Green Synthetic Fuels: Renewable Routes for the Conversion of Non-Fossil Feedstocks into Gaseous Fuels and Their End Uses





Abstract

1. Introduction
1.1. Background
1.2. Motivation of This Work
2. Heat-to-Gas Routes
2.1. Solar Water Thermolysis
- Microporous ceramic membranes made of refractory materials in sub-atmospheric pressure reactors that allow selective separation of a species [73].
- Membranes semipermeable to oxygen made by a high temperature solid electrolyte material (ZrO2, CaO, CeO2, Y2O3, perovskite) placed in a chemical potential gradient. The mixed ionic and electronic conducting (MIEC) membranes are realized with refractory oxides with a mixed type of electrical conductivity with ionic and electronic components. In the presence of a gradient of oxygen pressure, a chemical potential induces migration of oxygen ions across the membrane, and an electron current circulates in the opposite direction due to electronic conductivity of the material [74].
- A rotating cylindrical vessel in which the gas mixture could pass through. The centrifugal field of force modifies the equilibrium pressure of species, separating them [76].
- Supersonic jets generated by expanding a gas mixture rich of dense gases near the jet axis and with lighter gases on the external part. This distribution leads to beam deflection and gas separation due to centrifuge forces in the curved flow [77].
2.2. Thermochemical Cycles
2.2.1. Reactors
2.2.2. Technology Readiness Level
3. Power-to-Gas Routes
3.1. Electrolysis
3.1.1. Alkaline Electrolysis Cells
3.1.2. Polymer Electrolyte Membrane Electrolysis Cells
3.1.3. High-Temperature Electrolysis Cells
3.1.4. Microbial Electrolysis Cells
3.2. Photoelectrochemical Cell
3.2.1. Photocatalysts
- A broad spectral range for light absorption (the bandgap range should spread between 1.8 and 2.2 eV);
- Proper valence and conduction band energy level in function of the redox potential of water splitting;
- Effective electron–hole pairs separation;
- Effective charge and mass transport and long carrier diffusion length;
- High chemical, electrochemical and photoelectrochemical stability;
- Low cost and environmentally friendly.
3.2.2. Modification Techniques
3.3. Technology Readiness Level
3.3.1. Electrolysis
Alkaline Electrolysis
Proton Exchange Membrane Electrolysis Cells
Solid Oxide Electrolysis Cells
Microbial Electrolysis Cells
3.3.2. Photoelectrochemical Cells
4. Bio-to-Gas Routes
- Feedstock pre-treatment
- Pyrolysis
- Char gasification
- Syngas clean up.
4.1. Feedstock Pre-Treatment
4.2. Pyrolysis
4.2.1. Pyrolysis Process
4.2.2. Pyrolysis Reactors
4.3. Gasification
4.3.1. Gasification Process
4.3.2. Design of Gasifiers
- fixed-bed (or packed-bed) gasifiers;
- fluidized-bed gasifiers;
- entrained-flow gasifiers.
4.4. Syngas Clean Up
- Primary products: cellulose-derived products (levoglucosan, furfurals, hydroxy-acetaldehyde), hemicellulose-derived products and lignin-derived methoxyphenols;
- Secondary products: phenolics and olefins;
- Tertiary products: methyl derivates, toluene and indene;
- Condensed tertiary products: polycyclic aromatic hydrocarbon (benzene, naphthalene, pyrene, anthracene, acenaphthylene).
4.5. Technology Readiness Level
4.6. Catalytic Methanation
4.6.1. Reactors
4.6.2. Technology Readiness Level
5. Green Synthetic Fuels Supply Chain
5.1. Gas Quality and Interchangeability
5.2. Gas Network Injection
5.3. Combustion
5.4. Fuel Cells Gas Quality
5.5. Storage
5.5.1. Compressed Gas Storage
Natural Gas
Hydrogen
5.5.2. Liquefied Gas Storage
Natural Gas
Hydrogen
5.5.3. Gas Solid Storage
Natural Gas
Hydrogen
5.5.4. Underground Gas Storage
Natural Gas
Hydrogen
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- IEA. Global Energy & CO2 Status Report; IEA: Paris, France, 2019. [Google Scholar]
- International Energy Agency. Technology Roadmap—Hydrogen and Fuel Cells; International Energy Agency: Paris, France, 2015. [Google Scholar]
- Cochran, J.; Miller, M.; Zinaman, O.; Milligan, M.; Arent, D.; Palmintier, B.; Malley, M.O.; Mueller, S.; Lannoye, E.; Epri, A.T.; et al. Flexibility in 21st Century Power Systems; National Renewable Energy Lab: Golden, CO, USA, 2014.
- IEA. The Future of Hydrogen for G20; IEA: Paris, France, 2019. [Google Scholar]
- Teter, J.; Feuvre, P.L.; Gorner, M.; Scheffer, S. Tracking Transport. Available online: https://www.iea.org/reports/tracking-transport-2019 (accessed on 1 December 2019).
- IEA. Global Trends and Outlook for Hydrogen; IEA: Paris, France, 2017. [Google Scholar]
- Hannula, I.; Reiner, D.M. Near-term potential of biofuels, electrofuels, and battery electric vehicles in decarbonizing road transport. Joule 2019, 3, 2390–2402. [Google Scholar] [CrossRef]
- Council of the European Union, European Parliament. Directive 2003/30/EC of the European Parliament and of the Council of 8 May 2003 on the Promotion of the Use of Biofuels or Other Renewable Fuels for Transport; Publications Office of the European Union: Brusselsm, Belgium, 2003. [Google Scholar]
- European Parliament, Council of the European Union. Directive 2009/28/EC of the European Parliament and of the Council of 23 April 2009 on the Promotion of the Use of Energy from Renewable Sources and Amending and Subsequently Repealing Directives 2001/77/EC and 2003/30/EC2009; Publications Office of the European Union: Brusselsm, Belgium, 2009. [Google Scholar]
- Berndes, G.; Bird, N.; Cowie, A. Bioenergy, Land Use Change and Climate Change Mitigation—Background Technical Report; IEA: Paris, France, 2011. [Google Scholar]
- Delucchi, M.A.; Yang, C.; Burke, A.F.; Ogden, J.M.; Kurani, K.; Kessler, J.; Sperling, D. An assessment of electric vehicles: Technology, infrastructure requirements, greenhouse-gas emissions, petroleum use, material use, lifetime cost, consumer acceptance and policy initiatives. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372. [Google Scholar] [CrossRef] [PubMed]
- IRENA. Hydrogen: A Renewable Energy Perspective; IRENA: Abu Dhabi, UAE, 2019. [Google Scholar]
- Aarnes, J.; Eijgelaar, M.; Hektor, E. Hydrogen as an Energy Carrier; DNV GL: Oslo, Norway, 2018. [Google Scholar]
- Committee on Climate Change. Hydrogen in a Low-Carbon Economy; Committee on Climate Change: London, UK, 2018. [Google Scholar]
- Damen, K.; Van Troost, M.; Faaij, A.; Turkenburg, W. A comparison of electricity and hydrogen production systems with CO2 capture and storage. Part A: Review and selection of promising conversion and capture technologies. Prog. Energy Combust. Sci. 2006, 32, 215–246. [Google Scholar] [CrossRef]
- Hydrogen Council. Hydrogen Scaling Up; Hydrogen Council: Brussels, Belgium, 2017. [Google Scholar]
- Dincer, I.; Acar, C. Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrog. Energy 2014, 40, 11094–11111. [Google Scholar] [CrossRef]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Peña, M.A.; Gómez, J.P.; Fierro, J.L.G. New catalytic routes for syngas and hydrogen production. Appl. Catal. A Gen. 1996, 144, 7–57. [Google Scholar] [CrossRef]
- Levalley, T.L.; Richard, A.R.; Fan, M. The progress in water gas shift and steam reforming hydrogen production technologies—A review. Int. J. Hydrog. Energy 2014, 39, 16983–17000. [Google Scholar] [CrossRef]
- Gradisher, L.; Dutcher, B.; Fan, M. Catalytic hydrogen production from fossil fuels via the water gas shift reaction. Appl. Energy 2015, 139, 335–349. [Google Scholar] [CrossRef]
- Muradov, N. Low to near-zero CO2 production of hydrogen from fossil fuels: Status and perspectives. Int. J. Hydrog. Energy 2017, 42, 14058–14088. [Google Scholar] [CrossRef]
- Voldsund, M.; Jordal, K.; Anantharaman, R. Hydrogen production with CO2 capture. Int. J. Hydrog. Energy 2016, 41, 4969–4992. [Google Scholar] [CrossRef]
- Uçkun Kiran, E.; Trzcinski, A.P.; Ng, W.J.; Liu, Y. Bioconversion of food waste to energy: A review. Fuel 2014, 134, 389–399. [Google Scholar] [CrossRef]
- Nath, K.; Das, D. Biohydrogen production as a potential energy resource—Present state-of-art. J. Sci. Ind. Res. 2004, 63, 729–738. [Google Scholar]
- Hallenbeck, P.C.; Benemann, J.R. Biological hydrogen production; fundamentals and limiting processes. Int. J. Hydrog. Energy 2002, 27, 1185–1193. [Google Scholar] [CrossRef]
- Das, D.; Veziroǧlu, T.N. Hydrogen production by biological processes: A survey of literature. Int. J. Hydrog. Energy 2001, 26, 13–28. [Google Scholar] [CrossRef]
