scopelliti, R.; Zivkovic, I.; Mazzanti, M. Assembly of High-Spin [Fe3] Clusters by Ligand-Based Multielectron Reduction. J. Am. Chem. Soc. 2020, 142, 7301–7305. [Google Scholar] [CrossRef] [PubMed]
- Cometto, C.; Chen, L.; Mendoza, D.; Lassalle-Kaiser, B.; Lau, T.C.; Robert, M. An Iron Quaterpyridine Complex as Precursor for the Electrocatalytic Reduction of CO2 to Methane. ChemSusChem 2019, 12, 4500–4505. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef]
- Waldie, K.M.; Ostericher, A.L.; Reineke, M.H.; Sasayama, A.F.; Kubiak, C.P. Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction. ACS Catal. 2018, 8, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- Loewen, N.D.; Neelakantan, T.V.; Berben, L.A. Renewable Formate from C-H Bond Formation with CO2: Using Iron Carbonyl Clusters as Electrocatalysts. Acc. Chem. Res. 2017, 50, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Solis, B.H.; Maher, A.G.; Honda, T.; Powers, D.C.; Nocera, D.G.; Hammes-Schiffer, S. Theoretical analysis of cobalt hangman porphyrins: Ligand dearomatization and mechanistic implications for hydrogen evolution. ACS Catal. 2014, 4, 4516–4526. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Iron Complex | E1/2 (V) vs. Fc+/Fc, V (ΔE, mV) | λMAX FeI [a] | −iCO2/iN2 [b] | |
|---|---|---|---|---|
| FeIII/II | FeII/I | |||
| Fe(salophen)Cl [12] | −0.69 (84) | −2.00 [c] | 405 | −2.1 |
| Fe(salen)Cl | −0.77 (98) | −2.21 (89) | 354 | −4.2 |
| Fe(acacen)Cl | −0.93 (93) | −2.50 (68) | 288 | −5.6 |
| Fe(beacen)Cl | −0.83 (81) | −2.24 (93) | 344 | −5.3 |
| Fe(acacsalen)Cl | −0.83 (91) | −2.25 (131) | 308, 379 | −4.1 |
| Iron Complex | Proton Donor | Ep (V vs. Fc+/Fc) | −iCO2/iN2 [a] | FY, CO (FY, H2) [b] | CO vs. H2 Selectivity [b,c] |
|---|---|---|---|---|---|
| Fe(salophen)Cl [12] | PhOH 0.5 M [11] | −1.99 | −4.3 | 50 (1) | 98 |
| Fe(salen)Cl | PhOH 0.5 M | −2.25 | −7.6 | 40 (<0.5) | >98 [d] |
| TFE 0.3 M | −2.29 | −5.0 | 21 (<0.1) | >99 [e] | |
| Fe(acacen)Cl | PhOH 0.5 M | −2.31 | −9.7 [f] | 5.5 (13.7) | 28 |
| TFE 0.3 M | −2.31 | −10.0 | 13.5 (0.55) | 96 | |
| Fe(beacen)Cl | PhOH 0.5 M | −2.18 | −10.0 | 10 (14.4) | 41 |
| TFE 0.3 M | −2.15 | −7.8 | 42 (<0.1) | >99 | |
| Fe(acacsalen)Cl | PhOH 0.5 M | −2.19 | −7.4 | <1 (14.8) | <1 |
| TFE 0.3 M | −2.31 | −5.8 | 18 (6.6) | 73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonetto, R.; Civettini, D.; Crisanti, F.; Sartorel, A. FeI Intermediates in N2O2 Schiff Base Complexes: Effect of Electronic Character of the Ligand and of the Proton Donor on the Reactivity with Carbon Dioxide. Energies 2021, 14, 5723. https://doi.org/10.3390/en14185723
Bonetto R, Civettini D, Crisanti F, Sartorel A. FeI Intermediates in N2O2 Schiff Base Complexes: Effect of Electronic Character of the Ligand and of the Proton Donor on the Reactivity with Carbon Dioxide. Energies. 2021; 14(18):5723. https://doi.org/10.3390/en14185723
Chicago/Turabian StyleBonetto, Ruggero, Daniel Civettini, Francesco Crisanti, and Andrea Sartorel. 2021. "FeI Intermediates in N2O2 Schiff Base Complexes: Effect of Electronic Character of the Ligand and of the Proton Donor on the Reactivity with Carbon Dioxide" Energies 14, no. 18: 5723. https://doi.org/10.3390/en14185723
APA StyleBonetto, R., Civettini, D., Crisanti, F., & Sartorel, A. (2021). FeI Intermediates in N2O2 Schiff Base Complexes: Effect of Electronic Character of the Ligand and of the Proton Donor on the Reactivity with Carbon Dioxide. Energies, 14(18), 5723. https://doi.org/10.3390/en14185723