Ethanol Electrooxidation at Platinum-Rare Earth (RE = Ce, Sm, Ho, Dy) Binary Alloys

 ,
,  ,
,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Preparation of the Pt-RE Electrodes
2.2. Catalysts Characterization
2.3. Electrochemical Measurements
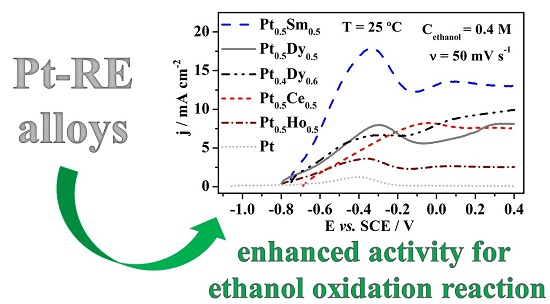
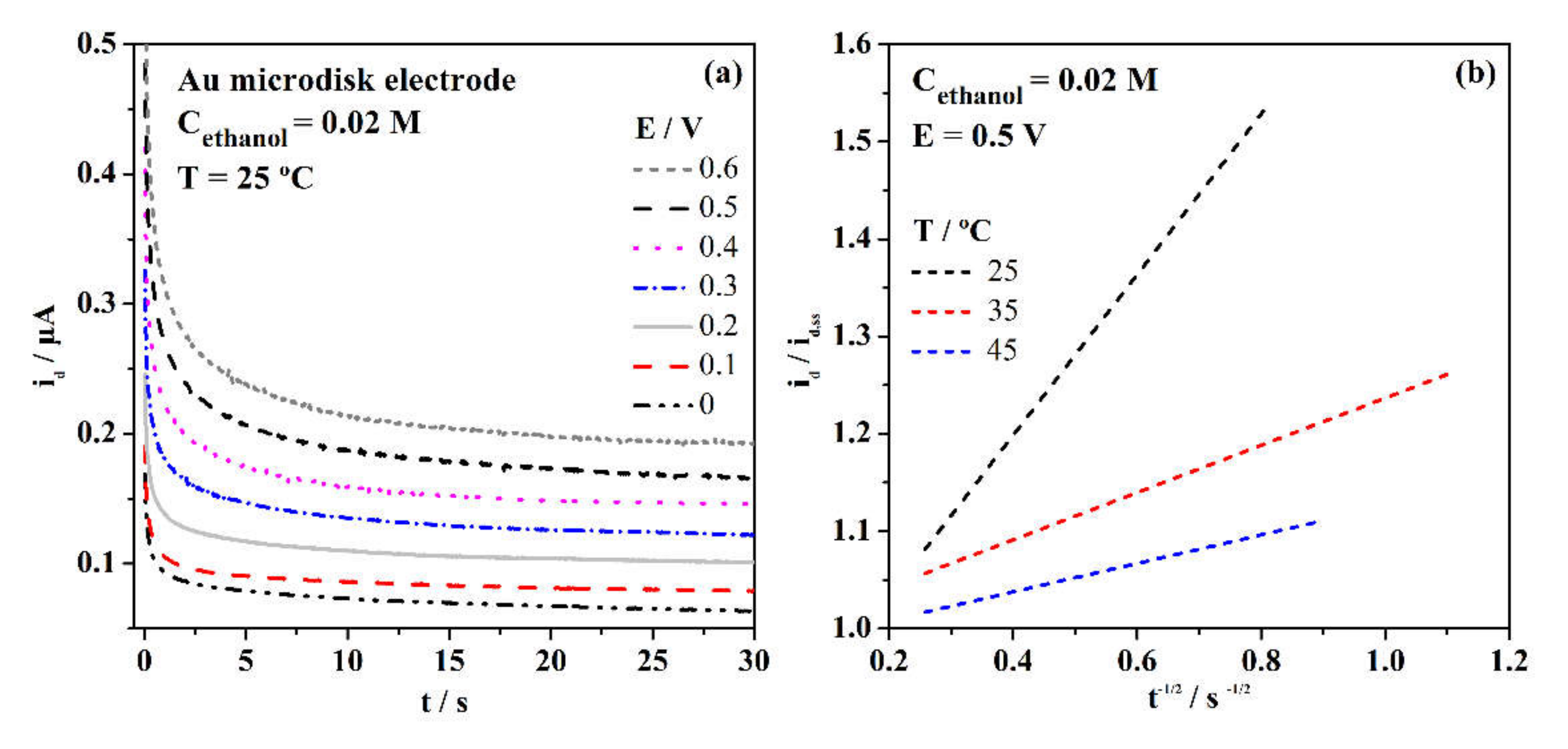
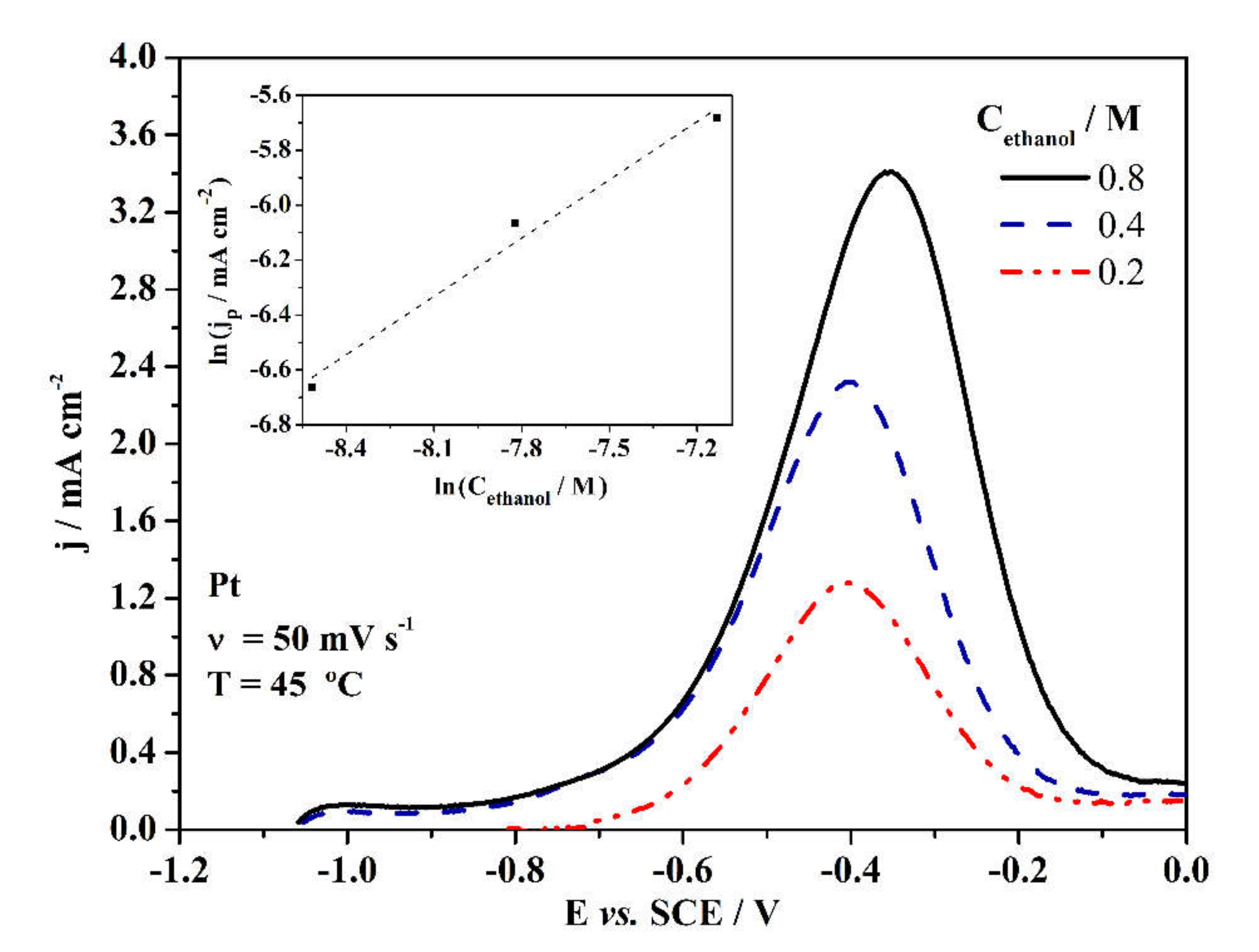
3. Results and Discussion
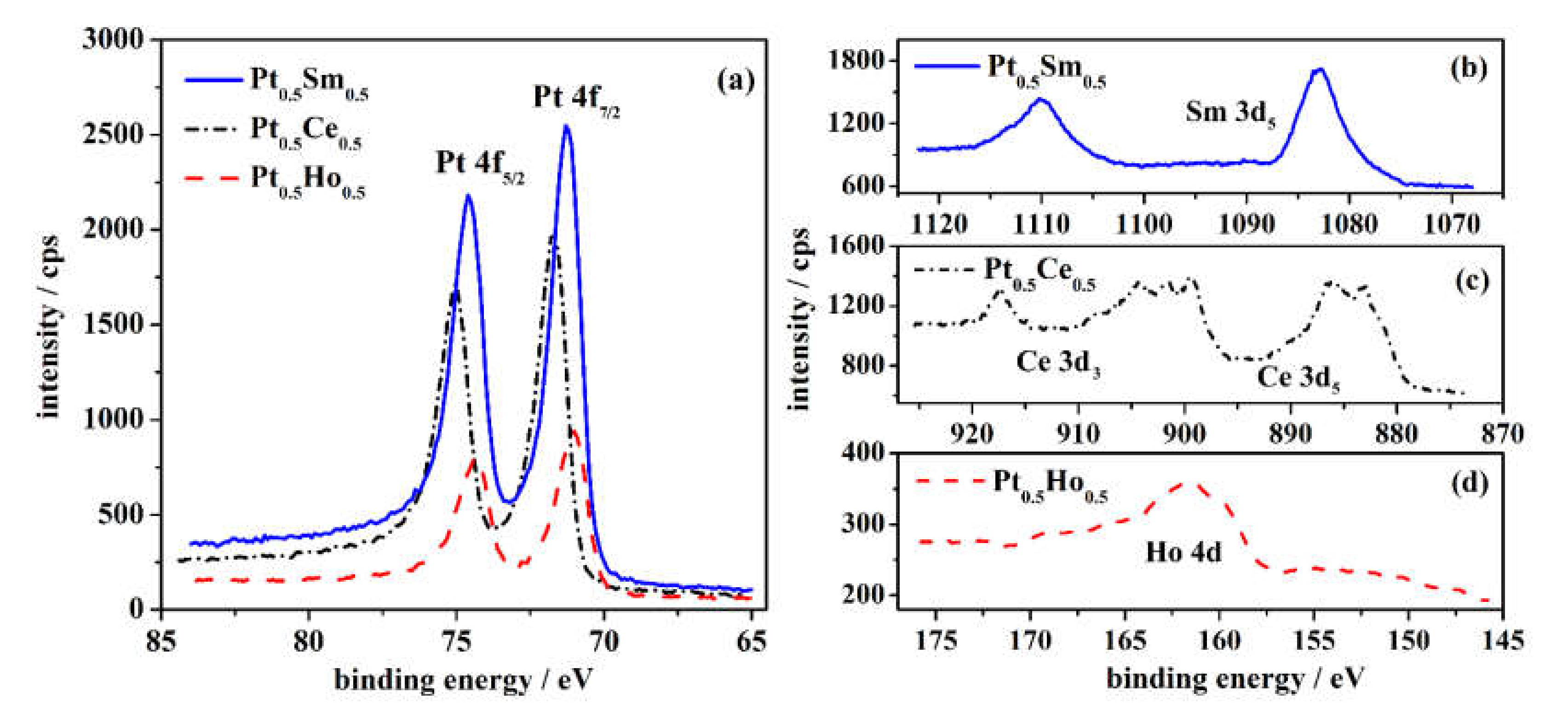
3.1. Characterization of the Pt-RE Alloys
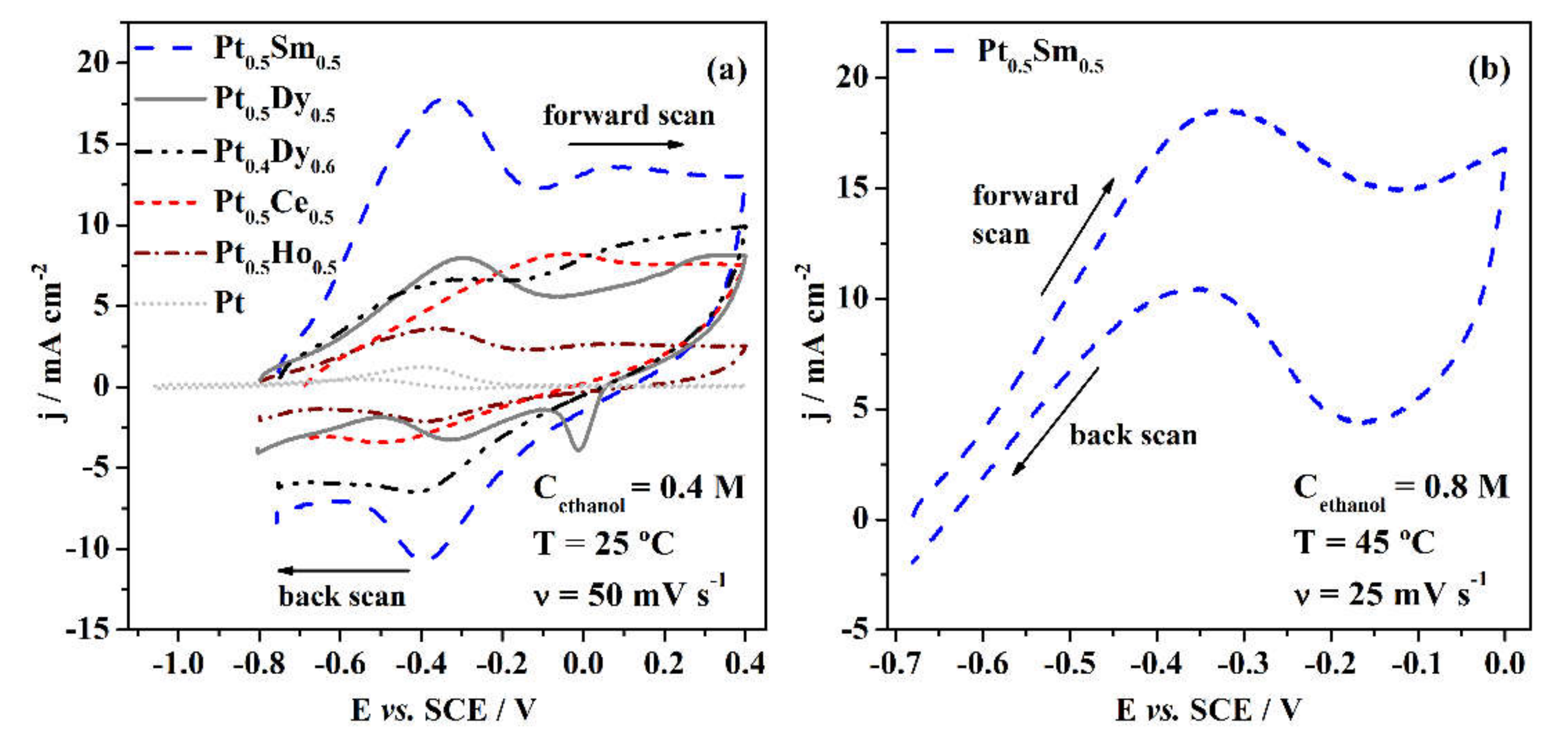
3.2. Electrochemical Properties
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dyer, C.K. Fuel cells for portable applications. J. Power Sources 2002, 106, 31–34. [Google Scholar] [CrossRef]
- Yao, L.; Chang, Y. Shaping China’s energy security: The impact of domestic reforms. Energy Policy 2015, 77, 131–139. [Google Scholar] [CrossRef]
- Töpler, J.; Lehmann, J. Hydrogen and Fuel Cell: Technologies and Market Perspectives; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Stolten, D.; Samsun, R.C.; Garland, N. Fuel Cells: Data, Facts and Figures; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016. [Google Scholar]
- Marken, F.; Fermin, D. Electrochemical Reduction of Carbon Dioxide: Overcoming the Limitations of Photosynthesis; RSC: Cambridge, UK, 2018. [Google Scholar]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Hirschenhofer, J.H.; Stanfer, D.B.; Engleman, R.R.; Klett, M.G. Fuel Cell Handbook, 4th ed.; FETC: Morgantown, WV, USA, 1998. [Google Scholar]
- Vielstich, W.; Lamm, A.; Gasteiger, H.A. Handbook of fuel Cells: Fundamentals, Technology and Applications; John Wiley & Sons Ltd.: Chichester, UK, 2003. [Google Scholar]
- Hartl, F.W.; Varela, H. The effect of solution pH and temperature on the oscillatory electro-oxidation of formic acid on platinum. Chem. Sel. 2017, 2, 8679–8685. [Google Scholar] [CrossRef]
- Maiyalagan, T.; Saji, V.S. Electrocatalysts for Low Temperature Fuel Cells: Fundamentals and Recent Trends; Wiley-VCH: Weinhein, Germany, 2017. [Google Scholar]
- Sarma, S.C.; Peter, S.C. Understanding small-molecule electro-oxidation on palladium based compounds—A feature on experimental and theoretical approaches. Dalton Trans. 2018, 47, 7864–7869. [Google Scholar] [CrossRef] [PubMed]
- Tripković, D.V.; Popović, K.D.; Jovanović, V.M.; Nogueira, J.A.; Varela, H.; Lopes, P.P.; Strmcnik, D.; Stamenkovic, V.R.; Markovic, N.M. Tuning of catalytic properties for electrooxidation of small organic molecules on Pt-based thin films via controlled thermal treatment. J. Catal. 2019, 371, 96–105. [Google Scholar] [CrossRef]
- Zheng, Y.; Wan, X.; Cheng, X.; Cheng, K.; Dai, Z.; Liu, Z. Advanced catalytic materials for ethanol oxidation in direct ethanol fuel cells. Catalysts 2020, 10, 166. [Google Scholar] [CrossRef]
- Yun, Y. Alcohol Fuels: Current status and future direction. IntechOpen 2020. Available online: https://www.intechopen.com/online-first/alcohol-fuels-current-status-and-future-direction (accessed on 29 January 2020). [CrossRef]
- Bahrami, H.; Faghai, A. Exergy analysis of a passive direct methanol fuel cell. J. Power Sources 2011, 196, 1191–1204. [Google Scholar] [CrossRef]
- Zhao, T.S.; Yang, W.W.; Chan, R.; Wu, Q.X. Towards operating direct methanol fuel cells with highly concentrated fuel. J. Power Sources 2010, 195, 3451–3462. [Google Scholar] [CrossRef]
- Yu, E.H.; Krewer, U.; Scott, K. Principles and materials aspects of direct alkaline alcohol fuel cells. Energies 2010, 3, 1499–1528. [Google Scholar] [CrossRef]
- Liang, Z.X.; Zha, T.S. Catalysts for Alcohol Fuelled Direct Oxidation Fuel Cells; RSC: Cambridge, UK, 2012. [Google Scholar]
- Yavari, Z.; Noroozifar, M.; Khorasani-Motlagh, M.; Roodbaneh, M.M.; Ajorlou, B. SrFeO3-x assisting with Pd nanoparticles on the performance of alcohols catalytic oxidation. Iran. J. Chem. Chem. Eng. 2017, 36, 21–37. [Google Scholar]