- Kumar Gupta, S.; Kumari, S.; Reddy, K.; Bux, F. Trends in biohydrogen production: Major challenges and state-of-the-art developments. Environ. Technol. 2013, 34, 1653–1670. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, R.; Man, Y.; Ren, J. Recent developments of hydrogen production from sewage sludge by biological and thermochemical process. Int. J. Hydrog. Energy 2019, 44, 19676–19697. [Google Scholar] [CrossRef]
- Xia, A.; Cheng, J.; Song, W.; Su, H.; Ding, L.; Lin, R.; Lu, H.; Liu, J.; Zhou, J.; Cen, K. Fermentative hydrogen production using algal biomass as feedstock. Renew. Sustain. Energy Rev. 2015, 51, 209–230. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Sheng, L.; Liu, X.; Lu, Y.; Li, W. A review on bio-hydrogen production technology. Int. J. Energy Res. 2018, 42, 3442–3453. [Google Scholar] [CrossRef]
- Sun, Y.; He, J.; Yang, G.; Sun, G.; Sage, V. A review of the enhancement of bio-hydrogen generation by chemicals addition. Catalysts 2019, 9, 353. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Christopher, K.; Dimitrios, R. A review on exergy comparison of hydrogen production methods from renewable energy sources. Energy Environ. Sci. 2012, 5, 6640–6651. [Google Scholar] [CrossRef]
- Koumi Ngoh, S.; Njomo, D. An overview of hydrogen gas production from solar energy. Renew. Sustain. Energy Rev. 2012, 16, 6782–6792. [Google Scholar] [CrossRef]
- Bičáková, O.; Straka, P. Production of hydrogen from renewable resources and its effectiveness. Int. J. Hydrog. Energy 2012, 37, 11563–11578. [Google Scholar] [CrossRef]
- Chaubey, R.; Sahu, S.; James, O.O.; Maity, S. A review on development of industrial processes and emerging techniques for production of hydrogen from renewable and sustainable sources. Renew. Sustain. Energy Rev. 2013, 23, 443–462. [Google Scholar] [CrossRef]
- Orhan, M.F.; Babu, B.S. Investigation of an integrated hydrogen production system based on nuclear and renewable energy sources: Comparative evaluation of hydrogen production options with a regenerative fuel cell system. Energy 2015, 88, 801–820. [Google Scholar] [CrossRef]
- Dutta, S. A review on production, storage of hydrogen and its utilization as an energy resource. J. Ind. Eng. Chem. 2014, 20, 1148–1156. [Google Scholar] [CrossRef]
- Bolat, P.; Thiel, C. Hydrogen supply chain architecture for bottom-up energy systems models. Part 1: Developing pathways. Int. J. Hydrog. Energy 2014, 39, 8881–8897. [Google Scholar] [CrossRef]
- Li, L.; Manier, H.; Manier, M.A. Hydrogen supply chain network design: An optimization-oriented review. Renew. Sustain. Energy Rev. 2019, 103, 342–360. [Google Scholar] [CrossRef]
- Balcombe, P.; Speirs, J.; Johnson, E.; Martin, J.; Brandon, N.; Hawkes, A. The carbon credentials of hydrogen gas networks and supply chains. Renew. Sustain. Energy Rev. 2018, 91, 1077–1088. [Google Scholar] [CrossRef]
- Sikarwar, V.S.; Zhao, M.; Fennell, P.S.; Shah, N.; Anthony, E.J. Progress in biofuel production from gasification. Prog. Energy Combust. Sci. 2017, 61, 189–248. [Google Scholar] [CrossRef]
- Göransson, K.; Söderlind, U.; He, J.; Zhang, W. Review of syngas production via biomass DFBGs. Renew. Sustain. Energy Rev. 2011, 15, 482–492. [Google Scholar] [CrossRef]
- Molino, A.; Chianese, S.; Musmarra, D. Biomass gasification technology: The state of the art overview. J. Energy Chem. 2016, 25, 10–25. [Google Scholar] [CrossRef]
- Babu, B.V. Biomass pyrolysis: A state-of-the-art review. Biofuels Bioprod. Biorefin. 2012, 6, 246–256. [Google Scholar] [CrossRef]
- Sikarwar, V.S.; Zhao, M.; Clough, P.; Yao, J.; Zhong, X.; Memon, M.Z.; Shah, N.; Anthony, E.J.; Fennell, P.S. An overview of advances in biomass gasification. Energy Environ. Sci. 2016, 9, 2939–2977. [Google Scholar] [CrossRef]
- Farzad, S.; Mandegari, M.A.; Görgens, J.F. A critical review on biomass gasification, co-gasification, and their environmental assessments. Biofuel Res. J. 2016, 3, 483–495. [Google Scholar] [CrossRef]
- Ren, J.; Cao, J.P.; Zhao, X.Y.; Yang, F.L.; Wei, X.Y. Recent advances in syngas production from biomass catalytic gasification: A critical review on reactors, catalysts, catalytic mechanisms and mathematical models. Renew. Sustain. Energy Rev. 2019, 116, 109426. [Google Scholar] [CrossRef]
- Mao, C.; Feng, Y.; Wang, X.; Ren, G. Review on research achievements of biogas from anaerobic digestion. Renew. Sustain. Energy Rev. 2015, 45, 540–555. [Google Scholar] [CrossRef]
- Sugumar, S.; Shanmuga Priyan, R.; Dinesh, S. A review on performance study of anaerobic digestion to enhance the biogas production. Int. J. Civ. Eng. Technol. 2016, 7, 202–206. [Google Scholar]
- Yang, Q.; Wu, B.; Yao, F.; He, L.; Chen, F.; Ma, Y.; Shu, X.; Hou, K.; Wang, D.; Li, X. Biogas production from anaerobic co-digestion of waste activated sludge: Co-substrates and influencing parameters. Rev. Environ. Sci. Biotechnol. 2019, 18, 771–793. [Google Scholar] [CrossRef]
- Kainthola, J.; Kalamdhad, A.S.; Goud, V.V. A review on enhanced biogas production from anaerobic digestion of lignocellulosic biomass by different enhancement techniques. Process Biochem. 2019, 84, 81–90. [Google Scholar] [CrossRef]
- Hagos, K.; Zong, J.; Li, D.; Liu, C.; Lu, X. Anaerobic co-digestion process for biogas production: Progress, challenges and perspectives. Renew. Sustain. Energy Rev. 2017, 76, 1485–1496. [Google Scholar] [CrossRef]
- Lora Grando, R.; de Souza Antune, A.M.; da Fonseca, F.V.; Sánchez, A.; Barrena, R.; Font, X. Technology overview of biogas production in anaerobic digestion plants: A European evaluation of research and development. Renew. Sustain. Energy Rev. 2017, 80, 44–53. [Google Scholar] [CrossRef]
- Deepanraj, B.; Sivasubramanian, V.; Jayaraj, S. Biogas generation through anaerobic digestion process-an overview. Res. J. Chem. Environ. 2014, 18, 80–94. [Google Scholar]
- Jankowska, E.; Sahu, A.K.; Oleskowicz-Popiel, P. Biogas from microalgae: Review on microalgae’s cultivation, harvesting and pretreatment for anaerobic digestion. Renew. Sustain. Energy Rev. 2017, 75, 692–709. [Google Scholar] [CrossRef]
- Leonzio, G. State of art and perspectives about the production of methanol, dimethyl ether and syngas by carbon dioxide hydrogenation. J. CO2 Util. 2018, 27, 326–354. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Sattler, C. A review on solar thermal syngas production via redox pair-based water/carbon dioxide splitting thermochemical cycles. Renew. Sustain. Energy Rev. 2015, 42, 254–285. [Google Scholar] [CrossRef]
- Loutzenhiser, P.G.; Meier, A.; Steinfeld, A. Review of the Two-Step H2O/CO2-Splitting solar thermochemical cycle based on Zn/ZnO redox reactions. Materials 2010, 3, 4922–4938. [Google Scholar] [CrossRef]
- Foit, S.R.; Vinke, I.C.; de Haart, L.G.J.; Eichel, R.A. Power-to-syngas: An enabling technology for the transition of the energy system? Angew. Chem Int. Ed. 2017, 56, 5402–5411. [Google Scholar] [CrossRef]
- Ghaib, K.; Ben-Fares, F.Z. Power-to-methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Power-to-gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–787. [Google Scholar] [CrossRef]
- Lecker, B.; Illi, L.; Lemmer, A.; Oechsner, H. Biological hydrogen methanation—A review. Bioresour. Technol. 2017, 245, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Villafán-Vidales, H.I.; Arancibia-Bulnes, C.A.; Riveros-Rosas, D.; Romero-Paredes, H.; Estrada, C.A. An overview of the solar thermochemical processes for hydrogen and syngas production: Reactors, and facilities. Renew. Sustain. Energy Rev. 2017, 75, 894–908. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar thermochemical production of hydrogen-a review. Sol. Energy 2005, 78, 603–615. [Google Scholar] [CrossRef]
- Ihara, S. Feasibility of hydrogen production by direct water splitting at high temperature. Int. J. Hydrog. Energy 1978, 3, 287–296. [Google Scholar] [CrossRef]
- Kogan, A. Direct solar thermcal on-site separation of the products-II. Experimental feasibility study. Int. J. Hydrog. Energy 1998, 23, 89–98. [Google Scholar] [CrossRef]
- Baykara, S.Z. Hydrogen production by direct solar thermal decomposition of water, possibilities for improvement of process efficiency. Int. J. Hydrog. Energy 2004, 29, 1451–1458. [Google Scholar] [CrossRef]
- Etiévant, C. Solar high-temperature direct water splitting—A review of experiments in France. Sol. Energy Mater. 1991, 24, 413–440. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S. Hydrogen membrane separation techniques. Ind. Eng. Chem. Res. 2006, 45, 875–881. [Google Scholar] [CrossRef]
