
Catalytic Hydrogenation of CO2 to Methanol over Cu/MgO Catalysts in a Semi-Continuous Reactor



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
- Calcination: To prepare the catalyst carrier MgO, MagGran© granulate was calcined in air in a muffle furnace (Heraeus M110) for five hours under mild conditions at 723 K followed by two hours at 823 K (Equation (7)).
- Impregnation: The calcined MgO granulate (13 g, white) was impregnated with 0.25 dm3 of an aqueous copper(II) nitrate solution (cCu = 35 g dm−3) in a water cooled flask under constant stirring. After two hours, the impregnated catalyst precursor (blue) was filtered off and dried overnight in a drying furnace at 303 K.
- Thermal decomposition: The impregnated dry catalyst precursor was calcined in the muffle furnace for one hour at 423 K followed by five hours at 723 K (Equation (8)). Calcination resulted in a change of color from blue to black.
- Reduction (catalyst activation): To generate the catalytically active Cu0 sites, the calcined CuO/MgO precursor was treated in H2 atmosphere for 3.5 h in the tank reactor that was also used for the hydrogenation experiments (Equation (9)). The activation of the catalyst was performed at the reaction conditions of the CO2 hydrogenation experiments at 573 K and 5 MPa.
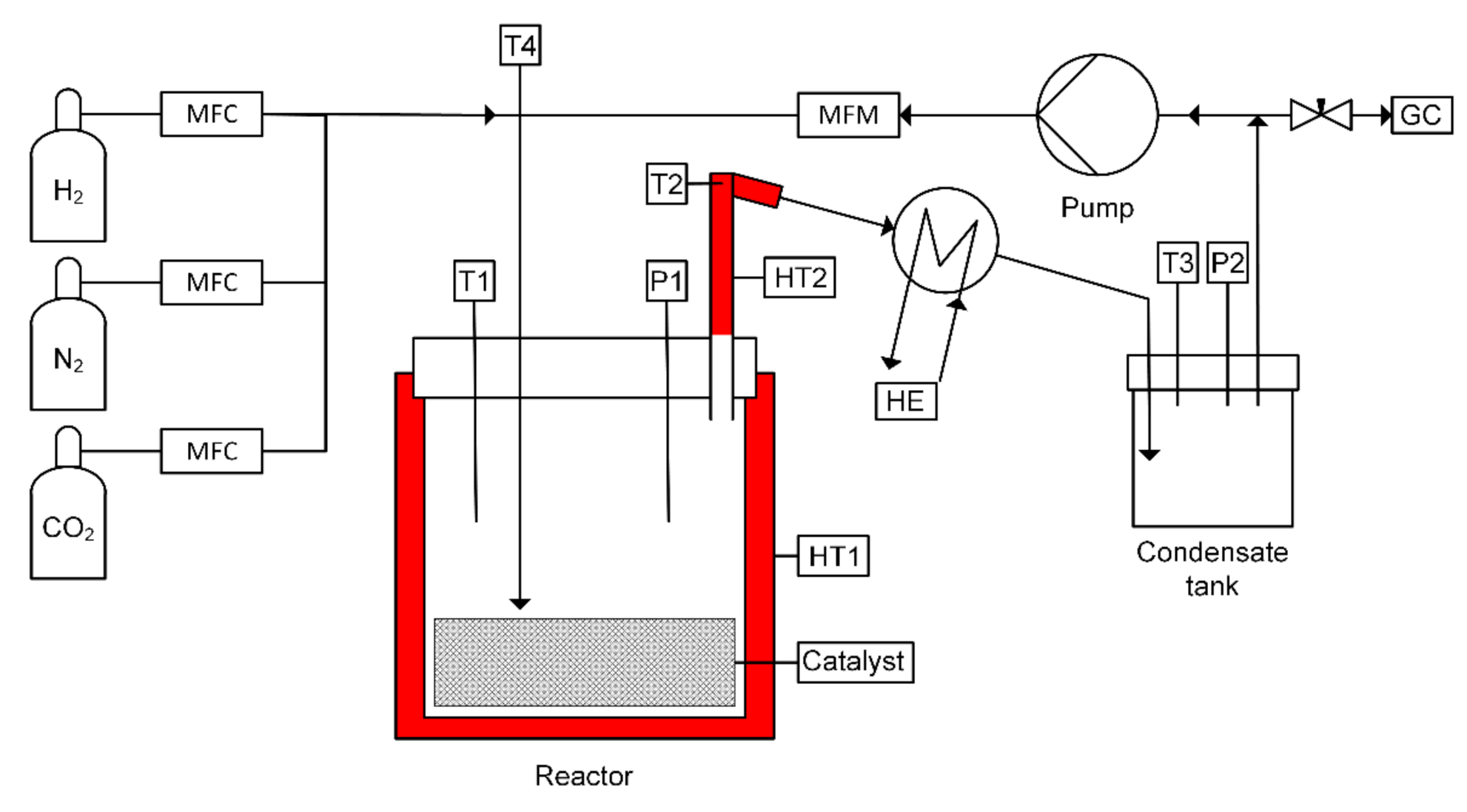
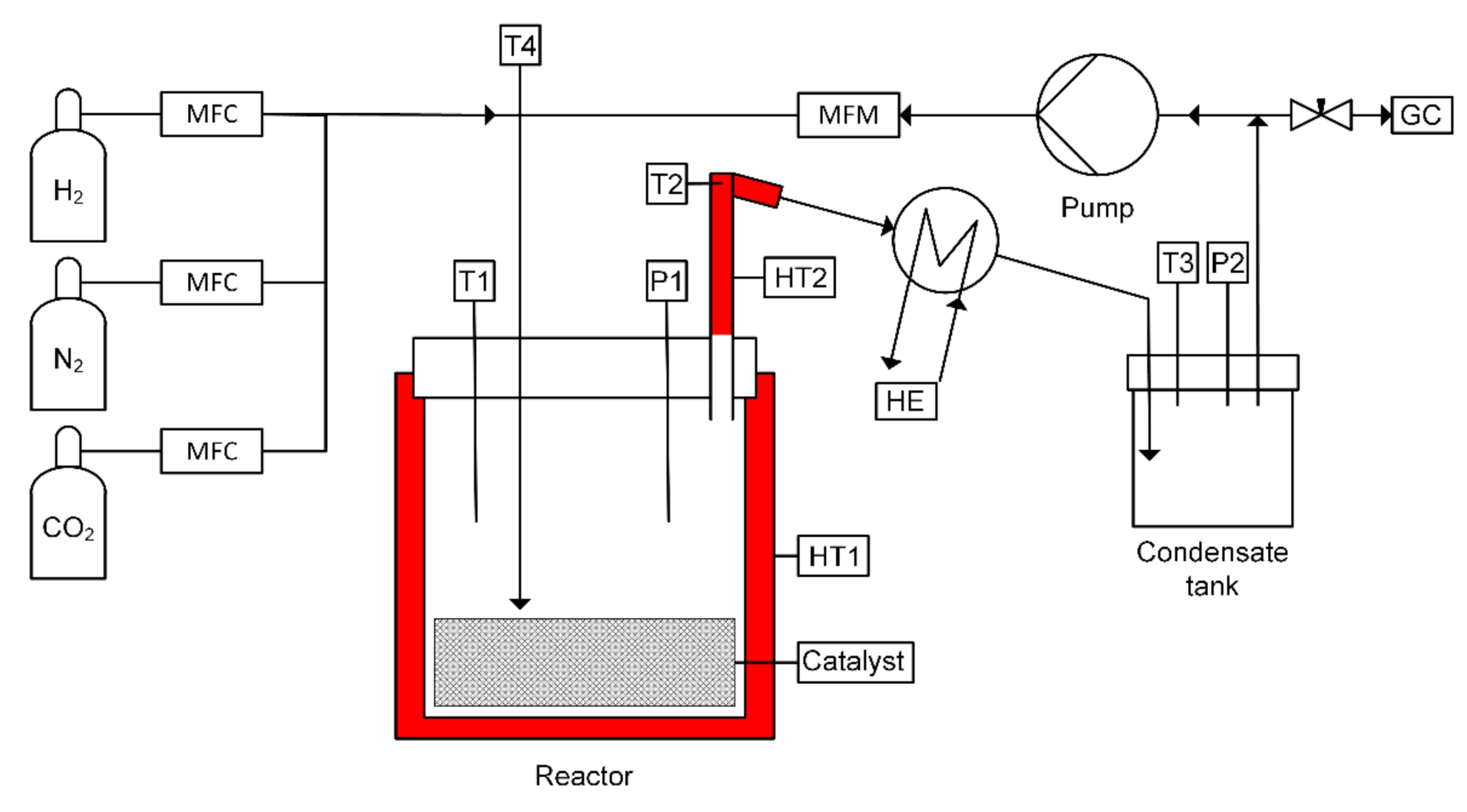
2.2. Experimental Setup and Procedure
2.3. Analysis