- Gurau, B.; Smotkin, E.S. Methanol crossover in direct methanol fuel cells: A link between power and energy density. J. Power Sources 2002, 112, 339–352. [Google Scholar] [CrossRef]
- Alizadeh, E.; Farhadi, M.; Sedighi, K.; Shakeri, M. The effect of cell temperature and channel geometry on the performance of a direct methanol fuel cell. J. Fuel Cell Sci. Technol. 2013, 10, 031002. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Yang, J. The effect of temperature on the output characteristics of micro direct methanol fuel cell. J. Power Sources 2015, 285, 318–324. [Google Scholar] [CrossRef]
- Badwal, S.P.S.; Giddy, S.; Kulkarni, A.; Goel, J.; Basu, S. Direct ethanol fuel cells for transport and stationary applications. A comprehensive review. Appl. Energy 2015, 145, 80–103. [Google Scholar] [CrossRef]
- Lamy, C.; Coutanceau, C. Electrocatalysis of alcohol oxidation reactions at platinum group metals. In Catalysts for Alcohol Fuelled Direct Oxidation Fuel Cells; Liang, Z.X., Zhao, T.S., Eds.; RSC: Cambridge, UK, 2012; pp. 1–70. [Google Scholar]
- Vigier, F.; Rousseau, S.; Coutanceau, C.; Leger, J.-M.; Lamy, C. Electrocatalysis for the direct alcohol fuel cell. Top. Catal. 2006, 40, 111–121. [Google Scholar] [CrossRef]
- Du, W.; Wang, Q.; Saxner, D.; Deskins, N.A.; Su, D.; Krzanowski, J.E.; Frenkel, A.I.; Teng, X. Highly active iridium/iridium à tin/tin oxide heterogeneous nanoparticles as alternative electrocatalysts for the ethanol oxidation reaction. J. Am. Chem. Soc. 2011, 133, 15172–15183. [Google Scholar] [CrossRef]
- Li, M.; Guo, W.; Jiang, R.; Zhao, L.; Shan, H. Decomposition of ethanol on Pd (111): A density functional theory study. Langmuir 2010, 26, 1879–1888. [Google Scholar] [CrossRef]
- Alcala, R.; Shabaker, J.W.; Huber, J.W.; Sanchez-Castillo, M.A.; Dumesic, J.A. Experimental and DFT studies of ethanol and acetic acid on Pt-Sn based catalysts. J. Phys. Chem. B 2005, 109, 2074–2085. [Google Scholar] [CrossRef]
- Alcala, R.; Mavrikakis, M.; Dumesic, J.A. DFT studies for cleavage of C-C and C-O bonds in surface species derived from ethanol on Pt (111). J. Catal. 2003, 218, 178–190. [Google Scholar] [CrossRef]
- Xin, H.; Holewinski, A.; Schweitzer, N.; Nikolla, E.; Linic, S. Electronic structure engineering in heterogeneous catalysis: Identified novel alloy catalysts based on rapid screening for materials with desired electronic properties. Top. Catal. 2012, 55, 376–390. [Google Scholar] [CrossRef]
- Ferrin, P.; Simonetti, D.; Kandoi, S.; Kunkes, E.; Dumesic, J.A.; Norskov, J.K.; Mavrikakis, M. Modeling ethanol decomposition on transition metals: A combined application of scaling and Bronsted-Evans-Polanyi relations. J. Am. Chem. Soc. 2009, 131, 5809–5815. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.I.; Sun, G.; Li, H.; Xin, Q. Carbon supported IrSn Catalysts for direct ethanol fuel cell. Fuel Cells Bull. 2007, 2007, 12–16. [Google Scholar] [CrossRef]
- Du, W.; Deskins, N.A.; Su, D.; Teng, X. Iridium-ruthenium alloyed nanoparticles for the ethanol oxidation. ACS Catal. 2012, 2, 1226–1231. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Z.; Song, S.; Li, W. Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B Environ. 2003, 46, 273–285. [Google Scholar] [CrossRef]
- Colmenares, L.; Wang, H.; Jusys, Z.; Jiang, J.; Yan, S.; Sun, G.Q.; Behm, R.J. Ethanol oxidation on novel carbon supported Pt alloy catalysts—Model studies under defined diffusion conditions. Electrochim. Acta 2006, 52, 221–233. [Google Scholar] [CrossRef]
- Lamy, C.; Rousseau, S.; Belgsir, E.M.; Coutanceau, C.; Leger, C. Recent Progress in the direct ethanol fuel cell: Development of new platinum-tin electrocatalysts. Electrochim. Acta 2004, 49, 3901–3908. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Leger, J.-H.; Perrard, A.; Belgsia, E.M.; Lamy, C. Development of anode catalysts for a direct etanol fuel cell. J. Appl. Electrochem. 2004, 34, 439–446. [Google Scholar] [CrossRef]