- Grosjean, R.; Delacroix, S.; Gouget, G.; Beaunier, P.; Ersen, O.; Ihiawakrim, D.; Kurakevych, O.; Portehault, D. High pressures pathway toward boron-based nanostructured solids. Dalton Trans. 2017, 47, 7634–7639. [Google Scholar] [CrossRef] [PubMed]
- Edlund, D.; Friesen, D.; Johnson, B.; Pledger, W. Hydrogen-permeable metal membranes for high-temperature gas separations. Gas Sep. Purif. 1994, 8, 131–136. [Google Scholar] [CrossRef]
- Calise, F.; D’Accadia, M.D.; Santarelli, M.; Lanzini, A.; Ferrero, D. Solar Hydrogen Production: Processes, Systems and Technologies; Elsevier Science: Amsterdam, The Netherlands, 2019. [Google Scholar]
- July, D. Direct-Thermal Solar Hydrogen Production from Water Using Nozzles/Skimmers and Glow Discharge in the Gas Phase at Low Pressure and High Temperature; H-Ion Solar Company: Richmond, CA, USA, 1994. [Google Scholar]
- Warner, J.W.; Stephen Berry, R. Injection quenching and the high temperature water-splitting reactor. Sol. Energy 1985, 35, 535–537. [Google Scholar] [CrossRef]
- Lapicque, F.; Lédé, J.; Villermaux, J. Design and optimization of a reactor for high temperature dissociation of water and carbon dioxide using solar energy. Chem. Eng. Sci. 1986, 41, 677–684. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Dey, S. Solar thermochemical splitting of water to generate hydrogen. Proc. Natl. Acad. Sci. USA 2017, 114, 13385–13393. [Google Scholar] [CrossRef]
- Kodama, T.; Gokon, N. Thermochemical cycles for high-temperature solar hydrogen production. Chem. Rev. 2007, 107, 4048–4077. [Google Scholar] [CrossRef]
- D’Souza, L. Thermochemical hydrogen production from water using reducible oxide materials: A critical review. Mater. Renew. Sustain. Energy 2013, 2, 7. [Google Scholar] [CrossRef]
- Bin, Z. CO2 valorisation based on Fe3O4/FeO thermochemical redox reactions using concentrated solar energy. Int. J. Energy Res. 2009, 31, 135–147. [Google Scholar]
- Chen, K.S.; Hogan, R.E. A two-phase model for solar thermochemical water splitting with FeO/Fe3O4. In Proceedings of the 3rd International Conference of Energy Sustainability, San Francisco, CA, USA, 19–23 July 2009; pp. 1–8. [Google Scholar]
- Steinfeld, A.; Sanders, S.; Palumbo, R. Design aspects of solar thermochemical engeneering-A case study: Two-step water-splitting cycle using the Fe3O4/FeO redox system. Sol. Energy 1999, 65, 43–53. [Google Scholar] [CrossRef]
- Stamatiou, A.; Loutzenhiser, P.G.; Steinfeld, A. Solar syngas production via H2O/CO2-splitting thermochemical cycles with Zn/ZnO and FeO/Fe3O4 redox reactions. Chem. Mater. 2010, 22, 851–859. [Google Scholar] [CrossRef]
- Steinfeld, A.; Palumbo, R. Solar thermochemical process technology. Encycl. Phys. Sci. Technol. 2003, 15, 237–256. [Google Scholar]
- Alonso, E.; Hutter, C.; Romero, M.; Steinfeld, A.; Gonzalez-Aguilar, J. Kinetics of Mn2O3-Mn3O4 and Mn3O4-MnO redox reactions performed under concentrated thermal radiative flux. Energy Fuels 2013, 27, 4884–4890. [Google Scholar] [CrossRef]
- Alonso, E.; Gallo, A.; Pérez-Rábago, C.; Fuentealba, E. Thermodynamic study of CuO/Cu2O and Co3O4/CoO redox pairs for solar energy thermochemical storage. In Proceedings of the AIP Conference Proceedings, Penang, Malaysia, 10–12 April 2018. [Google Scholar]
- Abanades, S.; Charvin, P.; Flamant, G.; Neveu, P. Screening of water-splitting thermochemical cycles potentially attractive for hydrogen production by concentrated solar energy. Energy 2006, 31, 2805–2822. [Google Scholar] [CrossRef]
- Wegner, K.; Ly, H.C.; Weiss, R.J.; Pratsinis, S.E.; Steinfeld, A. In situ formation and hydrolysis of Zn nanoparticles for H2 production by the 2-step ZnO/Zn water-splitting thermochemical cycle. Int. J. Hydrog. Energy 2006, 31, 55–61. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar hydrogen production via a two-step water-splitting thermochemical cycle based on Zn/ZnO redox reactions. Int. J. Hydrog. Energy 2002, 27, 611–619. [Google Scholar] [CrossRef]
- Abanades, S.; Charvin, P.; Lemont, F.; Flamant, G. Novel two-step SnO2/SnO water-splitting cycle for solar thermochemical production of hydrogen. Int. J. Hydrog. Energy 2008, 33, 6021–6030. [Google Scholar] [CrossRef]
- Abanades, S. CO2 and H2O reduction by solar thermochemical looping using SnO2/SnO redox reactions: Thermogravimetric analysis. Int. J. Hydrog. Energy 2012, 37, 8223–8231. [Google Scholar] [CrossRef]
- Bhosale, R.R.; Kumar, A.; Sutar, P. Thermodynamic analysis of solar driven SnO2/SnO based thermochemical water splitting cycle. Energy Convers. Manag. 2017, 135, 226–235. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.R.; Steinfeld, A. Syngas production by simultaneous splitting of H2O and CO2 via ceria redox reactions in a high-temperature solar reactor. Energy Environ. Sci. 2012, 5, 6098–6103. [Google Scholar] [CrossRef]
- Portarapillo, M.; Aronne, A.; Benedetto, A.D.; Imparato, C.; Landi, G.; Luciani, G. Syngas production through H2O/CO2 thermochemical splitting. Chem. Eng. Trans. 2019, 74, 43–48. [Google Scholar]
- Bowman, G. Interfacing Primary Heat Sources and Cycles for Thermochemical Hydrogen Production. In Hydrogen Energy Progress, Proceedings of the 3rd World Hydrogen Energy Conference, Tokyo, Japan, 23–26 June 1980; Pergamon Press: Oxford, UK; New York, NY, USA, 1981; Volume 1, pp. 335–344. [Google Scholar]
- Whaley, T.; Yudow, B.; Remick, R.; Pangborn, J.; Sammells, A. Status of the cadmium thermoelectrochemical hydrogen cycle. Int. J. Hydrog. Energy 1983, 8, 767–771. [Google Scholar] [CrossRef]
- Milshtein, J.D.; Gratz, E.; Basu, S.N.; Gopalan, S.; Pal, U.B. Study of the two-step W/WO3 solar to fuel conversion cycle for syngas production. J. Power Sources 2013, 236, 95–102. [Google Scholar] [CrossRef]
- Nakamura, T. Hydrogen production from water utilizing solar heat at high temperatures. Sol. Energy 1977, 19, 467–475. [Google Scholar] [CrossRef]
- Miller, J.E.; McDaniel, A.H.; Allendorf, M.D. Considerations in the design of materials for solar-driven fuel production using metal-oxide thermochemical cycles. Adv. Energy Mater. 2014, 4, 1–19. [Google Scholar] [CrossRef]
- Block, T.; Knoblauch, N.; Schmücker, M. The cobalt-oxide/iron-oxide binary system for use as high temperature thermochemical energy storage material. Thermochim. Acta 2014, 577, 25–32. [Google Scholar] [CrossRef]
- Gokon, N.; Mizuno, T.; Nakamuro, Y.; Kodama, T. Iron-containing yttria-stabilized zirconia system for two-step thermochemical water splitting. J. Sol. Energy Eng. Trans. ASME 2008, 130, 0110181–0110186. [Google Scholar] [CrossRef]
- Bhosale, R.R.; Kumar, A.; AlMomani, F.; Ghosh, U.; Sutar, P.; Takalkar, G.; Ashok, A.; Alxneit, I. Effectiveness of Ni incorporation in iron oxide crystal structure towards thermochemical CO2 splitting reaction. Ceram. Int. 2017, 43, 5150–5155. [Google Scholar] [CrossRef]
- Carrillo, A.J.; Serrano, D.P.; Pizarro, P.; Coronado, J.M. Understanding redox kinetics of iron-doped manganese oxides for high temperature thermochemical energy storage. J. Phys. Chem. C 2016, 120, 27800–27812. [Google Scholar] [CrossRef]
- Miller, J.E.; Allendorf, M.D.; Diver, R.B.; Evans, L.R.; Siegel, N.P.; Stuecker, J.N. Metal oxide composites and structures for ultra-high temperature solar thermochemical cycles. J. Mater. Sci. 2008, 43, 4714–4728. [Google Scholar] [CrossRef]
- Lorentzou, S.; Pagkoura, C.; Zygogianni, A.; Karagiannakis, G.; Konstandopoulos, A.G. Thermochemical cycles over redox structured reactors. Int. J. Hydrog. Energy 2017, 42, 19664–19682. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S.; Bion, N.; Le Mercier, T.; Harlé, V. Reactivity of doped ceria-based mixed oxides for solar thermochemical hydrogen generation via two-step water-splitting cycles. Energy Fuels 2013, 27, 6068–6078. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S. Dopant incorporation in ceria for enhanced water-splitting activity during solar thermochemical hydrogen generation. J. Phys. Chem. C 2012, 116, 13516–13523. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Jouannaux, J.; Drobek, M.; Ayral, A.; Julbe, A. Recent progress on ceria doping and shaping strategies for solar thermochemical water and CO2 splitting cycles. Aims Mater. Sci. 2019, 6, 657–684. [Google Scholar] [CrossRef]
- Muhich, C.; Steinfeld, A. Principles of doping ceria for the solar thermochemical redox splitting of H2O and CO2. J. Mater. Chem. A 2017, 5, 15578–15590. [Google Scholar] [CrossRef]
- Muhich, C.L.; Blaser, S.; Hoes, M.C.; Steinfeld, A. Comparing the solar-to-fuel energy conversion efficiency of ceria and perovskite based thermochemical redox cycles for splitting H2O and CO2. Int. J. Hydrog. Energy 2018, 43, 18814–18831. [Google Scholar] [CrossRef]