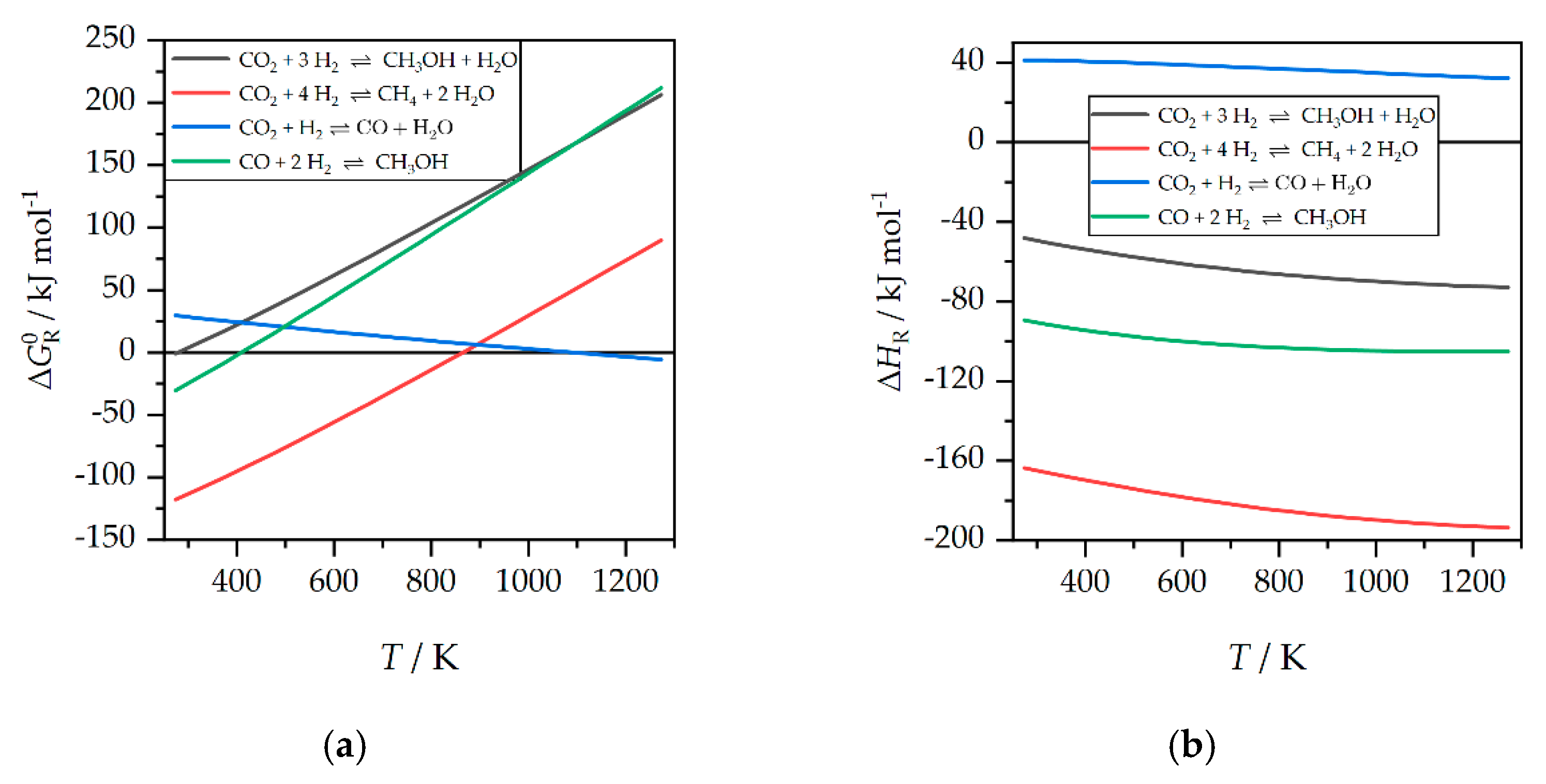
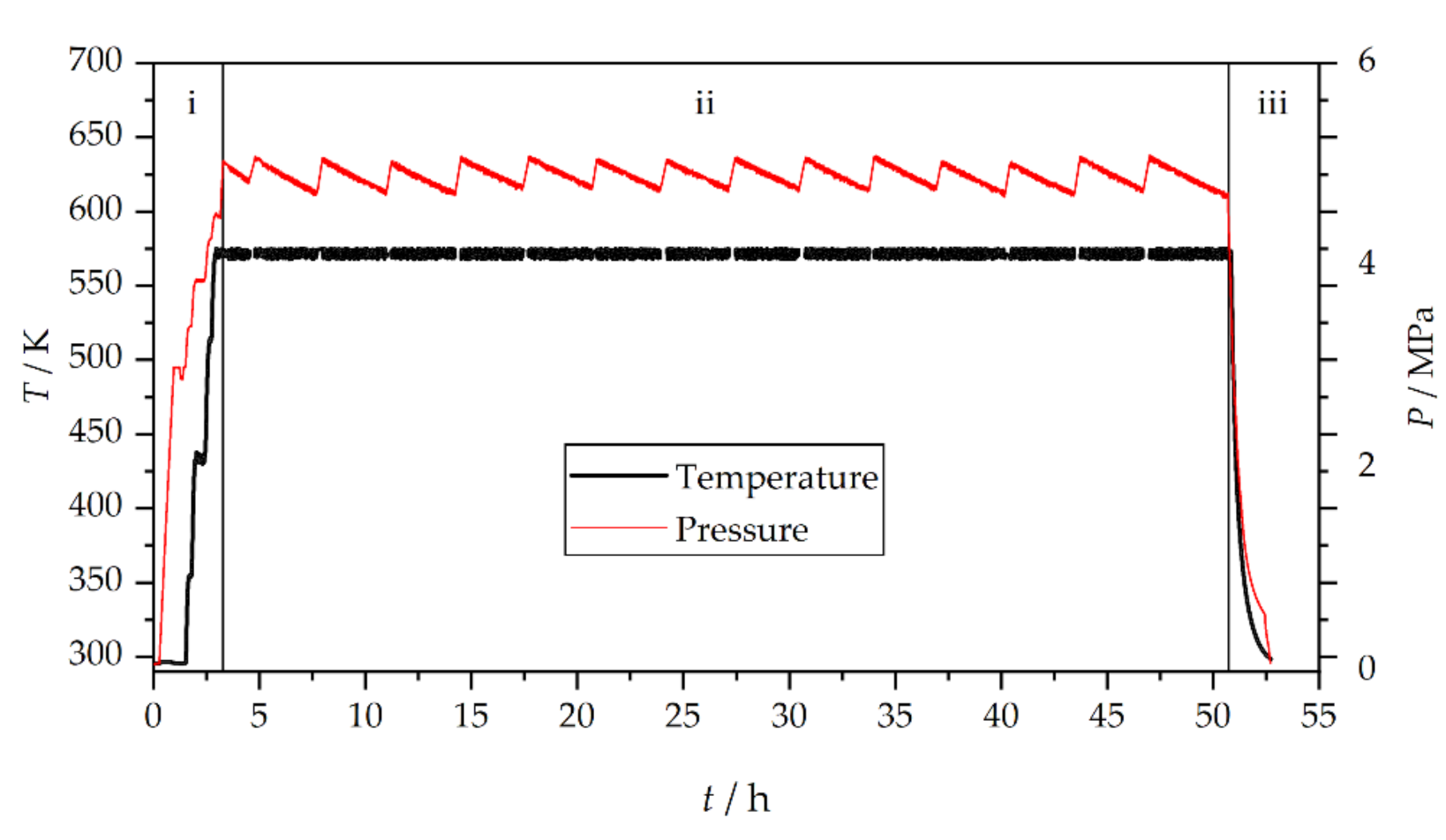
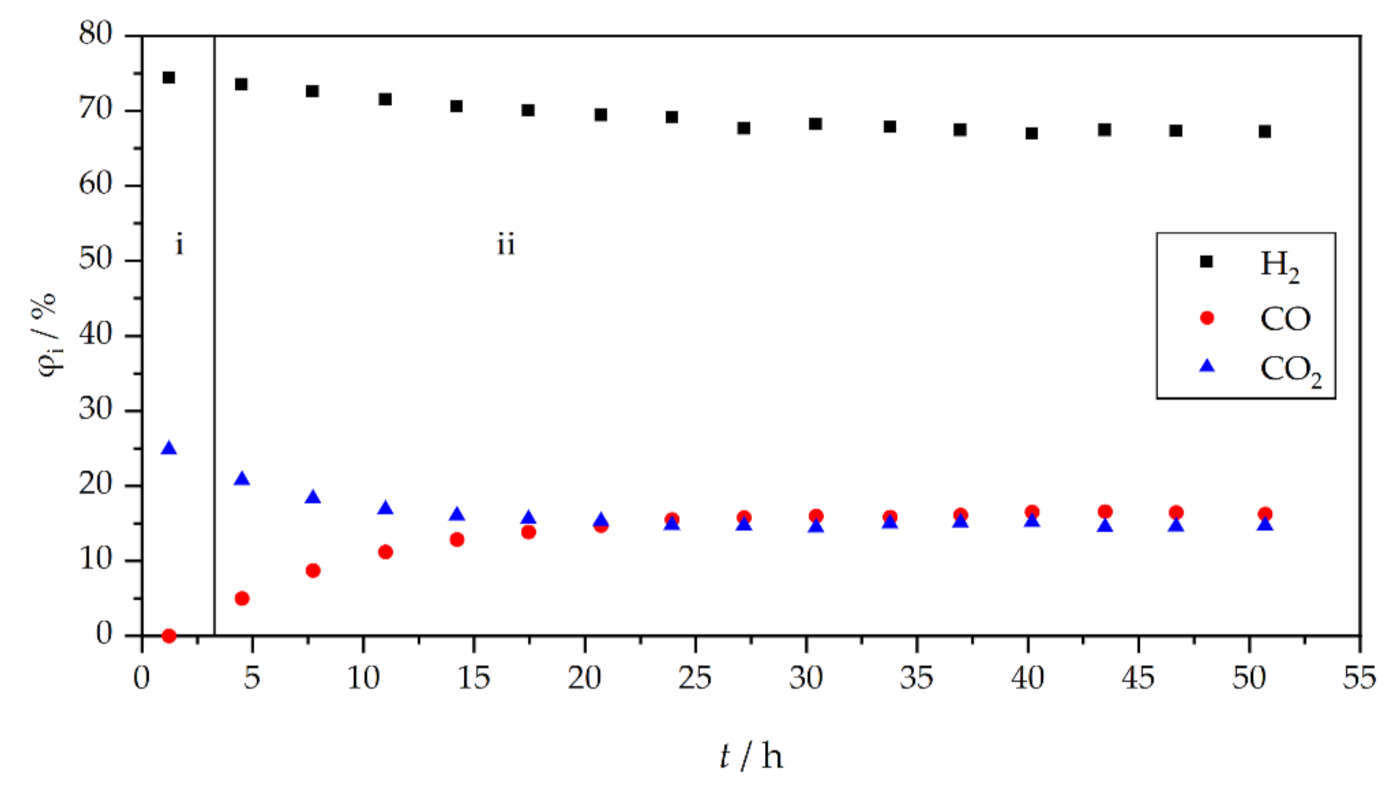
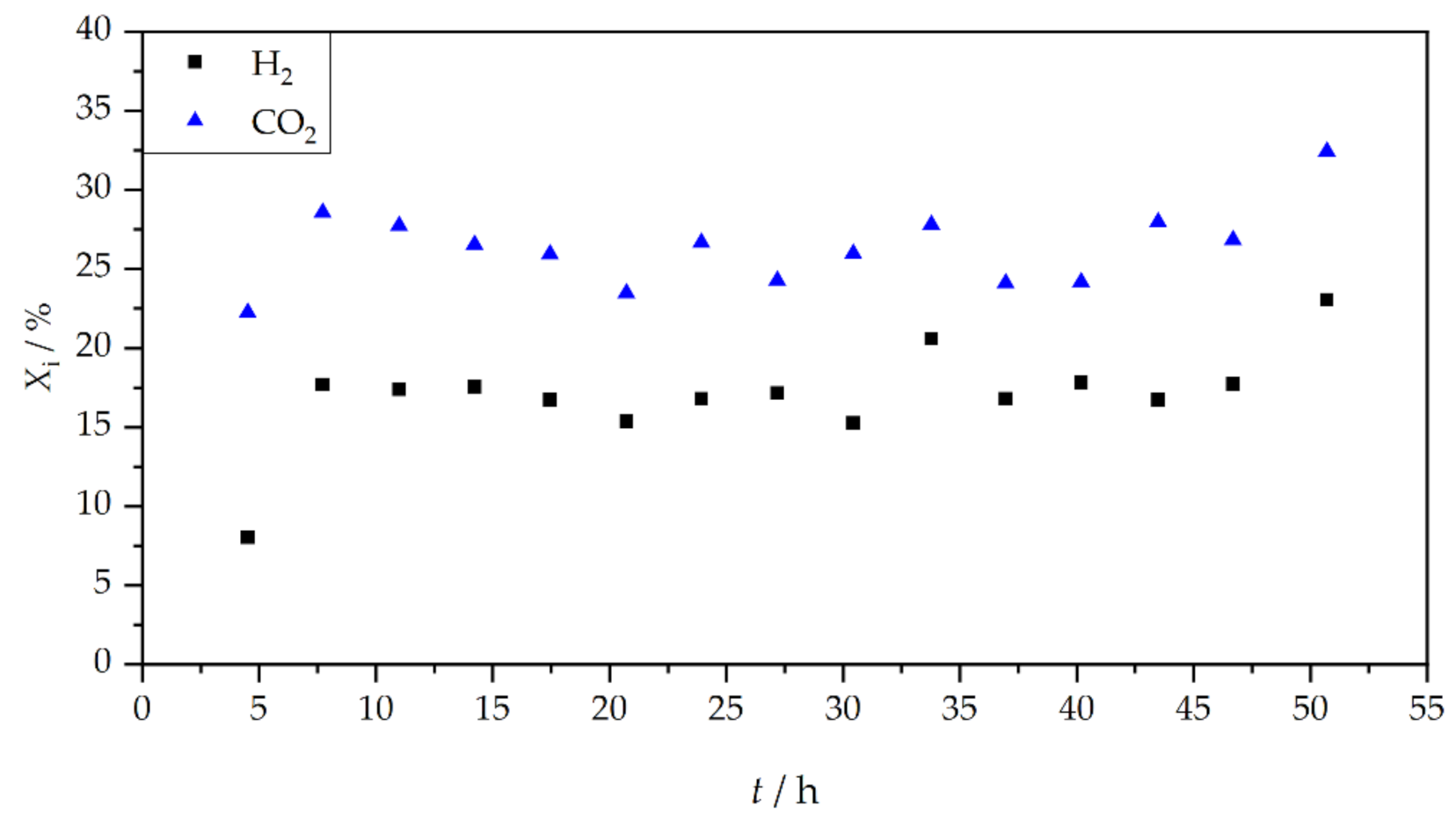
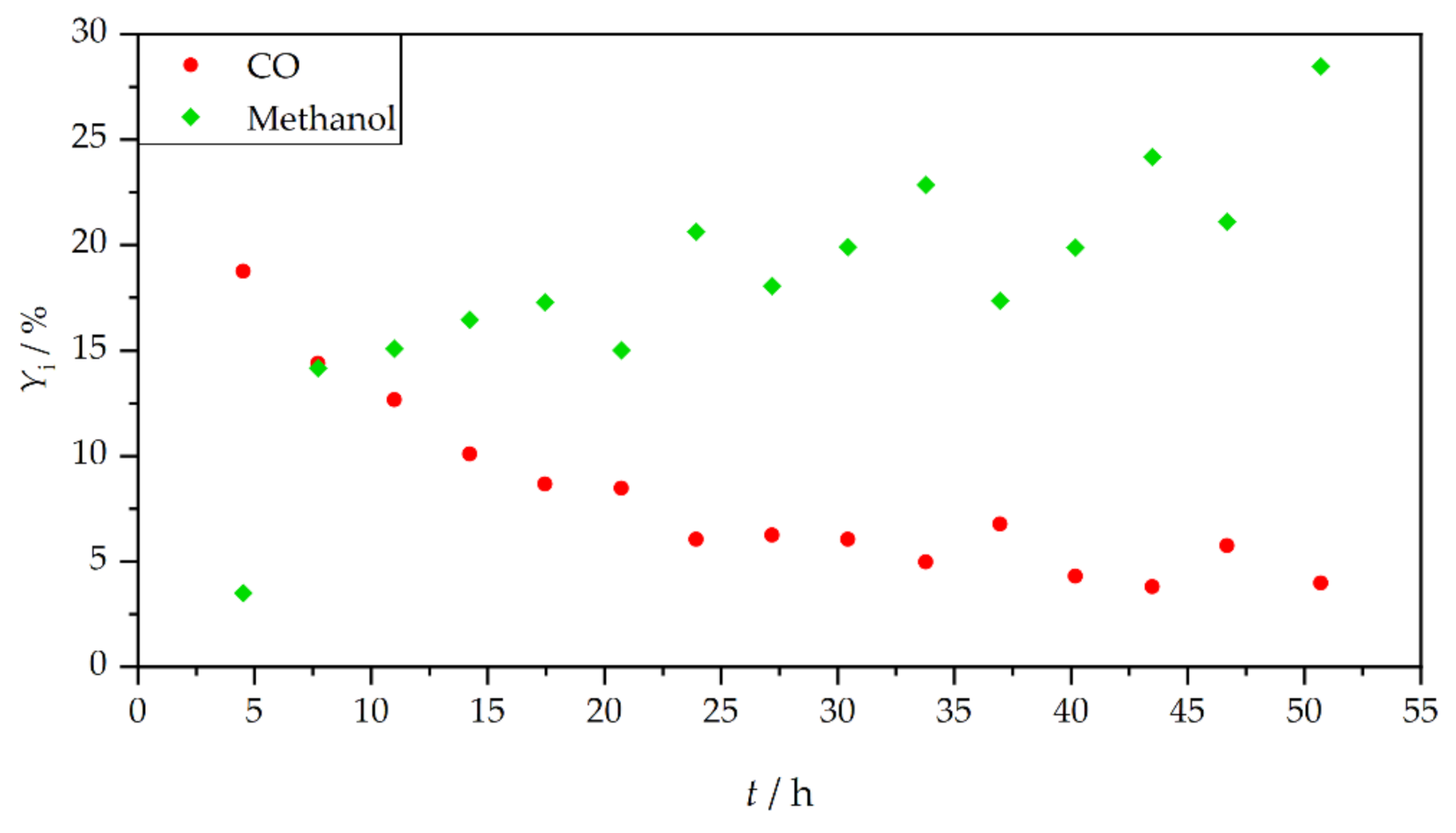
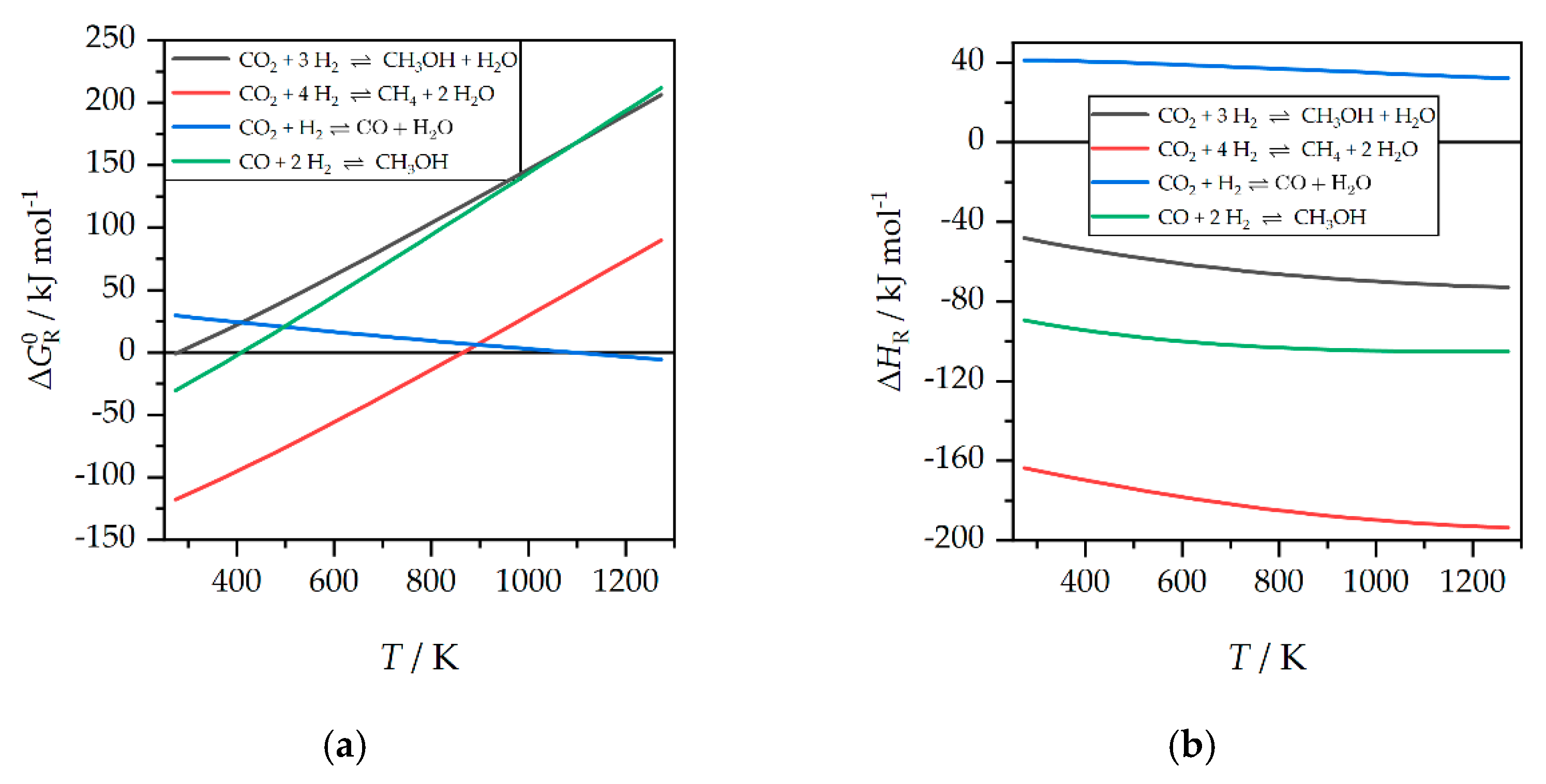
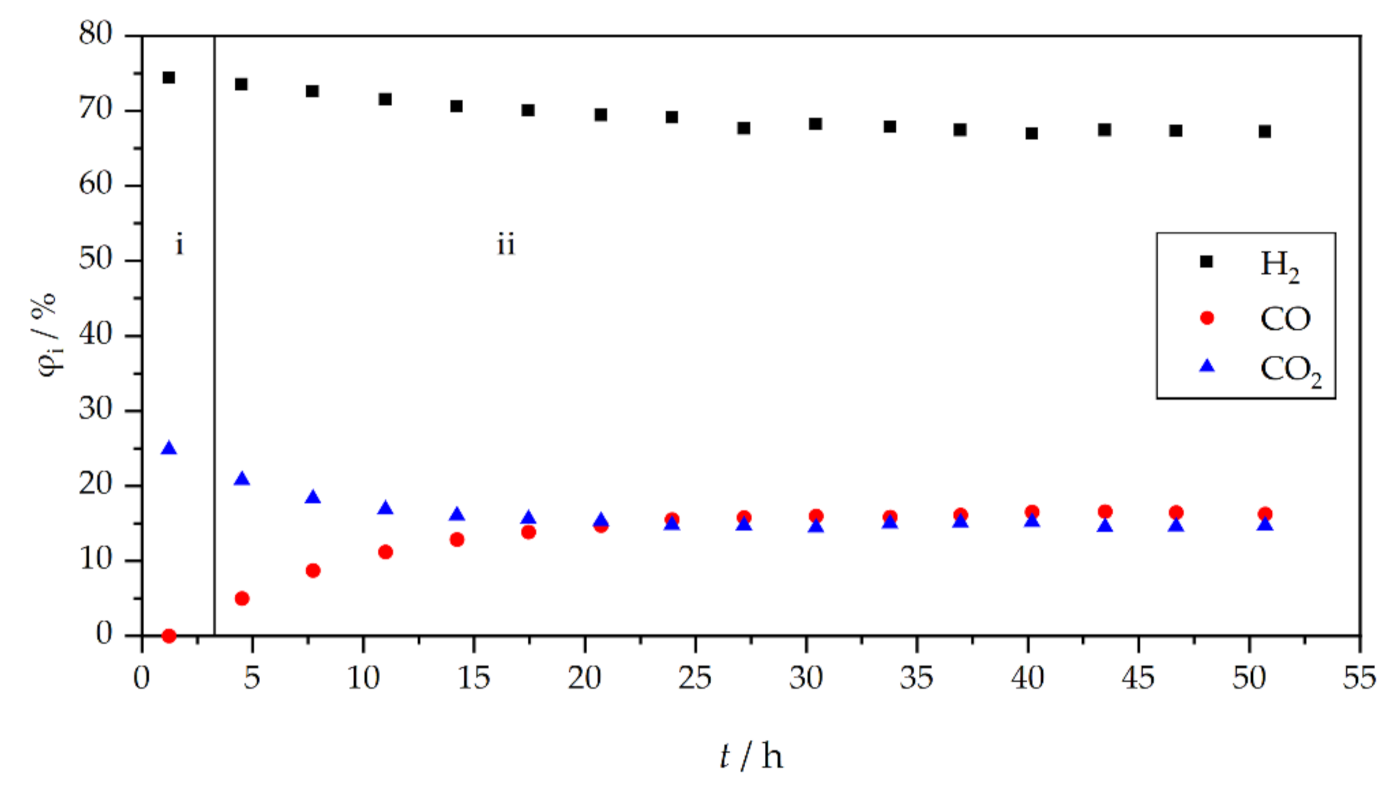
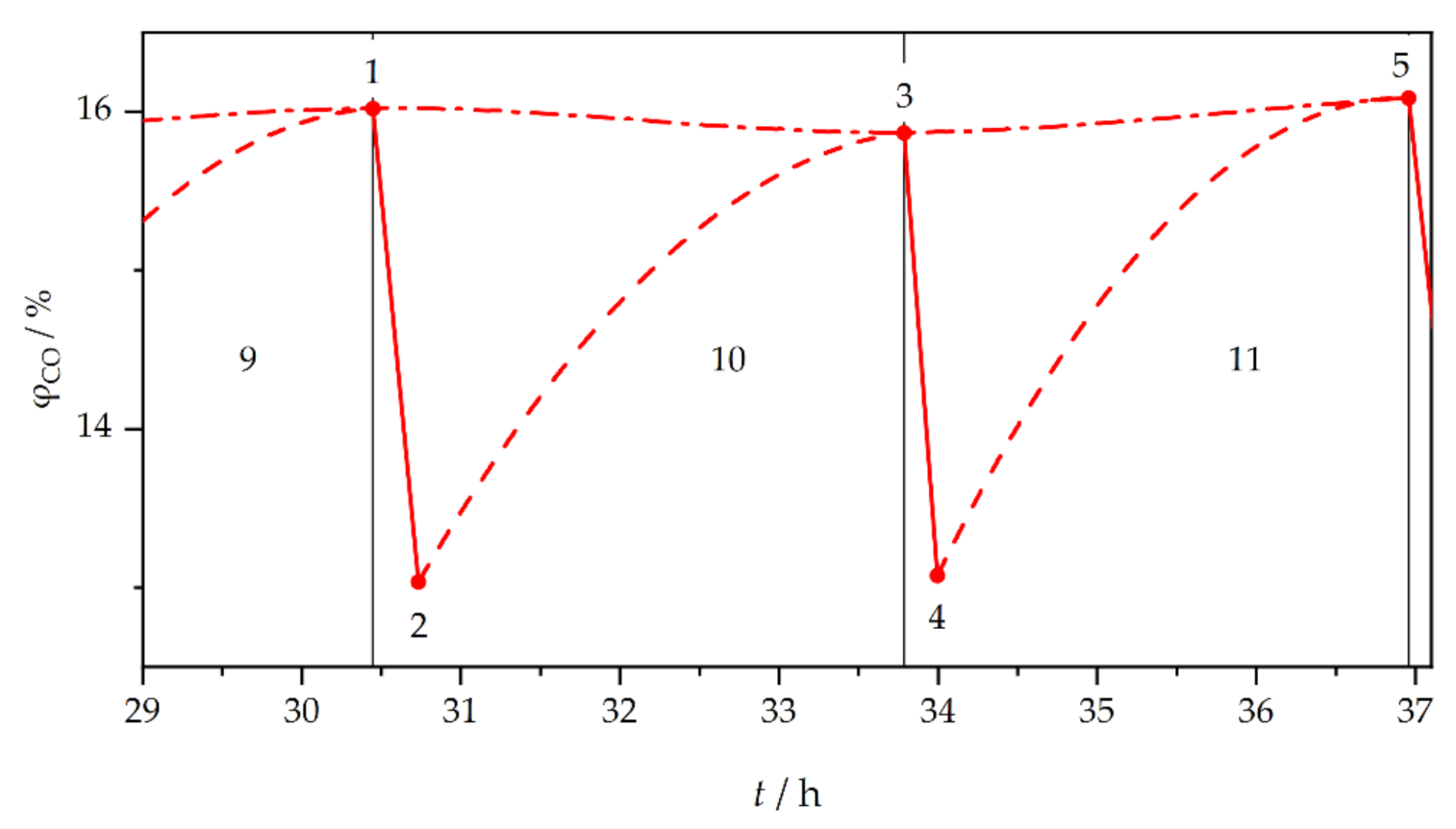
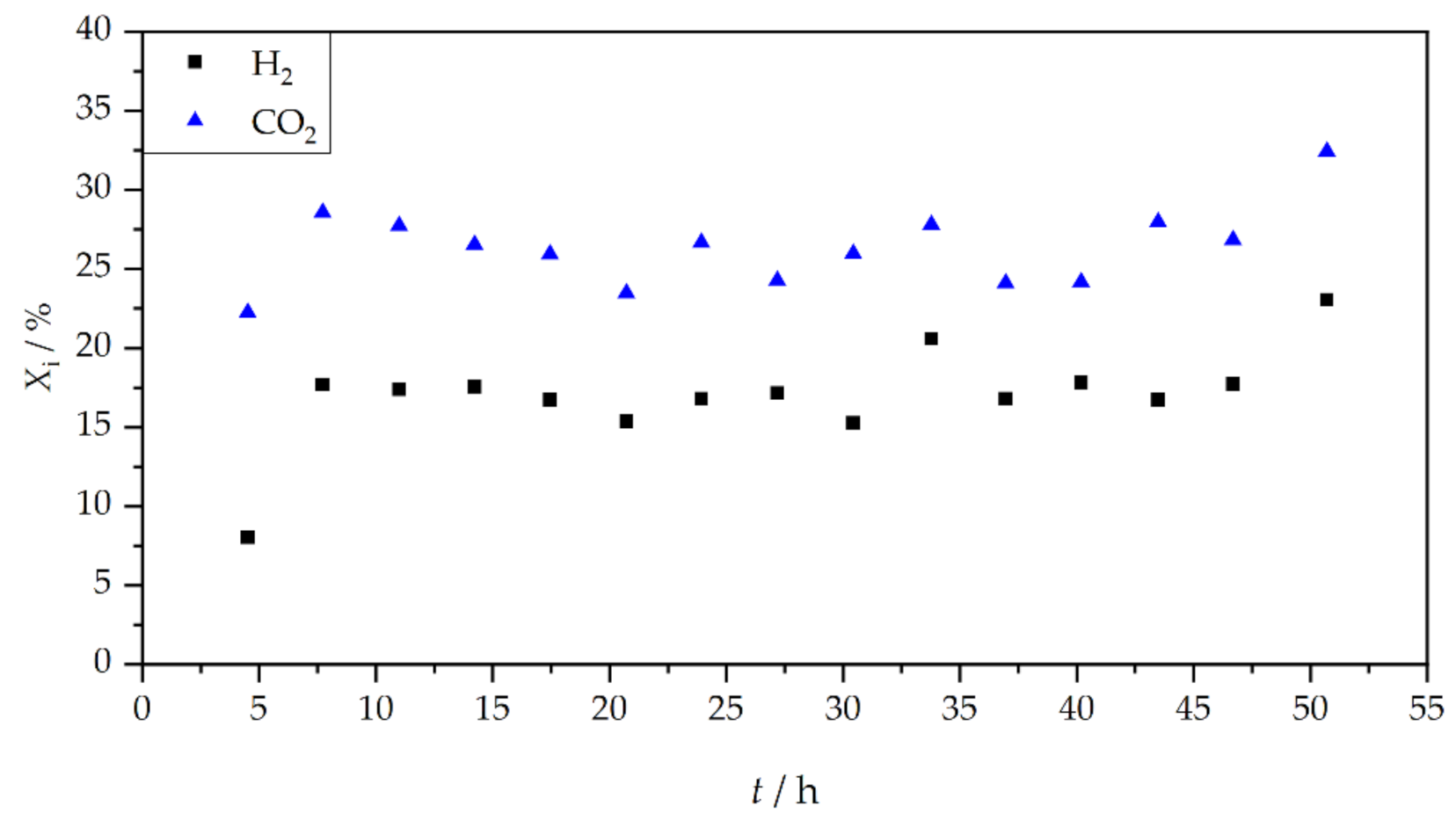
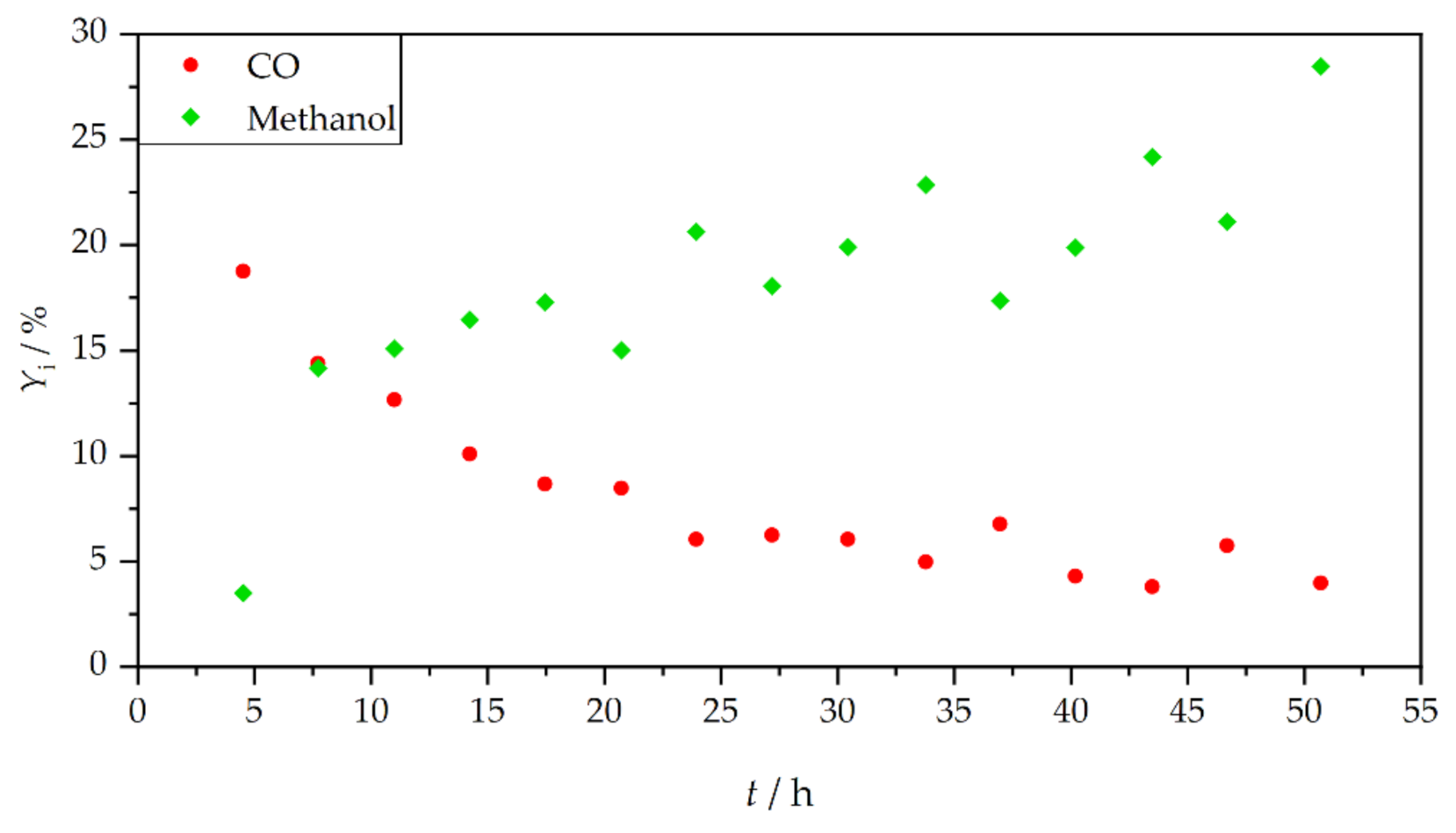
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Friedlingstein, P.; Jones, M.W.; O’Sullivan, M.; Andrew, R.M.; Hauck, J.; Peters, G.P.; Peters, W.; Pongratz, J.; Sitch, S.; le Quéré, C.; et al. Global Carbon Budget 2019. Earth Syst. Sci. Data 2019, 11, 1783–1838. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Din, I.U.; Shaharun, M.S.; Alotaibi, M.A.; Alharthi, A.I.; Naeem, A. Recent developments on heterogeneous catalytic CO2 reduction to methanol. J. CO2 Util. 2019, 34, 20–33. [Google Scholar] [CrossRef]
- Dang, S.; Yang, H.; Gao, P.; Wang, H.; Li, X.; Wei, W.; Sun, Y. A review of research progress on heterogeneous catalysts for methanol synthesis from carbon dioxide hydrogenation. Catal. Today 2019, 330, 61–75. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Yang, Y.; Mims, C.; Peden, C.H.; Li, J.; Mei, D. Insight into methanol synthesis from CO2 hydrogenation on Cu(111): Complex reaction network and the effects of H2O. J. Catal. 2011, 281, 199–211. [Google Scholar] [CrossRef]