- Yovanovich, M.; Piasentin, R.M.; Ayoub, J.M.S.; Nandenha, J.; Fontes, E.H.; De Souza, R.F.B.; Buzzo, G.S.; Silva, J.C.M.; Spinace, E.V.; Assumpção, M.H.M.T.; et al. Pt Bi/C electrocatalysts for formic acid electrooxidation in acid and alkaline electrolytes. Int. J. Electrochem. Sci. 2015, 10, 4801–4811. [Google Scholar]
- Wang, Y.; Zou, S.; Cai, W.B. Recent advances on electrooxidation of ethanol on Pt- and Pd-based electrocatalysts: From reaction mechanisms to catalytic materials. Catalysts 2015, 5, 1507–1534. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, F.; Wang, Z.; Yang, M.; Xia, J.; Gui, R.; Xia, Y. Facile preparation of PtPdPt/graphene nanocomposites with ultrahigh electrocatalytic performance for methanol oxidation. J. Electroanal. Chem. 2016, 761, 55–61. [Google Scholar] [CrossRef]
- Soares, L.A.; Morais, C.; Napporn, T.W.; Kokoh, K.B.; Olivi, P. Beneficial effects of rhodium and tin oxide on carbon supported platinum catalysts for ethanol electrooxidation. J. Power Sources 2016, 315, 47–55. [Google Scholar] [CrossRef]
- Ocampo-Restrepo, V.K.; Calderón-Cárdenas, A.; Lizcano-Valbuena, W.H. Catalytic activity of Pt-based nanoparticles with Ni and Co for ethanol and acetaldehyde electrooxidation in alkaline medium. Electrochim. Acta 2017, 246, 475–483. [Google Scholar] [CrossRef]
- He, C.; Desai, S.; Brown, G.; Bollepallic, S. PEM fuel cell catalysts: Cost, performance and durability. Electrochem. Soc. Interfaces 2005, 14, 41–44. [Google Scholar]
- Li, B.; Qiao, J.; Yang, D.; Lin, R.; Lv, H.; Wang, H.; Ma, J. Effect of metal particle size and Nafion content on performance of MEA using Ir-V/C as anode catalyst. Int. J. Hydrog. Energy 2010, 35, 5528–5538. [Google Scholar] [CrossRef]
- De Souza, J.P.I.; Queiroz, S.L.; Bergamaski, K.; Gonzalez, E.R.; Nart, F.C.; De Sa, U. SpC Electrooxidation of ethanol on Pt, Rh and PtRh electrodes. A Study using DEMS and in-situ FTIR techniques. J. Phys. Chem. B 2002, 106, 9825–9830. [Google Scholar] [CrossRef]
- Alayoglu, S.; Nilekar, A.U.; Mavrikakis, M.; Eichhorn, B. Ru-Pt core-shell nanoparticles for preferential oxidation of carbon monoxide in hydrogen. Nat. Mater. 2008, 7, 333–338. [Google Scholar] [CrossRef]
- Su, H.-Y.; Gu, X.-K.; Ma, X.; Zhao, Y.-X.; Bao, X.-H.; Li, W.-X. Structure evolution of Pt-3d transition metal alloys under reductive and oxidizing conditions and effect on the CO oxidation: A first principles study. Catal. Today 2011, 165, 89–95. [Google Scholar] [CrossRef]
- Antolini, E. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Leger, J.; Rousseau, S.; Coutanceau, C.; Hahn, F.; Lamy, C. How bimetallic electrocatalysts does work for reaction involved in fuel cells? Example of ethanol oxidation and comparison to methanol. Electrochim. Acta 2005, 50, 5118–5125. [Google Scholar] [CrossRef]
- Ju, K.-S.; Pak, S.-N.; Ri, C.-N.; Ryo, H.-S.; Kim, K.-I.; So, S.-R.; Ri, C.-K.; Ri, S.-P.; Nam, K.-W.; Pak, K.-S.; et al. Ethanol electrooxidation on carbon-supported Pt1Mn3 catalyst investigated by pinhole on-line electrochemical mass spectrometry. Chem. Phys. Lett. 2019, 727, 78–84. [Google Scholar] [CrossRef]
- Matin, M.A.; Kumar, A.; Saad, M.A.H.S.; Al-Marri, M.J.; Suslou, S. Zn-enriched PtZn nanoparticle electrocatalysts synthesized by solution combustion for ethanol oxidation reaction in an alkaline medium. MRS Commun. 2018, 8, 411–419. [Google Scholar] [CrossRef]
- Raoof, J.-B.; Hosseini, S.R.; Rezaee, S. Preparation of Pt/Poly (2-Methoxyaniline)—Sodium dodecyl sulfate composite and its application for electrocatalytic oxidation of methanol and formaldehyde. Electrochim. Acta 2014, 141, 340–348. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, G.; Shu, H.; Oyama, H.; Liu, X.; He, Y. Synthesis of Pt-Pd bimetallic nanoparticles anchored on graphene for highly active methanol electrooxidation. J. Power Sources 2014, 262, 278–285. [Google Scholar] [CrossRef]