- Lapp, J.; Davidson, J.H.; Lipiński, W. Efficiency of two-step solar thermochemical non-stoichiometric redox cycles withheat recovery. Energy 2012, 37, 591–600. [Google Scholar] [CrossRef]
- Perkins, C.; Weimer, A.W. Likely near-term solar-thermal water splitting technologies. Int. J. Hydrog. Energy 2004, 29, 1587–1599. [Google Scholar] [CrossRef]
- Charvin, P.; Stéphane, A.; Florent, L.; Gilles, F. Analysis of solar chemical processes for hydrogen production from water splitting thermochemical cycles. Energy Convers. Manag. 2008, 49, 1547–1556. [Google Scholar] [CrossRef]
- Palumbo, R.; Lédé, J.; Boutin, O.; Ricart, E.E.; Steinfeld, A.; Möller, S.; Weidenkaff, A.; Fletcher, E.A.; Bielicki, J. The production of Zn from ZnO in a high-temperature solar decomposition quench process—I. The scientific framework for the process. Chem. Eng. Sci. 1998, 53, 2503–2517. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Beche, E.; Lemont, F.; Flamant, G. Hydrogen production from mixed cerium oxides via three-step water-splitting cycles. Solid State Ion. 2009, 180, 1003–1010. [Google Scholar] [CrossRef]
- Liberatore, R.; Lanchi, M.; Giaconia, A.; Tarquini, P. Energy and economic assessment of an industrial plant for the hydrogen production by water-splitting through the sulfur-iodine thermochemical cycle powered by concentrated solar energy. Int. J. Hydrog. Energy 2012, 37, 9550–9565. [Google Scholar] [CrossRef]
- Caple, K.; Kreider, P.; Auyeung, N.; Yokochi, A. Experimental modeling of hydrogen producing steps in a novel sulfur-sulfur thermochemical water splitting cycle. Int. J. Hydrog. Energy 2015, 40, 2484–2492. [Google Scholar] [CrossRef]
- Yilmaz, F.; Selbaş, R. Thermodynamic performance assessment of solar based sulfur-iodine thermochemical cycle for hydrogen generation. Energy 2017, 140, 520–529. [Google Scholar] [CrossRef]
- Naterer, G.F.; Suppiah, S.; Stolberg, L.; Lewis, M.; Wang, Z.; Daggupati, V.; Gabriel, K.; Dincer, I.; Rosen, M.A.; Spekkens, P.; et al. Canada’s program on nuclear hydrogen production and the thermochemical Cu-Cl cycle. Int. J. Hydrog. Energy 2010, 35, 10905–10926. [Google Scholar] [CrossRef]
- Wang, Z.L.; Naterer, G.F.; Gabriel, K.S.; Gravelsins, R.; Daggupati, V.N. Comparison of sulfur-iodine and copper-chlorine thermochemical hydrogen production cycles. Int. J. Hydrog. Energy 2010, 35, 4820–4830. [Google Scholar] [CrossRef]
- Wang, Z.; Naterer, G.F.; Gabriel, K.S.; Secnik, E.; Gravelsins, R.; Daggupati, V. Thermal design of a solar hydrogen plant with a copper-chlorine cycle and molten salt energy storage. Int. J. Hydrog. Energy 2011, 36, 11258–11272. [Google Scholar] [CrossRef]
- Naterer, G.F.; Gabriel, K.; Wang, Z.L.; Daggupati, V.N.; Gravelsins, R. Thermochemical hydrogen production with a copper-chlorine cycle. I: Oxygen release from copper oxychloride decomposition. Int. J. Hydrog. Energy 2008, 33, 5439–5450. [Google Scholar] [CrossRef]
- Balta, M.T.; Dincer, I.; Hepbasli, A. Energy and exergy analyses of a new four-step copper-chlorine cycle for geothermal-based hydrogen production. Energy 2010, 35, 3263–3272. [Google Scholar] [CrossRef]
- Simpson, M.F.; Herrmann, S.D.; Boyle, B.D. A hybrid thermochemical electrolytic process for hydrogen production based on the reverse deacon reaction. In Proceedings of the 2004 AIChE Spring National Meeting, New Orleans, LA, USA, 25–29 April 2004; Volume 31, pp. 2700–2704. [Google Scholar]
- Ozcan, H.; Dincer, I. Performance investigation of magnesium-chloride hybrid thermochemical cycle for hydrogen production. Int. J. Hydrog. Energy 2014, 39, 76–85. [Google Scholar] [CrossRef]
- Ozcan, H.; Dincer, I. Energy and exergy analyses of a solar driven Mg-Cl hybrid thermochemical cycle for co-production of power and hydrogen. Int. J. Hydrog. Energy 2014, 39, 15330–15341. [Google Scholar] [CrossRef]
- Balta, M.T.; Dincer, I.; Hepbasli, A. Energy and exergy analyses of magnesium-chlorine (Mg-Cl) thermochemical cycle. Int. J. Hydrog. Energy 2012, 37, 4855–4862. [Google Scholar] [CrossRef]
- Tamaura, Y.; Steinfeld, A.; Kuhn, P.; Ehrensberger, K. Production of solar hydrogen by a novel, 2-step, water-splitting thermochemical cycle. Energy 1995, 20, 325–330. [Google Scholar] [CrossRef]
- Gokon, N.; Takahashi, S.; Yamamoto, H.; Kodama, T. Thermochemical two-step water-splitting reactor with internally circulating fluidized bed for thermal reduction of ferrite particles. Int. J. Hydrog. Energy 2008, 33, 2189–2199. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Konstandopoulos, A.G.; Nalbandian, L.; Zaspalis, V.T.; Sattler, C.; Stobbe, P.; Steele, A.M. Solar water splitting for hydrogen production with monolithic reactors. Sol. Energy 2005, 79, 409–421. [Google Scholar] [CrossRef]
- Dahl, J.K.; Buechler, K.J.; Weimer, A.W.; Lewandowski, A.; Bingham, C. Solar-thermal dissociation of methane in a fluid-wall aerosol flow reactor. Int. J. Hydrog. Energy 2004, 29, 725–736. [Google Scholar] [CrossRef]
- Wyss, J.; Martinek, J.; Kerins, M.; Dahl, J.K.; Weimer, A.; Lewandowski, A.; Bingham, C. Rapid solar-thermal decarbonization of methane in a fluid-wall aerosol flow reactor—Fundamentals and application. Int. J. Chem. React. Eng. 2007, 5. [Google Scholar] [CrossRef]
- Tapia, E.; González-Pardo, A.; Iranzo, A.; Romero, M.; González-Aguilar, J.; Vidal, A.; Martín-Betancourt, M.; Rosa, F. Multi-tubular reactor for hydrogen production: CFD thermal design and experimental testing. Processes 2019, 7, 31. [Google Scholar] [CrossRef]
- Furler, P.; Steinfeld, A. Heat transfer and fluid flow analysis of a 4kW solar thermochemical reactor for ceria redox cycling. Chem. Eng. Sci. 2015, 137, 373–383. [Google Scholar] [CrossRef]
- Kaneko, H.; Miura, T.; Fuse, A.; Ishihara, H.; Taku, S.; Fukuzumi, H.; Naganuma, Y.; Tamaura, Y. Rotary-type solar reactor for solar hydrogen production with two-step water splitting process. Energy Fuels 2007, 21, 2287–2293. [Google Scholar] [CrossRef]
- Schunk, L.O.; Haeberling, P.; Wept, S.; Wuillemin, D.; Meier, A.; Steinfeld, A. A receiver-reactor for the solar thermal dissociation of zinc oxide. J. Sol. Energy Eng. Trans. ASME 2008, 130, 1–6. [Google Scholar] [CrossRef]
- Diver, R.B.; Miller, J.E.; Allendorf, M.D.; Siegel, N.P.; Hogan, R.E. Solar thermochemical water-splitting ferrite-cycle heat engines. J. Sol. Energy Eng. Trans. ASME 2008, 130, 041001. [Google Scholar] [CrossRef]
- Nalbandian, L.; Evdou, A.; Zaspalis, V. La1-xSrxMO3 (M=Mn, Fe) perovskites as materials for thermochemical hydrogen production in conventional and membrane reactors. Int. J. Hydrog. Energy 2009, 34, 7162–7172. [Google Scholar] [CrossRef]
- Kaneko, H.; Lee, C.; Ishikawa, Y.; Hosogoe, K.; Tamaura, Y. Solar H2 production with Tokyo Tech rotary-type solar reactor to be tested using solar concentration system at CSIRO in Australia. In Proceedings of the 3rd International Conference of Energy Sustainability, San Francisco, CA, USA, 19–23 July 2009; pp. 1–6. [Google Scholar]
- Gonzalez-Pardo, A.; Denk, T.; Vidal, A. Thermal tests of a multi-tubular reactor for hydrogen production by using mixed ferrites thermochemical cycle. In Proceedings of the AIP Conference Proceedings, Penang, Malaysia, 10–12 April 2018; pp. 1–8. [Google Scholar]
- Villasmil, W.; Brkic, M.; Wuillemin, D.; Meier, A.; Steinfeld, A. Pilot scale demonstration of a 100-kWth solar thermochemical plant for the thermal dissociation of ZnO. J. Sol. Energy Eng. Trans. ASME 2014, 136, 1–11. [Google Scholar] [CrossRef]
- Koepf, E.; Villasmil, W.; Meier, A. Pilot-scale solar reactor operation and characterization for fuel production via the Zn/ZnO thermochemical cycle. Appl. Energy 2016, 165, 1004–1023. [Google Scholar] [CrossRef]
- Roeb, M.; Monnerie, N.; Schmitz, M.; Sattler, C.; Konstandopoulos, A.G.; Agrafiotis, C.; Zaspalis, V.T. Thermo-chemical production of hydrogen from water by metal oxides fixed on ceramic substrates. In Proceedings of the World Hydrogen Energy Conference, Lyon, France, 13–16 June 2006; pp. 1–12. [Google Scholar]
- Roeb, M.; Säck, J.P.; Rietbrock, P.; Prahl, C.; Schreiber, H.; Neises, M.; de Oliveira, L.; Graf, D.; Ebert, M.; Reinalter, W.; et al. Test operation of a 100 kW pilot plant for solar hydrogen production from water on a solar tower. Sol. Energy 2011, 85, 634–644. [Google Scholar] [CrossRef]
- Säck, J.; Breuer, S.; Cotelli, P.; Houaijia, A.; Lange, M.; Wullenkord, M.; Spenke, C.; Roeb, M.; Sattler, C. High temperature hydrogen production: Design of a 750 KW demonstration plant for a two step thermochemical cycle. Sol. Energy 2016, 135, 232–241. [Google Scholar] [CrossRef]