- Deutschmann, O.; Knözinger, H.; Kochloefl, K.; Turek, T. Heterogeneous Catalysis and Solid Catalysts, 3. Industrial Applications. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chichester, UK, 2010. [Google Scholar]
- Gurau, B.; Smotkin, E.S. Methanol crossover in direct methanol fuel cells: A link between power and energy density. J. Power Sources 2002, 112, 339–352. [Google Scholar] [CrossRef]
- Breeze, P.A. Power Generation Technologies, 3rd ed.; Newnes: Kidlington, UK; Oxford, UK, 2019. [Google Scholar]
- Day, W.H. Methanol Fuel in Commercial Operation on Land and Sea. Gas Turbine World. 2016. Available online: https://www.methanol.org/wp-content/uploads/2016/12/Methanol-Nov-Dec-2016-GTW-.pdf (accessed on 7 June 2021).
- Bertau, M.; Offermanns, H.; Plass, L.; Schmidt, F.; Wernicke, H.-J. Methanol: The Basic Chemical and Energy Feedstock of the Future; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Ott, J.; Gronemann, V.; Pontzen, F.; Fiedler, E.; Grossmann, G.; Kersebohm, D.B.; Weiss, G.; Witte, C. Methanol. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chichester, UK, 2010; pp. 1–27. [Google Scholar]
- Outotec Technologies. HSC Chemistry® Software; Outotec Technologies: Helsinki, Finland, 2018. [Google Scholar]
- Raudaskoski, R.; Turpeinen, E.; Lenkkeri, R.; Pongrácz, E.; Keiski, R.L. Catalytic activation of CO2: Use of secondary CO2 for the production of synthesis gas and for methanol synthesis over copper-based zirconia-containing catalysts. Catal. Today 2009, 144, 318–323. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spandaro, L. How oxide carriers control the catalytic functionality of the Cu-ZnO system in the hydrogenation of CO2 to methanol. Catal. Today 2013, 210, 39–46. [Google Scholar] [CrossRef]
- Qi, G.-X.; Zheng, X.-M.; Fei, J.-H.; Hou, Z.-Y. Low-temperature methanol synthesis catalyzed over Cu/γ-Al2O3-TiO2 for CO2 hydrogenation. Catal. Lett. 2001, 72, 191–196. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Denny, P.J.; Parker, D.G.; Spencer, M.S.; Whan, D.A. Mechanism of methanol synthesis from CO2/CO/H2 mixtures over copper/zinc oxide/alumina catalysts: Use of 14C-labelled reactants. Appl. Catal. 1987, 30, 333–338. [Google Scholar] [CrossRef]
- Bowker, M. The mechanism of methanol synthesis on copper/zinc oxide/alumina catalysts. J. Catal. 1988, 109, 263–273. [Google Scholar] [CrossRef]
- Portha, J.-F.; Parkhomenko, K.; Kobl, K.; Roger, A.-C.; Arab, S.; Commenge, J.-M.; Falk, L. Kinetics of Methanol Synthesis from Carbon Dioxide Hydrogenation over Copper-Zinc Oxide Catalysts. Ind. Eng. Chem. Res. 2017, 56, 13133–13145. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Razali, N.A.M.; Lee, K.T.; Bhatia, S.; Mohamed, A.R. Heterogeneous catalysts for production of chemicals using carbon dioxide as raw material: A review. Renew. Sustain. Energy Rev. 2012, 16, 4951–4964. [Google Scholar] [CrossRef]