- Zhu, H.; Li, G.; Lv, X.; Zhao, Y.; Huang, T.; Liu, H.; Li, J. Controlled synthesis of hierarchical tetrapod Pd nanocrystal and their enhanced electrocatalytic properties. RSC Adv. 2014, 4, 6535–6539. [Google Scholar] [CrossRef]
- Hathoot, A.A. Electrooxidation of iodide ion at poly 8-(3-acetylimino-6 methyl-2,4-dioxopyran)-1-aminonaphthalene modified electrode in aqueous solution. Croat. Chem. Acta 2011, 84, 469–473. [Google Scholar] [CrossRef]
- Hathoot, A.A.; Yousef, U.S.; Shatla, A.S.; Abdel-Azzem, M. Simultaneous determination of ascorbic acid, uric acid and dopamine at modified electrode based on hybrid nickel hexacyanoferrate/poly (1,5 diaminonaphtalene). J. Iran. Chem. Soc. 2017, 14, 1789–1799. [Google Scholar] [CrossRef]
- Liu, L.; Pippel, E.; Scholz, R.; Gosele, U. Nanoporous Pt-Co alloy nanowires: Fabrication, characterization and electrocatalytic properties. Nano Lett. 2009, 9, 4352–4358. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Alloy catalysts designed from first principles. Nat. Mater. 2004, 3, 810–815. [Google Scholar] [CrossRef]
- Nilekar, A.U.; Alayoglu, S.; Eichhorn, B.; Mavrikakis, M. Preferential CO oxidation in hydrogen: Reactivity of core-shell nanoparticles. J. Am. Chem. Soc. 2010, 132, 7418–7428. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.M.; Ianniello, R.; Pastor, E.; González, S. Electrochemical reactivity of ethanol on porous Pt and PtRu: Oxidation/reduction reactions in 1 M HClO4. J. Phys. Chem. 1996, 100, 17901–17908. [Google Scholar] [CrossRef]
- Colmati, F.; Antolini, E.; Gonzalez, E.R. Effect of temperature on the mechanism of ethanol oxidation on carbon supported Pt, PtRu and Pt3Sn electrocatalysts. J. Power Sources 2006, 157, 98–103. [Google Scholar] [CrossRef]
- Rutkowska, I.A.; Koster, M.D.; Blanchard, G.J.; Kulesza, P.J. Enhancement of ethanol oxidation at Pt and PtRu nanoparticles dispersed over hybrid zirconia-rhodium supports. J. Power Sources 2014, 272, 681–688. [Google Scholar] [CrossRef]
- Rutkowska, I.A.; Kulesza, P.J. Electroanalysis of ethanol oxidation reactivity of platinum-ruthenium catalysts supported onto nanostructured titanium dioxide matrices. J. Electrochem. Soc. 2016, 163, H3052–H3060. [Google Scholar] [CrossRef]
- Asgardi, J.; Calderón, J.C.; Alcaide, F.; Querejeta, A.; Calvillo, L.; Lázaro, M.J.; Garcia, J.; Pastor, E. Carbon monoxide and ethanol oxidation on PtSn supported catalysts: Effect of the nature of the carbon support and Pt: Sn composition. Appl. Catal. B Environ. 2015, 168, 33–41. [Google Scholar] [CrossRef]
- Rizo, R.; Sebastian, D.; Lazaro, M.J.; Pastor, E. On the design of Pt-Sn efficient catalyst for carbon monoxide and ethanol oxidation in acid and alkaline media. Appl. Catal. B Environ. 2017, 200, 246–254. [Google Scholar] [CrossRef]
- Zhou, W.J.; Li, W.Z.; Song, S.Q.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Poulianitis, K.; Kontou, S.; Tsiakaras, P. Bi-and tri-metallic Pt-based anode catalysts for direct ethanol fuel cells. J. Power Sources 2004, 131, 217–223. [Google Scholar] [CrossRef]
- Ghavidel, M.Z.; Easton, E.B. Improving the ethanol oxidation activity of Pt-Mn alloys through the use of additives during deposition. Catalysts 2015, 5, 1016–1033. [Google Scholar] [CrossRef]
- Zhang, Q.; Mellinger, Z.J.; Jiang, Z.; Chen, X.; Wang, B.; Tian, B.; Liang, Z.; Chen, J.G. Palladium-modified tungsten carbide for ethanol electrooxidation: From surface science studies to electrochemical evaluation. J. Electrochem. Soc. 2018, 165, J3031–J3038. [Google Scholar] [CrossRef]
- Mukherjee, P.; Bagchi, J.; Dutta, S.; Bhattacharia, S.K. The nickel supported platinum catalyst for anodic oxidation of ethanol in alkaline medium. Appl. Catal. A-Gen. 2015, 506, 220–227. [Google Scholar] [CrossRef]
- Ottoni, C.A.; Ramos, C.E.D.; de Souza, R.F.B.; da Silva, S.G.; Spinace, E.V.; Neto, A.O. Glycerol and ethanol oxidation in alkaline medium using PtCu/C electrocatalysts. Int. J. Electrochem. Sci. 2018, 13, 1893–1904. [Google Scholar] [CrossRef]