- Siegel, N.P.; Miller, J.E.; Ermanoski, I.; Diver, R.B.; Stechel, E.B. Factors affecting the efficiency of solar driven metal oxide thermochemical cycles. Ind. Eng. Chem. Res. 2013, 52, 3276–3286. [Google Scholar] [CrossRef]
- Riahi, A.; Atashkari, K.; Mahmoudimehr, J.; Rodat, S. The influences of major geometrical parameters on detailed radiative performance of a multi-tubular solar thermochemical reactor. Appl. Therm. Eng. 2019, 159, 113793. [Google Scholar] [CrossRef]
- Schiebahn, S.; Grube, T.; Robinius, M.; Tietze, V.; Kumar, B.; Stolten, D. Power to gas: Technological overview, systems analysis and economic assessment for a case study in Germany. Int. J. Hydrog. Energy 2015, 40, 4285–4294. [Google Scholar] [CrossRef]
- Ursúa, A.; Gandía, L.M.; Sanchis, P. Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE 2011, 100, 410–426. [Google Scholar] [CrossRef]
- Sapountzi, F.M.; Gracia, J.M.; Weststrate, C.J.; Kee, J.; Fredriksson, H.O.A.; Niemantsverdriet, J.W. Electrocatalysts for the generation of hydrogen, oxygen and synthesis gas. Prog. Energy Combust. Sci. 2017, 58, 1–35. [Google Scholar] [CrossRef]
- Buttler, A.; Spliethoff, H. Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review. Renew. Sustain. Energy Rev. 2018, 82, 2440–2454. [Google Scholar] [CrossRef]
- Rashid, M.M.; Mesfer, M.K.; Naseem, H.; Danish, M. Hydrogen production by water electrolysis: A review of alkaline water electrolysis, PEM water electrolysis and high temperature water electrolysis. Int. J. Eng. Adv. Technol. 2015, 4, 2249–8958. [Google Scholar]
- Malkow, T.; Pilenga, A.; Tsotridis, G.; De Marco, G. EU Harmonised Polarisation Curve Test Method for Low Temperature Water Electrolysis; Publications Office of the European Union: Brusselsm, Belgium, 2018. [Google Scholar]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Gong, X.; Guo, Z. The intensification technologies to water electrolysis for hydrogen production-A review. Renew. Sustain. Energy Rev. 2014, 29, 573–588. [Google Scholar] [CrossRef]
- Marini, S.; Salvi, P.; Nelli, P.; Pesenti, R.; Villa, M.; Berrettoni, M.; Zangari, G.; Kiros, Y. Advanced alkaline water electrolysis. Electrochim. Acta 2012, 82, 384–391. [Google Scholar] [CrossRef]
- Feng, Q.; Yuan, X.Z.; Liu, G.; Wei, B.; Zhang, Z.; Li, H.; Wang, H. A review of proton exchange membrane water electrolysis on degradation mechanisms and mitigation strategies. J. Power Sources 2017, 366, 33–55. [Google Scholar] [CrossRef]
- Scott, K. Process intensification: An electrochemical perspective. Renew. Sustain. Energy Rev. 2018, 81, 1406–1426. [Google Scholar] [CrossRef]
- Tijani, A.S.; Rahim, A.H.A. Numerical modeling the effect of operating variables on Faraday efficiency in PEM electrolyzer. Procedia Technol. 2016, 26, 419–427. [Google Scholar] [CrossRef]
- Kai, J.; Saito, R.; Terabaru, K.; Li, H.; Nakajima, H.; Ito, O. Effect of temperature on the performance of polymer electrolyte membranewater electrolysis: Numerical analysis of electrolysis voltage considering gas/liquid two-phase flow. J. Electrochem. Soc. 2019, 166, F246–F254. [Google Scholar] [CrossRef]
- Buelvas, W.L.; Ávila, K.C.P.; Jiménez, Á.R. Temperature as a factor determining on water electrolysis. Int. J. Eng. Trends Technol. 2014, 7, 5–9. [Google Scholar] [CrossRef]
- Roy, A.; Watson, S.; Infield, D. Comparison of electrical energy efficiency of atmospheric and high-pressure electrolysers. Int. J. Hydrog. Energy 2006, 31, 1964–1979. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C.; Figueiredo, J.L. Hydrogen production by alkaline water electrolysis. Quim. Nova 2013, 36, 1176–1193. [Google Scholar] [CrossRef]
- Bodner, M.; Hofer, A.; Hacker, V. H2 generation from alkaline electrolyzer. Wiley Interdiscip. Rev. Energy Environ. 2015, 4, 365–381. [Google Scholar] [CrossRef]
- Zeng, K.; Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 2010, 36, 307–326. [Google Scholar] [CrossRef]
- Colli, A.N.; Girault, H.H.; Battistel, A. Non-precious electrodes for practical alkaline water electrolysis. Materials 2019, 12, 1336. [Google Scholar] [CrossRef]
- Schalenbach, M.; Zeradjanin, A.R.; Kasian, O.; Cherevko, S.; Mayrhofer, K.J.J. A perspective on low-temperature water electrolysis—Challenges in alkaline and acidic technology. Int. J. Electrochem. Sci. 2018, 13, 1173–1226. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, L.; Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 2019, 12, 2620–2645. [Google Scholar] [CrossRef]
- Letcher, T. Storing energy, with special reference to renewable energy sources. Chem. Int. 2016, 38, 28. [Google Scholar]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Ulleberg, Ø. Modeling of advanced alkaline electrolyzers a system. Hydrog. Energy 2003, 28, 21–33. [Google Scholar] [CrossRef]
- Faid, A.Y.; Barnett, A.O.; Seland, F.; Sunde, S. Highly active nickel-based catalyst for hydrogen evolution in anion exchange membrane electrolysis. Catalysts 2018, 8, 614. [Google Scholar] [CrossRef]
- Phillips, R.; Dunnill, C.W. Zero gap alkaline electrolysis cell design for renewable energy storage as hydrogen gas. RSC Adv. 2016, 6, 100643–100651. [Google Scholar] [CrossRef]
- Pletcher, D.; Li, X. Prospects for alkaline zero gap water electrolysers for hydrogen production. Int. J. Hydrog. Energy 2011, 36, 15089–15104. [Google Scholar] [CrossRef]
- David, M.; Ocampo-Martínez, C.; Sánchez-Peña, R. Advances in alkaline water electrolyzers: A review. J. Energy Storage 2019, 23, 392–403. [Google Scholar] [CrossRef]
- Diéguez, P.M.; Ursúa, A.; Sanchis, P.; Sopena, C.; Guelbenzu, E.; Gandía, L.M. Thermal performance of a commercial alkaline water electrolyzer: Experimental study and mathematical modeling. Int. J. Hydrog. Energy 2008, 33, 7338–7354. [Google Scholar] [CrossRef]
- Ganley, J.C. High temperature and pressure alkaline electrolysis. Int. J. Hydrog. Energy 2009, 34, 3604–3611. [Google Scholar] [CrossRef]
- Allebrod, F.; Chatzichristodoulou, C.; Mogensen, M.B. Alkaline electrolysis cell at high temperature and pressure of 250 °C and 42 bar. J. Power Sources 2013, 229, 22–31. [Google Scholar] [CrossRef]
- Mahmood, N.; Yao, Y.; Zhang, J.W.; Pan, L.; Zhang, X.; Zou, J.J. Electrocatalysts for hydrogen evolution in alkaline electrolytes: Mechanisms, challenges, and prospective solutions. Adv. Sci. 2018, 5, 1700464. [Google Scholar] [CrossRef]
- Gong, M.; Wang, D.Y.; Chen, C.C.; Hwang, B.J.; Dai, H. A mini review on nickel-based electrocatalysts for alkaline hydrogen evolution reaction. Nano Res. 2016, 9, 28–46. [Google Scholar] [CrossRef]
- Mauer, A.E.; Kirk, D.W.; Thorpe, S.J. The role of iron in the prevention of nickel electrode deactivation in alkaline electrolysis. Electrochim. Acta 2007, 52, 3505–3509. [Google Scholar] [CrossRef]
- Cardoso, D.S.P.; Amaral, L.; Santos, D.M.F.; Šljukić, B.; Sequeira, C.A.C.; Macciò, D.; Saccone, A. Enhancement of hydrogen evolution in alkaline water electrolysis by using nickel-rare earth alloys. Int. J. Hydrog. Energy 2015, 40, 4295–4302. [Google Scholar] [CrossRef]
- Safizadeh, F.; Ghali, E.; Houlachi, G. Electrocatalysis developments for hydrogen evolution reaction in alkaline solutions—A review. Int. J. Hydrog. Energy 2015, 40, 256–274. [Google Scholar] [CrossRef]
- Brown, D.E.; Mahmood, M.N.; Man, M.C.M.; Turner, A.K. Preparation and characterization of low overvoltage transition metal alloy electrocatalysts for hydrogen evolution in alkaline solutions. Electrochim. Acta 1984, 29, 1551–1556. [Google Scholar] [CrossRef]
- Arul Raj, I. Nickel-based, binary-composite electrocatalysts for the cathodes in the energy-efficient industrial production of hydrogen from alkaline-water electrolytic cells. J. Mater. Sci. 1993, 28, 4375–4382. [Google Scholar]
- Kim, J.E.; Bae, K.K.; Park, C.S.; Jeong, S.U.; Baik, K.H.; Kim, J.W.; Kim, Y.H.; Kang, K.S.; Lee, K.B. Data on the characterization of Raney nickel powder and Raney-nickel-coated electrodes prepared by atmospheric plasma spraying for alkaline water electrolysis. Data Brief 2018, 21, 2059–2062. [Google Scholar] [CrossRef]
- Shetty, S.; Mohamed Jaffer Sadiq, M.; Bhat, D.K.; Hegde, A.C. Electrodeposition and characterization of Ni-Mo alloy as an electrocatalyst for alkaline water electrolysis. J. Electroanal. Chem. 2017, 796, 57–65. [Google Scholar] [CrossRef]
- Ganci, F.; Baguet, T.; Aiello, G.; Cusumano, V.; Mandin, P.; Sunseri, C.; Inguanta, R. Nanostructured Ni based anode and cathode for alkaline water electrolyzers. Energies 2019, 12, 3669. [Google Scholar] [CrossRef]