- Agny, R.M.; Takoudis, C.G. Synthesis of methanol from carbon monoxide and hydrogen over a copper-zinc oxide-alumina catalyst. Ind. Eng. Chem. Prod. Res. Dev. 1985, 24, 50–55. [Google Scholar] [CrossRef]
- Henrici-Olivé, G.; Olivé, S. Mechanistic reflections on the methanol synthesis with Cu/Zn catalysts. J. Mol. Catal. 1982, 17, 89–92. [Google Scholar] [CrossRef]
- Fujitani, T.; Saito, M.; Kanai, Y.; Kakumoto, T.; Watanabe, T.; Nakamura, J.; Uchijima, T. The role of metal oxides in promoting a copper catalyst for methanol synthesis. Catal. Lett. 1994, 25, 271–276. [Google Scholar] [CrossRef]
- Robbins, J.L.; Iglesia, E.; Kelkar, C.P.; DeRites, B. Methanol synthesis over Cu/SiO2 catalysts. Catal. Lett. 1991, 10, 1–10. [Google Scholar] [CrossRef]
- Baltes, C.; Vukojevic, S.; Schuth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Nielsen, N.D.; Thrane, J.; Jensen, A.D.; Christensen, J.M. Bifunctional Synergy in CO Hydrogenation to Methanol with Supported Cu. Catal. Lett. 2020, 150, 1427–1433. [Google Scholar] [CrossRef]
- Ren, H.; Xu, C.-H.; Zhao, H.-Y.; Wang, Y.-X.; Liu, J.; Liu, J.-Y. Methanol synthesis from CO2 hydrogenation over Cu/γ-Al2O3 catalysts modified by ZnO, ZrO2 and MgO. J. Ind. Eng. Chem. 2015, 28, 261–267. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Neja, S.Š.; Likozar, B. Correlation between synthesis pH, structure and Cu/MgO/Al2O3 eterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol. J. CO2 Util. 2018, 28, 189–199. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Štefančič, N.S.; Huš, M.; Likozar, B. Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction. Fuel 2018, 233, 103–112. [Google Scholar] [CrossRef]