- Delpeuch, A.B.; Maillard, F.; Chatenet, M.; Soudant, P.; Cremers, C. Ethanol oxidation reaction (EOR) investigation on Pt/C, Rh/C, and Pt-based bi- and tri-metallic electrocatalysts: A DEMS and in situ FTIR study. Appl. Catal. B Environ. 2016, 181, 672–680. [Google Scholar] [CrossRef]
- Hassan, K.M.; Hathoot, A.A.; Maher, R.; Azzen, M.A. Electrocatalytic oxidation of ethanol at Pd, Pt, Pd/Pt and Pt/Pd nanoparticles supported on poly 1,8 diaminonaphthalene film in alkaline medium. RSC Adv. 2018, 8, 15417–15426. [Google Scholar] [CrossRef]
- Amorim, A.; Parreira, L.S.; Silva, J.C.M.; dos Santos, M.C. Electrooxidation of mixed ethanol and methanol solutions on PtSn/C electrocatalysts prepared by the polymeric precursor method. J. Braz. Chem. Soc. 2017, 28, 1091–1097. [Google Scholar] [CrossRef]
- Gu, Z.; Li, S.; Xiong, Z.; Xu, H.; Gao, F.; Du, Y. Rapid synthesis of platinum-ruthenium bimetallic nanoparticles dispersed on carbon support as improved electrocatalysts for ethanol oxidation. J. Coll. Interface Sci. 2018, 521, 111–118. [Google Scholar] [CrossRef]
- Marinkovic, N.S.; Li, M.; Adzic, R.R. Pt-based catalysts for electrochemical oxidation of ethanol. Top. Curr. Chem. 2019, 377, 11. [Google Scholar] [CrossRef]
- Bai, J.; Liu, D.; Yang, J.; Chen, Y. Nanocatalysts for electrocatalytic oxidation of ethanol. ChemSusChem 2019, 12, 2117–2132. [Google Scholar] [CrossRef]
- Tripkovic, A.V.; Lovic, J.D.; Popovic, K.D. Comparative study of ethanol oxidation at Pt-based nanoalloys and UDP-modified Pt nanoparticles. J. Serb. Chem. Soc. 2010, 75, 1559–1574. [Google Scholar] [CrossRef]
- Li, M.; Kowal, A.; Sasaki, K.; Marinkovic, N.; Su, D.; Korach, E.; Liu, P.; Adzic, R.R. Ethanol oxidation on the ternary Pt-Rh-SnO2/C electrocatalysts with varied Pt: Rh: Sn ratios. Electrochim. Acta 2010, 55, 4331–4338. [Google Scholar] [CrossRef]
- Kowal, A.; Li, M.; Shao, M.; Sasaki, K.; Vukmirovic, M.; Zhang, J.; Marinkovic, N.; Liu, P.; Frenkel, A.; Adzic, R.R. Ternary Pt/Rh/SnO2 electrocatalysts for oxidizing ethanol to CO2. Nat. Mater. 2009, 8, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Kutz, R.B.; Braunschweig, B.; Mukherjee, P.; Behrens, R.L.; Dlott, D.D.; Wieckowski, A. Reaction pathways of ethanol electrooxidation on polycrystalline platinum catalysts in acidic electrolytes. J. Catal. 2011, 278, 181–188. [Google Scholar] [CrossRef]
- Santasalo-Aarnio, A.; Tuomi, S.; Jalkanen, K.; Kontturi, K.; Kallio, T. The correlation of electrochemical and fuel cell results for alcohol oxidation in acidic and alkaline media. Electrochim. Acta 2013, 87, 730–738. [Google Scholar] [CrossRef]
- Ma, L.; He, H.; Hsu, A.; Chen, R. PdRu/C catalysts for ethanol oxidation in anion-exchange membrane direct ethanol fuel cells. J. Power Sources 2013, 241, 696–702. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Wang, Q.; Lin, J.L.; Tian, N.; Sun, S.G. In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 2010, 55, 7995–7999. [Google Scholar] [CrossRef]
- Lai, S.C.S.; Koper, M.T.M. Ethanol electro-oxidation on platinum in alkaline media. Phys. Chem. Chem. Phys. 2009, 11, 10446–10456. [Google Scholar] [CrossRef]
- García, G.; Koper, M.T.M. Stripping voltammetry of carbon monoxide oxidation on stepped platinum single-crystal electrodes in alkaline solution. Phys. Chem. Chem. Phys. 2008, 10, 3802–3811. [Google Scholar] [CrossRef]
- Kutz, R.B.; Braunschweig, B.; Mukherjee, P.; Dlott, D.D.; Wieckowski, A. Study of ethanol electrooxidation in alkaline electrolytes with isotope labels and sum-frequency generation. J. Phys. Chem. Lett. 2011, 2, 2236–2240. [Google Scholar] [CrossRef]
- Almeida, T.S.; van Wassen, R.R.; van Dover, R.B.; de Andrade, A.R.; Abruña, H.D. Combinatorial PtSnM (M = Fe, Ni, Ru and Pd) nanoparticle catalyst library toward ethanol electrooxidation. J. Power Sources 2015, 284, 623–630. [Google Scholar] [CrossRef]