- Pomerantseva, E.; Resini, C.; Kovnir, K.; Kolen’ko, Y.V. Emerging nanostructured electrode materials for water electrolysis and rechargeable beyond Li-ion batteries. Adv. Phys. X 2017, 2, 211–253. [Google Scholar] [CrossRef]
- Ganci, F.; Lombardo, S.; Sunseri, C.; Inguanta, R. Nanostructured electrodes for hydrogen production in alkaline electrolyzer. Renew. Energy 2018, 123, 117–124. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.-C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)₂-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrog. Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Grubb, W.T. Ionic migration in ion-exchange membranes. J. Phys. Chem. 1959, 63, 55–58. [Google Scholar] [CrossRef]
- Gahleitner, G. Hydrogen from renewable electricity: An international review of power-to-gas pilot plants for stationary applications. Int. J. Hydrog. Energy 2013, 38, 2039–2061. [Google Scholar] [CrossRef]
- Shiva Kumar, S.; Himabindu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Siracusano, S.; Baglio, V.; Van Dijk, N.; Merlo, L.; Aricò, A.S. Enhanced performance and durability of low catalyst loading PEM water electrolyser based on a short-side chain perfluorosulfonic ionomer. Appl. Energy 2017, 192, 477–489. [Google Scholar] [CrossRef]
- Wang, R.; Liu, S.; Wang, L.; Li, M.; Gao, C. Understanding of nanophase separation and hydrophilic morphology in Nafion and SPEEK membranes: A combined experimental and theoretical studies. Nanomaterials 2019, 9, 869. [Google Scholar] [CrossRef]
- Devanathan, R.; Venkatnathan, A.; Rousseau, R.; Dupuis, M.; Frigato, T.; Gu, W.; Helms, V. Atomistic simulation of water percolation and proton hopping in Nafion fuel cell membrane. J. Phys. Chem. B 2010, 114, 13681–13690. [Google Scholar] [CrossRef]
- Sun, C.W.; Hsiau, S.S. Effect of electrolyte concentration difference on hydrogen production during PEM electrolysis. J. Electrochem. Sci. Technol. 2018, 9, 99–108. [Google Scholar] [CrossRef]
- Ayers, K.E.; Anderson, E.B.; Capuano, C.B.; Carter, B.D.; Dalton, L.T.; Hanlon, G.; Manco, J.; Niedzwiecki, M. Research advances towards low cost, high efficiency PEM electrolysis. ECS Trans. 2010, 33, 3–15. [Google Scholar]
- Schalenbach, M.; Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. Pressurized PEM water electrolysis: Efficiency and gas crossover. Int. J. Hydrog. Energy 2013, 38, 14921–14933. [Google Scholar] [CrossRef]
- Rasten, E.; Hagen, G.; Tunold, R. Electrocatalysis in water electrolysis with solid polymer electrolyte. Electrochim. Acta 2003, 48, 3945–3952. [Google Scholar] [CrossRef]
- Xu, W.; Scott, K. The effects of ionomer content on PEM water electrolyser membrane electrode assembly performance. Int. J. Hydrog. Energy 2010, 35, 12029–12037. [Google Scholar] [CrossRef]
- Miles, M.H.; Thomason, M.A. Periodic variations of overvoltages for water electrolysis in acid solutions from cyclic voltammetric studies. J. Electrochem. Soc. 1976, 123, 1459–1461. [Google Scholar] [CrossRef]
- Bessarabov, D.G.; Wang, H.H.; Li, H.; Zhao, N. PEM Electrolysis for Hydrogen Production: Principles and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Chisholm, G.; Kitson, P.J.; Kirkaldy, N.D.; Bloor, L.G.; Cronin, L. 3D printed flow plates for the electrolysis of water: An economic and adaptable approach to device manufacture. Energy Environ. Sci. 2014, 7, 3026–3032. [Google Scholar] [CrossRef]
- Mo, J.; Kang, Z.; Retterer, S.T.; Cullen, D.A.; Toops, T.J.; Green, J.B.; Mench, M.M.; Zhang, F.Y. Discovery of true electrochemical reactions for ultrahigh catalyst mass activity in water splitting. Sci. Adv. 2016, 2, 1–7. [Google Scholar] [CrossRef]
- Nikiforov, A.; Petrushina, I.M.; Jensen, J.O.; Bjerrum, N.J.; Christensen, E. Advanced Construction Materials for High Temperature Steam PEM Electrolysers. In Electrolysis; Linkov, V., Kleperis, J., Eds.; IntechOpen: London, UK, 2012; pp. 61–86. [Google Scholar]
- Millet, P.; Ngameni, R.; Grigoriev, S.A.; Mbemba, N.; Brisset, F.; Ranjbari, A.; Etiévant, C. PEM water electrolyzers: From electrocatalysis to stack development. Int. J. Hydrog. Energy 2010, 35, 5043–5052. [Google Scholar] [CrossRef]
- Stempien, J.P.; Sun, Q.; Chan, S.H. Solid oxide electrolyzer cell modeling: A review. J. Power Technol. 2013, 93, 216–246. [Google Scholar]
- Wang, Y.; Liu, T.; Lei, L.; Chen, F. High temperature solid oxide H2O/CO2 co-electrolysis for syngas production. Fuel Process. Technol. 2017, 161, 248–258. [Google Scholar] [CrossRef]
- Bo, Y.; Wenqiang, Z.; Jingming, X.; Jing, C. Status and research of highly efficient hydrogen production through high temperature steam electrolysis at INET. Int. J. Hydrog. Energy 2010, 35, 2829–2835. [Google Scholar] [CrossRef]
- Pandiyan, A.; Uthayakumar, A.; Subrayan, R.; Cha, S.W.; Krishna Moorthy, S.B. Review of solid oxide electrolysis cells: A clean energy strategy for hydrogen generation. Nanomater. Energy 2019, 8, 2–22. [Google Scholar] [CrossRef]
- Hu, L.; Lindbergh, G.; Lagergren, C. Electrode kinetics of the NiO porous electrode for oxygen production in the molten carbonate electrolysis cell (MCEC). Faraday Discuss. 2015, 182, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Brisse, A.; Schefold, J.; Zahid, M. High temperature water electrolysis in solid oxide cells. Int. J. Hydrog. Energy 2008, 33, 5375–5382. [Google Scholar] [CrossRef]
- Laguna-Bercero, M.A.; Skinner, S.J.; Kilner, J.A. Performance of solid oxide electrolysis cells based on scandia stabilised zirconia. J. Power Sources 2009, 192, 126–131. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.H.; Leung, D.Y.C. Technological development of hydrogen production by solid oxide electrolyzer cell (SOEC). Int. J. Hydrog. Energy 2008, 33, 2337–2354. [Google Scholar] [CrossRef]
- Han, M.; Tang, X.; Yin, H.; Peng, S. Fabrication, microstructure and properties of a YSZ electrolyte for SOFCs. J. Power Sources 2007, 165, 757–763. [Google Scholar] [CrossRef]
- Rathod, S.G.; Bhajantri, R.F.; Ravindrachary, V.; Pujari, P.K.; Nagaraja, G.K.; Naik, J.; Hebbar, V.; Chandrappa, H. Temperature-dependent ionic conductivity and transport properties of LiClO4-doped PVA/modified cellulose composites. Bull. Mater. Sci. 2015, 38, 1213–1221. [Google Scholar] [CrossRef]
- Bocanegra-bernal, M.H.; Díaz de la torre, S. Phase transitions in zirconium dioxide and related materials for high performance engineering ceramics. J. Mater. Sci. 2002, 37, 4947–4971. [Google Scholar] [CrossRef]
- Badwal, S.P.S. Zirconia-based solid electrolytes: Microstructure, stability and ionic conductivity. Solid State Ion. 1992, 52, 23–32. [Google Scholar] [CrossRef]
- Butler, E.P.; Bonanos, N. The characterization of ZrO2 engineering ceramics by A.C. impedance spectroscopy. Mater. Sci. Eng. 1985, 71, 49–56. [Google Scholar] [CrossRef]
- Kilner, J.A. Fast anion transport in solids. Solid State Ion. 1983, 8, 201–207. [Google Scholar] [CrossRef]
- Biswas, M.; Sadanala, K.C. Electrolyte materials for solid oxide fuel cell. J. Powder Metall. Min. 2013, 2, 10–15. [Google Scholar] [CrossRef]
- Ivanov, V.; Shkerin, S.; Rempel, A.; Khrustov, V.; Lipilin, A.; Nikonov, A. The grain size effect on the yttria stabilized Zirconia grain boundary conductivity. J. Nanosci. Nanotechnol. 2010, 10, 7411–7415. [Google Scholar] [CrossRef] [PubMed]
- Omar, S. Doped Ceria for Solid Oxide Fuel Cells. In Cerium Oxide—Applications and Attributes; Khan, S.B., Akhtar, K., Eds.; IntechOpen: London, UK, 2019; p. 17. [Google Scholar]
- Artini, C.; Pani, M.; Carnasciali, M.M.; Plaisier, J.R.; Costa, G.A. Lu-, Sm-, and Gd-doped ceria: A comparative approach to their structural properties. Inorg. Chem. 2016, 55, 10567–10579. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kanoh, M.; Quan, C.J.; Inaba, H.; Wang, S.; Dokiya, M.; Tagawa, H. Thermal expansion of Gd-doped ceria and reduced ceria. Solid State Ion. 2000, 132, 227–233. [Google Scholar] [CrossRef]
- Mangalaraja, R.V.; Ananthakumar, S.; Paulraj, M.; Pesenti, H.; López, M.; Camurri, C.P.; Barcos, L.A.; Avila, R.E. Electrical and thermal characterization of Sm3+ doped ceria electrolytes synthesized by combustion technique. J. Alloys Compd. 2011, 510, 134–140. [Google Scholar] [CrossRef]
- Wang, D.Y.; Park, D.S.; Griffith, J.; Nowick, A.S. Oxigen-ion conductivity and defect interactions in yttria-doped ceria. Solid State Ion. 1981, 2, 95–105. [Google Scholar] [CrossRef]