- Zhan, H.; Li, F.; Gao, P.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Methanol synthesis from CO2 hydrogenation over La–M–Cu–Zn–O (M = Y, Ce, Mg, Zr) catalysts derived from perovskite-type precursors. J. Power Sources 2014, 251, 113–121. [Google Scholar] [CrossRef]
- Liu, C.; Guo, X.; Guo, Q.; Mao, D.; Yu, J.; Lu, G. Methanol synthesis from CO2 hydrogenation over copper catalysts supported on MgO-modified TiO2. J. Mol. Catal. A Chem. 2016, 425, 86–93. [Google Scholar] [CrossRef]
- Zander, S.; Kunkes, E.L.; Schuster, M.E.; Schumann, J.; Weinberg, G.; Teschner, D.; Jacobsen, N.; Schlögl, R.; Behrens, M. The role of the oxide component in the development of copper composite catalysts for methanol synthesis. Angew. Chem. Int. Ed. 2013, 52, 6536–6540. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Y.; Iwama, Y.; Tsubaki, N. Mechanistic study of a new low-temperature methanol synthesis on Cu/MgO catalysts. Appl. Catal. A Gen. 2005, 288, 126–133. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.M.; Park, J.-N. Bifunctional Mechanism of CO2 Methanation on Pd-MgO/SiO2 Catalyst: Independent Roles of MgO and Pd on CO2 Methanation. J. Phys. Chem. C 2010, 114, 7128–7131. [Google Scholar] [CrossRef]