- Zhu WKe, J.; Wang, S.-B.; Ren, J.; Wang, H.-H.; Zhou, Z.-Y.; Si, R.; Zhang, Y.-W.; Yan, C.-H. Shaping single-crystalline trimetallic Pt-Pd-Rh nanocrystals toward high efficiency C-C splitting of ethanol in conversion to CO2. ACS Catal. 2015, 5, 1995–2008. [Google Scholar]
- Maillard, F.; Dubau, L.; Durst, J.; Chatenet, M.; André, J.; Rossinot, E. Durability of Pt3Co/C nanoparticles in a proton-exchange membrane fuel cell: Direct evidence of bulk Co segregation to the surface. Electrochem. Commun. 2010, 12, 1161–1164. [Google Scholar] [CrossRef]
- Dubau, L.; Maillard, F.; Chatenet, M.; Guetaz, L.; André, J.; Rossinot, E. Durability of Pt3Co/C cathodes in a 16 cell PEMFC stack: Macro/microstructural changes and degradation mechanisms. J. Electrochem. Soc. 2010, 157, B1887–B1895. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; Pergamon Press: Brussels, Belgium, 1974. [Google Scholar]
- Vej-Hansen, U.G.; Rossmeisl, J.; Stephens, I.E.L.; Schiotz, J. Correlation between diffusion barriers and alloying energy in binary alloys. Phys. Chem. Chem. Phys. 2016, 18, 3302–3307. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Norskof, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Stephens, I.E.L.; Bondarenko, A.S.; Gronbjerg, U.; Rossmeisl, J.; Chorkendorff, I. Understanding the electrocatalysis of oxygen reduction on platinum and its alloys. Energy Environ. Sci. 2012, 5, 6744–6762. [Google Scholar] [CrossRef]
- Stephens, I.E.L.; Bondarenko, A.S.; Bech, L.; Chorkendorff, I. Oxygen electroreduction activity and X-ray photoelectron spectroscopy of platinum and early transition metal alloys. ChemCatChem 2012, 4, 341–349. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Verdaguer-Casadevall, A.; Malacrida, P.; Gronbjerg, U.; Knudsen, B.P.; Jepsen, A.K.; Rossmeisl, J.; Stephens, I.E.L.; Chorkendorff, I. Pt5Gd as a highly active and stable catalyst for oxygen electroreduction. J. Am. Chem. Soc. 2012, 134, 16476–16479. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Malacrida, P.; Hansen, M.H.; Vej-Hansen, U.G.; Velasquez-Palenzuela, A.; Tripkov, V.; Schiotz, J.; Rossmeisl, J.; Stephens, I.E.L.; Chorkendorff, I. Tuning the activity of Pt alloy electrocatalysts by means of the lanthanide contraction. Science 2016, 352, 73–76. [Google Scholar] [CrossRef]
- Malacrida, P.; Escudero-Escribano, M.; Verdaguer-Casadevall, A.; Stephens, I.E.L.; Chorkendorff, I. Enhanced activity and stability of Pt-La and Pt-Ce alloys for oxygen electroreduction: The elucidation of the active surface phase. J. Mater. Chem. A 2014, 2, 4234–4243. [Google Scholar] [CrossRef]
- Hultgren, R. Selected Values of the Thermodynamic Properties of Binary Alloys; American Society for Metals: Metals Park, OH, USA, 1973. [Google Scholar]
- Kleykamp, H. Thermodynamics of the systems of the platinum metals with other transition metals: I. Integral data. J. Nucl. Mater. 1993, 201, 193–217. [Google Scholar] [CrossRef]
- Walker, R.; Darby, J. Thermodynamic properties of solid nickel-platinum alloys. Acta Metall. 1970, 18, 1261–1266. [Google Scholar] [CrossRef]
- Pretorius, R.; Morais, T.; Theron, C. Thin film compound phase formation sequence: An effective heat of formation model. Mater. Sci. Rep. 1993, 10, 1–83. [Google Scholar] [CrossRef]
- Jacob, K.; Waseda, Y. Gibbs energies of formation of rare earth MPt5 compounds. Thermochim. Acta 1990, 165, 223–233. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Norskov, J.K.; Barteau, M.A.; Chem, J.G. Modification of the surface electronic and chemical properties of Pt(111) by subsurface 3d transition metals. J. Chem. Phys. 2004, 120, 10240–10246. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Laltice-strain control of the activity in dealloyed core-shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, J.; Marguis, D. Mechanism of hollow nanoparticle formation due to shape fluctuations. Phys. Rev. Lett. 2014, 112, 155505. [Google Scholar] [CrossRef]