- Gerhardt, R.; Lee, W.-K.; Nowick, A.S. Anelastic and dielectric relaxation of scandia-doped ceria. J. Phys. Chem. Solids 1987, 48, 563–569. [Google Scholar] [CrossRef]
- Sachdeva, A.; Chavan, S.V.; Goswami, A.; Tyagi, A.K.; Pujari, P.K. Positron annihilation spectroscopic studies on Nd-doped ceria. J. Solid State Chem. 2005, 178, 2062–2066. [Google Scholar] [CrossRef]
- Eguchi, K.; Setoguchi, T.; Inoue, T.; Arai, H. Electrical properties of ceria-based oxides and their application to solid oxide fuel cells. Solid State Ion. 1992, 52, 165–172. [Google Scholar] [CrossRef]
- Singh, M.; Singh, A.K. Studies on structural, morphological, and electrical properties of Ga3+ and Cu2+ co-doped ceria ceramics as solid electrolyte for IT-SOFCs. Int. J. Hydrog. Energy 2019, 1–12. [Google Scholar] [CrossRef]
- Wang, F.Y.; Chen, S.; Cheng, S. Gd3+ and Sm3+ co-doped ceria based electrolytes for intermediate temperature solid oxide fuel cells. Electrochem. Commun. 2004, 6, 743–746. [Google Scholar] [CrossRef]
- Tadokoro, S.K.; Muccillo, E.N.S. Effect of Y and Dy co-doping on electrical conductivity of ceria ceramics. J. Eur. Ceram. Soc. 2007, 27, 4261–4264. [Google Scholar] [CrossRef]
- Omar, S.; Wachsman, E.D.; Nino, J.C. Higher conductivity Sm3+ and Nd3+ co-doped ceria-based electrolyte materials. Solid State Ion. 2008, 178, 1890–1897. [Google Scholar] [CrossRef]
- Ishihara, T.; Shibayama, T.; Honda, M.; Nishiguchi, H.; Takita, Y. Intermediate temperature solid oxide fuel cells using LaGaO3 electrolyte II. Improvement of oxide ion conductivity and power density by doping Fe for Ga site of LaGaO3. J. Electrochem. Soc. 2000, 147, 1332–1337. [Google Scholar] [CrossRef]
- Yamaji, K.; Horita, T.; Ishikawa, M.; Sakai, N.; Yokokawa, H. Chemical stability of the La0.9Sr0.1Ga0.8Mg0.2O2.85 electrolyte in a reducing atmosphere. Solid State Ion. 1999, 121, 217–224. [Google Scholar] [CrossRef]
- Bozza, F.; Polini, R.; Traversa, E. Electrophoretic deposition of dense La0.8Sr0.2Ga0.8Mg0.115Co0.085O3−δ electrolyte films from single-phase powders for intermediate temperature solid oxide fuel cells. J. Am. Ceram. Soc. 2009, 92, 1999–2004. [Google Scholar] [CrossRef]
- Peña-Martínez, J.; Marrero-López, D.; Pérez-Coll, D.; Ruiz-Morales, J.C.; Núñez, P. Performance of XSCoF (X = Ba, La and Sm) and LSCrX′ (X′ = Mn, Fe and Al) perovskite-structure materials on LSGM electrolyte for IT-SOFC. Electrochim. Acta 2007, 52, 2950–2958. [Google Scholar] [CrossRef]
- Hossain, S.; Abdalla, A.M.; Jamain, S.N.B.; Zaini, J.H.; Azad, A.K. A review on proton conducting electrolytes for clean energy and intermediate temperature-solid oxide fuel cells. Renew. Sustain. Energy Rev. 2017, 79, 750–764. [Google Scholar] [CrossRef]
- Bi, L.; Da’as, E.H.; Shafi, S.P. Proton-conducting solid oxide fuel cell (SOFC) with Y-doped BaZrO3 electrolyte. Electrochem. Commun. 2017, 80, 20–23. [Google Scholar] [CrossRef]
- Xie, K.; Zhang, Y.; Meng, G.; Irvine, J.T.S. Electrochemical reduction of CO2 in a proton conducting solid oxide electrolyser. J. Mater. Chem. 2011, 21, 195–198. [Google Scholar] [CrossRef]
- Peters, R.; Deja, R.; Blum, L.; Nguyen, V.N.; Fang, Q.; Stolten, D. Influence of operating parameters on overall system efficiencies using solid oxide electrolysis technology. Int. J. Hydrog. Energy 2015, 40, 7103–7113. [Google Scholar] [CrossRef]
- Kim, J.; Jun, A.; Gwon, O.; Yoo, S.; Liu, M.; Shin, J.; Lim, T.H.; Kim, G. Hybrid-solid oxide electrolysis cell: A new strategy for efficient hydrogen production. Nano Energy 2018, 44, 121–126. [Google Scholar] [CrossRef]
- Tietz, F. Thermal expansion of SOFC materials. Ionics 1999, 5, 129–139. [Google Scholar] [CrossRef]
- Xu, X.; Wang, C.; Fronzi, M.; Liu, X.; Bi, L. Modification of a first-generation solid oxide fuel cell cathode with Co3O4 nanocubes having selectively exposed crystal planes. Mater. Renew. Sustain. Energy 2019, 8, 1–8. [Google Scholar] [CrossRef]
- Huang, K.; Feng, M.; Goodenough, J.B.; Schmerling, M. Characterization of Sr-doped LaMnO3 and LaCoO3 as cathode materials for a doped LaGaO3 ceramic fuel cell. J. Electrochem. Soc. 1996, 143, 3630–3636. [Google Scholar] [CrossRef]
- Maguire, E.; Gharbage, B.; Marques, F.M.B.; Labrincha, J.A. Cathode materials for intermediate temperature SOFCs. Solid State Ion. 2000, 127, 329–335. [Google Scholar] [CrossRef]
- Nerat, M.; Juričić, Đ. Modelling of anode delamination in solid oxide electrolysis cell and analysis of its effects on electrochemical performance. Int. J. Hydrog. Energy 2018, 43, 8179–8189. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, J.; Yu, B.; Zhang, W.; Chen, J.; Qiao, J.; Zhang, J. A review of high temperature co-electrolysis of H2O and CO2 to produce sustainable fuels using solid oxide electrolysis cells (SOECs): Advanced materials and technology. Chem. Soc. Rev. 2017, 46, 1427–1463. [Google Scholar] [CrossRef]
- Ivers-Tiffée, E.; Weber, A.; Herbstritt, D. Materials and technologies for SOFC-components. J. Eur. Ceram. Soc. 2001, 21, 1805–1811. [Google Scholar] [CrossRef]
- Marina, O.A.; Pederson, L.R.; Williams, M.C.; Coffey, G.W.; Meinhardt, K.D.; Nguyen, C.D.; Thomsen, E.C. Electrode performance in reversible solid oxide fuel cells. J. Electrochem. Soc. 2007, 154, 452–459. [Google Scholar] [CrossRef]
- Kong, J.; Zhang, Y.; Deng, C.; Xu, J. Synthesis and electrochemical properties of LSM and LSF perovskites as anode materials for high temperature steam electrolysis. J. Power Sources 2009, 186, 485–489. [Google Scholar] [CrossRef]
- Jiang, S.P. Challenges in the development of reversible solid oxide cell technologies: A mini review. Asia-Pac. J. Chem. Eng. 2016, 11, 386–391. [Google Scholar] [CrossRef]
- Lessing, P.A. A review of sealing technologies applicable to solid oxide electrolysis cells. J. Mater. Sci. 2007, 42, 3465–3476. [Google Scholar] [CrossRef]
- Redissi, Y.; Bouallou, C. Valorization of carbon dioxide by co-electrolysis of CO2/H2O at high temperature for syngas production. Energy Procedia 2013, 37, 6667–6678. [Google Scholar] [CrossRef]
- Chen, X.; Guan, C.; Xiao, G.; Du, X.; Wang, J.Q. Syngas production by high temperature steam/CO2 coelectrolysis using solid oxide electrolysis cells. Faraday Discuss. 2015, 182, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Ni, M. An electrochemical model for syngas production by co-electrolysis of H2O and CO2. J. Power Sources 2012, 202, 209–216. [Google Scholar] [CrossRef]
- Sun, X.; Chen, M.; Jensen, S.H.; Ebbesen, S.D.; Graves, C.; Mogensen, M. Thermodynamic analysis of synthetic hydrocarbon fuel production in pressurized solid oxide electrolysis cells. Int. J. Hydrog. Energy 2012, 37, 17101–17110. [Google Scholar] [CrossRef]
- Hansen, J.B. Solid oxide electrolysis—A key enabling technology for sustainable energy scenarios. Faraday Discuss. 2015, 182, 9–48. [Google Scholar] [CrossRef]
- Becker, W.L.; Braun, R.J.; Penev, M.; Melaina, M. Production of Fischer-Tropsch liquid fuels from high temperature solid oxide co-electrolysis units. Energy 2012, 47, 99–115. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Küngas, R.; Lin, T.E.; Diethelm, S.; Maréchal, F.; Van herle, J. Power-to-fuels via solid-oxide electrolyzer: Operating window and techno-economics. Renew. Sustain. Energy Rev. 2019, 110, 174–187. [Google Scholar] [CrossRef]
- Bian, L.Z.; Chen, Z.Y.; Wang, L.J.; Li, F.S.; Chou, K.C. Electrochemical performance and carbon deposition of anode-supported solid oxide fuel cell exposed to H2/CO fuels. Int. J. Hydrog. Energy 2017, 42, 14246–14252. [Google Scholar] [CrossRef]
- Skafte, T.L.; Blennow, P.; Hjelm, J.; Graves, C. Carbon deposition and sulfur poisoning during CO2 electrolysis in nickel-based solid oxide cell electrodes. J. Power Sources 2018, 373, 54–60. [Google Scholar] [CrossRef]
- Graves, C.; Ebbesen, S.D.; Mogensen, M. Co-electrolysis of CO2 and H2O in solid oxide cells: Performance and durability. Solid State Ion. 2011, 192, 398–403. [Google Scholar] [CrossRef]
- Ebbesen, S.D.; Graves, C.; Hauch, A.; Jensen, S.H.; Mogensen, M. Poisoning of solid oxide electrolysis cells by impurities. J. Electrochem. Soc. 2010, 157, B1419–B1429. [Google Scholar] [CrossRef]
- Kadier, A.; Simayi, Y.; Abdeshahian, P.; Azman, N.F.; Chandrasekhar, K.; Kalil, M.S. A comprehensive review of microbial electrolysis cells (MEC) reactor designs and configurations for sustainable hydrogen gas production. Alex. Eng. J. 2016, 55, 427–443. [Google Scholar] [CrossRef]
- Rozendal, R.A.; Buisman, C.J.N. Process for Producing Hydrogen; Patent and Trademark Office: Washington, DC, USA, 2005; pp. 1–13.