- Loder, A.; Siebenhofer, M.; Lux, S. The reaction kinetics of CO2 methanation on a bifunctional Ni/MgO catalyst. J. Ind. Eng. Chem. 2020, 85, 196–207. [Google Scholar] [CrossRef]
- Seeger, M.; Otto, W.; Flick, W.; Bickelhaupt, F.; Akkerman, O.S. Magnesium Compounds. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chichester, UK, 2010. [Google Scholar]
- Jin, F.; Al-Tabbaa, A. Thermogravimetric study on the hydration of reactive magnesia and silica mixture at room temperature. Thermochim. Acta 2013, 566, 162–168. [Google Scholar] [CrossRef]
- Yang, N.; Ning, P.; Li, K.; Wang, J. MgO-based adsorbent achieved from magnesite for CO2 capture in simulate wet flue gas. J. Taiwan Inst. Chem. Eng. 2018, 86, 73–80. [Google Scholar] [CrossRef]
- Salomão, R.; Pandolfelli, V.C. Magnesia sinter hydration–dehydration behavior in refractory castables. Ceram. Int. 2008, 34, 1829–1834. [Google Scholar] [CrossRef]
- Toth, A.; Schnedl, S.; Painer, D.; Siebenhofer, M.; Lux, S. Interfacial Catalysis in Biphasic Carboxylic Acid Esterification with a Nickel-Based Metallosurfactant. ACS Sustain. Chem. Eng. 2019, 7, 18547–18553. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Likozar, B. The role of copper oxidation state in Cu/ZnO/Al2O3 catalysts in CO2 hydrogenation and methanol productivity. Renew. Energy 2019, 140, 452–460. [Google Scholar] [CrossRef]