- Han, B.; Carlton, C.E.; Suntivich, J.; Xu, Z.; Shao-Horn, Y. Oxygen reduction activity and stability trends of bimetallic Pt0.5 M0.5 nanoparticle in acid. J. Phys. Chem. C 2015, 119, 3971–3978. [Google Scholar] [CrossRef]
- Johansson, T.P.; Ulrikkeholm, E.T.; Hernandez-Fernandez, P.; Escudero-Escribano, M.; Malacrida, P.; Stephens, I.E.L.; Chorkendorff, I. Towards the elucidation of the high oxygen electroreduction activity of PtxY: Surface science and electrochemical studies of Y/Pt (111). Phys. Chem. Chem. Phys. 2014, 16, 13718–13725. [Google Scholar] [CrossRef]
- Ulrikkeholm, E.T.; Pedersen, A.F.; Vej-Hansen, U.G.; Escudero-Escribano, M.; Stephens, I.E.; Friebel, D.; Mehta, A.; Schiotz, J.; Feidenhansl, R.K.; Nilsson, A.; et al. PtxGd alloy formation on Pt (111): Preparation and structural characterization. Surf. Sci. 2016, 652, 114–122. [Google Scholar] [CrossRef]
- Pedersen, A.F.; Ulrikkeholm, E.T.; Escudero-Escribano, M.; Johansson, T.P.; Malacrida, P.; Pedersen, C.M.; Hansen, M.H.; Jensen, K.D.; Rossmeisl, J.; Friebel, D.; et al. Probing the nanoscale structure of the catalytically active overlayer on Pt alloys with rare earths. Nano Energy 2016, 29, 249–260. [Google Scholar] [CrossRef]
- Paulo, M.J.; Venancio, R.H.D.; Freitas, R.G.; Pereira, E.C.; Tavares, A.C. Investigation of the electrocatalytic activity for ethanol oxidation of Pt nanoparticles modified with small amount (5 wt%) of CeO2. J. Electroanal. Chem. 2019, 840, 367–375. [Google Scholar] [CrossRef]
- Jacob, J.; Corradini, P.; Antolini, E.; Santos, N.A.; Perez, J. Electro-oxidation of ethanol on ternary Pt-Sn-Ce/C catalysts. Appl. Catal. B Environ. 2015, 165, 176–184. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z.; An, S.; Wang, R.; Wang, Y.; Xu, T. Effect of CeO2–ZrO2 on Pt/C electrocatalysts for alcohols oxidation. J. Rare Earths 2016, 34, 276–282. [Google Scholar] [CrossRef]
- Corddacho-Garcia, J.; Betancourt, L.E.; Velez, C.A.; Senanayake, S.D.; Stacchiola, D.; Sasaki, K.; Guinel, M.J.-F.; Zhou, Y.; Cheung, C.L.; Cabrera, C.R. Cerium oxide as a promoter for the electrooxidation reaction of ethanol: In situ XAFS characterization of the Pt nanoparticles supported on CeO2 nanoparticles and nanorods. Phys. Chem. Chem. Phys. 2015, 17, 32251–32256. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Wang, L.; Huang, C.-C.; Fan, Y.-Z.; Tang, H.-G.; Wei, L.; Sun, S.-G. Concave cubic PtLa alloy nanocrystals with high-index facets: Controllable synthesis in deep eutectic solvents and their superior electrocatalytic properties for ethanol oxidation. J. Power Sources 2018, 399, 422–428. [Google Scholar] [CrossRef]
- Corradini, P.G.; Santos, N.A.; Perez, J. Pt-Sn-Eu/C catalysts: Application of rare earth metals as anodes in direct ethanol fuel cells. Fuel Cells 2018, 18, 73–81. [Google Scholar] [CrossRef]
- Corradini, P.G.; Antolini, E.; Perez, J. Electrooxidation of ethanol on ternary non-alloyed Pt-Sn-Pr/C catalysts. J. Power Sources 2015, 275, 377–383. [Google Scholar] [CrossRef]
- Corradini, P.G.; Antolini, E.; Perez, J. Activity, short-term stability (poisoning tolerance) and durability of carbon supported Pt-Pr catalysts for ethanol oxidation. J. Power Sources 2014, 251, 402–410. [Google Scholar] [CrossRef]
- Ma, Y.; Du, Y.; Ye, W.; Su, B.; Yang, M.; Wang, C. Electrocatalytic oxidation of ethanol on platinum electrode decorated with Nd-Fe-Mo hybrid-metallic cyano-bridged mixing coordination polymer in weak acidic medium. Int. J. Electrochem. Sci. 2012, 7, 2654–2679. [Google Scholar]
- Song, S.; Tsiakaras, P. Recent progress in direct ethanol proton exchange membrane fuel cells (DE-PEMFCs). Appl. Catal. B Environ. 2006, 63, 187–193. [Google Scholar] [CrossRef]
- Abdullah, S.; Kamarudin, S.K.; Hasran, U.A.; Masdar, M.S.; Daud, W.R.W. Development of a conceptual design model of a direct ethanol fuel cell (DEFC). Int. J. Hydrog. Energy 2015, 40, 11943–11948. [Google Scholar] [CrossRef]
- Duan, J.J.; Feng, J.J.; Zhang, L.; Yuan, J.; Zhang, Q.L.; Wang, A.J. Facile one-pot aqueous fabrication of interconnected ultrathin PtPbPd nanowires as advanced electrocatalysts for ethanol oxidation and oxygen reduction reactions. Int. J. Hydrog. Energy 2019, 44, 27455–27464. [Google Scholar] [CrossRef]
- Carrera-Cerritos, R.; Fuentes-Ramírez, R.; Cuevas-Muñiz, F.M.; Ledesma-García, J.; Arriaga, L.G. Performance and stability of Pd nanostructures in an alkaline direct ethanol fuel cell. J. Power Sources 2014, 269, 370–378. [Google Scholar] [CrossRef]
- Cardoso, D.S.P.; Santos, D.M.F.; Šljukić, B.; Sequeira, C.A.C.; Macciò, D.; Saccone, A. Platinum-rare earth cathodes for direct borohydride-peroxide fuel cells. J. Power Sources 2016, 307, 251–258. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Saturnino, P.G.; Macciò, D.; Saccone, A.; Sequeira, C.A.C. Platinum rare-earth intermetallic alloys as anode electrocatalysts for borohydride oxidation. Catal. Today 2011, 170, 134–140. [Google Scholar] [CrossRef]
- Šljukić, B.; Milikić, J.; Santos, D.M.F.; Sequeira, C.A.C.; Macciò, D.; Saccone, A. Electrocatalytic performance of Pt-Dy alloys for direct borohydride fuel cells. J. Power Sources 2014, 272, 335–343. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Šljukić, B.; Sequeira, C.A.C.; Macciò, D.; Saccone, A.; Figueiredo, J.L. Electrocatalytic approach for the efficiency increase of electrolytic hydrogen production: Proof-of-concept using Pt-Dy. Energy 2013, 50, 486–492. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C.; Macciò, D.; Saccone, A.; Figueiredo, J.L. Platinum rare earth electrodes for hydrogen evolution in alkaline water electrolysis. Int. J. Hydrog. Energy 2013, 38, 3137–3145. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy, 2nd ed.; PHI: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Xie, S.W.; Chen, S.; Liu, Z.Q.; Xu, C.W. Comparison of alcohol electrooxidation on Pt and Pd electrodes in alkaline medium. Int. J. Electrochem. Soc. 2011, 6, 882–888. [Google Scholar]
- Brownson, D.A.C.; Banks, C.E. Interpreting electrochemistry. In The Handbook of Graphene Electrochemistry; Springer: London, UK, 2014; pp. 23–77. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Potential sweep methods. In Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2001; pp. 226–260. [Google Scholar]
- Denuault, G.; Mirkin, M.V.; Bard, A.J. Direct determination of diffusion coefficients by chronoamperometry at microdisk electrodes. J. Electroanal. Chem. 1991, 308, 27–38. [Google Scholar] [CrossRef]
- Wang, K.; Lu, J.; Zhuang, L. Direct determination of diffusion coefficient for borohydride anions in alkaline solutions using chronoamperometry with spherical Au electrodes. J. Electroanal. Chem. 2005, 585, 191–196. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, D.M.F.; Lourenço, J.R.B.; Macciò, D.; Saccone, A.; Sequeira, C.A.C.; Figueiredo, J.L. Ethanol Electrooxidation at Platinum-Rare Earth (RE = Ce, Sm, Ho, Dy) Binary Alloys. Energies 2020, 13, 1658. https://doi.org/10.3390/en13071658
Santos DMF, Lourenço JRB, Macciò D, Saccone A, Sequeira CAC, Figueiredo JL. Ethanol Electrooxidation at Platinum-Rare Earth (RE = Ce, Sm, Ho, Dy) Binary Alloys. Energies. 2020; 13(7):1658. https://doi.org/10.3390/en13071658
Chicago/Turabian StyleSantos, D.M.F., J.R.B. Lourenço, D. Macciò, A. Saccone, C.A.C. Sequeira, and J.L. Figueiredo. 2020. "Ethanol Electrooxidation at Platinum-Rare Earth (RE = Ce, Sm, Ho, Dy) Binary Alloys" Energies 13, no. 7: 1658. https://doi.org/10.3390/en13071658
APA StyleSantos, D. M. F., Lourenço, J. R. B., Macciò, D., Saccone, A., Sequeira, C. A. C., & Figueiredo, J. L. (2020). Ethanol Electrooxidation at Platinum-Rare Earth (RE = Ce, Sm, Ho, Dy) Binary Alloys. Energies, 13(7), 1658. https://doi.org/10.3390/en13071658