- Girod, K.; Lohmann, H.; Kaluza, S. Methanol Synthesis with Steel Mill Gases: Performance Investigations in an On-Site Technical Center. Chem. Ing. Tech. 2021, 93, 850–855. [Google Scholar] [CrossRef]
- Bos, M.J.; Brilman, D. A novel condensation reactor for efficient CO2 to methanol conversion for storage of renewable electric energy. Chem. Eng. J. 2015, 278, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Bos, M.J.; Kersten, S.; Brilman, D. Wind power to methanol: Renewable methanol production using electricity, electrolysis of water and CO2 air capture. Appl. Energy 2020, 264, 114672. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleiber, S.; Pallua, M.; Siebenhofer, M.; Lux, S. Catalytic Hydrogenation of CO2 to Methanol over Cu/MgO Catalysts in a Semi-Continuous Reactor. Energies 2021, 14, 4319. https://doi.org/10.3390/en14144319
Kleiber S, Pallua M, Siebenhofer M, Lux S. Catalytic Hydrogenation of CO2 to Methanol over Cu/MgO Catalysts in a Semi-Continuous Reactor. Energies. 2021; 14(14):4319. https://doi.org/10.3390/en14144319
Chicago/Turabian StyleKleiber, Sascha, Moritz Pallua, Matthäus Siebenhofer, and Susanne Lux. 2021. "Catalytic Hydrogenation of CO2 to Methanol over Cu/MgO Catalysts in a Semi-Continuous Reactor" Energies 14, no. 14: 4319. https://doi.org/10.3390/en14144319
APA StyleKleiber, S., Pallua, M., Siebenhofer, M., & Lux, S. (2021). Catalytic Hydrogenation of CO2 to Methanol over Cu/MgO Catalysts in a Semi-Continuous Reactor. Energies, 14(14), 4319. https://doi.org/10.3390/en14144319