- Liu, H.; Grot, S.; Logan, B.E. Electrochemically assisted microbial production of hydrogen from acetate. Environ. Sci. Technol. 2005, 39, 4317–4320. [Google Scholar] [CrossRef]
- Escapa, A.; San-Martín, M.I.; Morán, A. Potential use of microbial electrolysis cells in domestic wastewater treatment plants for energy recovery. Front. Energy Res. 2014, 2, 19. [Google Scholar] [CrossRef]
- Waqas, M.; Rehan, M.; Aburiazaiza, A.S.; Nizami, A.-S. Wastewater Biorefinery Based on the Microbial Electrolysis Cell: Opportunities and Challenges. In Progress and Recent Trends in Microbial Fuel Cells; Elsevier: Amsterdam, The Netherlands, 2018; pp. 347–374. [Google Scholar]
- Chen, Y.; Chen, M.; Shen, N.; Zeng, R.J. H2 production by the thermoelectric microconverter coupled with microbial electrolysis cell. Int. J. Hydrog. Energy 2016, 41, 22760–22768. [Google Scholar] [CrossRef]
- Santoro, C.; Arbizzani, C.; Erable, B.; Ieropoulos, I. Microbial fuel cells: From fundamentals to applications. A review. J. Power Sources 2017, 356, 225–244. [Google Scholar] [CrossRef]
- Feng, C.; Li, J.; Qin, D.; Chen, L.; Zhao, F.; Chen, S.; Hu, H.; Yu, C.P. Characterization of exoelectrogenic bacteria enterobacter strains isolated from a microbial fuel cell exposed to copper shock load. PLoS ONE 2014, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.B.; Bond, D.R.; Lovley, D.R. Graphite electrodes as electron donors for anaerobic respiration. Environ. Microbiol. 2004, 6, 596–604. [Google Scholar] [CrossRef]
- Rathinam, N.K.; Salem, D.R.; Sani, R.K. Biofilm Engineering for Improving the Performance of Microbial Electrochemical Technologies. In Microbial Electrochemical Technology; CRC: Boca Raton, FL, USA, 2019; pp. 315–338. [Google Scholar]
- Mathuriya, A.S. Inoculum selection to enhance performance of a microbial fuel cell for electricity generation during wastewater treatment. Environ.Technol. 2013, 34, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Logan, B.E.; Call, D.; Cheng, S.; Hamelers, H.V.M.; Sleutels, T.H.J.A.; Jeremiasse, A.W.; Rozendal, R.A. Microbial electrolysis cells for high yield hydrogen gas production from organic matter. Environ. Sci. Technol. 2008, 42, 8630–8640. [Google Scholar] [CrossRef] [PubMed]
- Yasri, N.; Roberts, E.P.L.; Gunasekaran, S. The electrochemical perspective of bioelectrocatalytic activities in microbial electrolysis and microbial fuel cells. Energy Rep. 2019, 5, 1116–1136. [Google Scholar] [CrossRef]
- Xie, X.; Criddle, C.; Cui, Y. Design and fabrication of bioelectrodes for microbial bioelectrochemical systems. Energy Environ. Sci. 2015, 8, 3418–3441. [Google Scholar] [CrossRef]
- Logan, B.E.; Hamelers, B.; Rozendal, R.; Schröder, U.; Keller, J.; Freguia, S.; Aelterman, P.; Verstraete, W.; Rabaey, K. Microbial fuel cells: Methodology and technology. Environ. Sci. Technol. 2006, 40, 5181–5192. [Google Scholar] [CrossRef]
- Hindatu, Y.; Annuar, M.S.M.; Gumel, A.M. Mini-review: Anode modification for improved performance of microbial fuel cell. Renew. Sustain. Energy Rev. 2017, 73, 236–248. [Google Scholar] [CrossRef]
- Kundu, A.; Sahu, J.N.; Redzwan, G.; Hashim, M.A. An overview of cathode material and catalysts suitable for generating hydrogen in microbial electrolysis cell. Int. J. Hydrog. Energy 2013, 38, 1745–1757. [Google Scholar] [CrossRef]
- Kadier, A.; Logroño, W.; Rai, P.K.; Kalil, M.S.; Mohamed, A.; Hasan, H.A.; Hamid, A.A. None-platinum electrode catalysts and membranes for highly efficient and inexpensive H2 production in microbial electrolysis cells (MECs): A review. Iran. J. Catal. 2017, 7, 89–102. [Google Scholar]
- Rozendal, R.A.; Hamelers, H.V.M.; Molenkamp, R.J.; Buisman, C.J.N. Performance of single chamber biocatalyzed electrolysis with different types of ion exchange membranes. Water Res. 2007, 41, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Call, D.; Logan, B.E. Hydrogen production in a single chamber microbial electrolysis cell lacking a membrane. Environ. Sci. Technol. 2008, 42, 3401–3406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Quan, X.; Chen, S.; Afzal, S. Enhanced anaerobic digestion of organic contaminants containing diverse microbial population by combined microbial electrolysis cell (MEC) and anaerobic reactor under Fe(III) reducing conditions. Bioresour. Technol. 2013, 136, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Wrana, N.; Sparling, R.; Cicek, N.; Levin, D.B. Hydrogen gas production in a microbial electrolysis cell by electrohydrogenesis. J. Clean. Prod. 2010, 18, S105–S111. [Google Scholar] [CrossRef]
- Cheng, S.; Xing, D.; Call, D.F.; Logan, B.E. Direct biological conversion of electrical current into methane by electromethanogenesis. Environ. Sci. Technol. 2009, 43, 3953–3958. [Google Scholar] [CrossRef] [PubMed]
- Clauwaert, P.; Tolêdo, R.; van der Ha, D.; Crab, R.; Verstraete, W.; Hu, H.; Udert, K.M.; Rabaey, K. Combining biocatalyzed electrolysis with anaerobic digestion. Water Sci. Technol. 2008, 57, 575–579. [Google Scholar] [CrossRef]
- Nelabhotla, A.B.T.; Dinamarca, C. Bioelectrochemical CO2 reduction to methane: MES integration in biogas production processes. Appl. Sci. 2019, 9, 1056. [Google Scholar] [CrossRef]
- Li, S.; Chen, G. Factors affecting the effectiveness of bioelectrochemical system applications: Data synthesis and meta-analysis. Batteries 2018, 4, 34. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Zhang, L.; Xu, T.; Ding, K. Influence of initial anolyte pH and temperature on hydrogen production through simultaneous saccharification and fermentation of lignocellulose in microbial electrolysis cell. Int. J. Hydrog. Energy 2017, 42, 22663–22670. [Google Scholar] [CrossRef]
- Andrei, V.; Reuillard, B.; Reisner, E. Bias-free solar syngas production by integrating a molecular cobalt catalyst with perovskite–BiVO4 tandems. Nat. Mater. 2019. [Google Scholar] [CrossRef]
- Liao, C.H.; Huang, C.W.; Wu, J.C.S. Hydrogen production from semiconductor-based photocatalysis via water splitting. Catalysts 2012, 2, 490–516. [Google Scholar] [CrossRef]
- Li, Z.; Luo, W.; Zhang, M.; Feng, J.; Zou, Z. Photoelectrochemical cells for solar hydrogen production: Current state of promising photoelectrodes, methods to improve their properties, and outlook. Energy Environ. Sci. 2013, 6, 347–370. [Google Scholar] [CrossRef]
- Ahmad, H.; Kamarudin, S.K.; Minggu, L.J.; Kassim, M. Hydrogen from photo-catalytic water splitting process: A review. Renew. Sustain. Energy Rev. 2015, 43, 599–610. [Google Scholar] [CrossRef]
- Shi, X.; Cai, L.; Ma, M.; Zheng, X.; Park, J.H. General Characterization Methods for Photoelectrochemical Cells for Solar Water Splitting. ChemSusChem 2015, 8, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Tembhurne, S.; Nandjou, F.; Haussener, S. A thermally synergistic photo-electrochemical hydrogen generator operating under concentrated solar irradiation. Nat. Energy 2019, 4, 399–407. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Sharma, G.P.; Upadhyay, A.P.; Behara, D.K.; Sivakumar, S.; Pala, R.G.S. Fundamental Aspect of Photoelectrochemical Water Splitting. In The Water-Food-Energy Nexus: Processes, Technologies, and Challenges; Mujtaba, I.M., Srinivasan, R., Elbashir, N.O., Eds.; CRC: Boca Raton, FL, USA, 2017; pp. 677–689. [Google Scholar]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Energy Mater. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Maeda, K.; Domen, K. New non-oxide photocatalysts designed for overall water splitting under visible light. J. Phys. Chem. C 2007, 111, 7851–7861. [Google Scholar] [CrossRef]
- Li, J.; Wu, N. Semiconductor-based photocatalysts and photoelectrochemical cells for solar fuel generation: A review. Catal. Sci. Technol. 2015, 5, 1360–1384. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; Lai, T.-H.; Kuo, M.-Y.; Hsieh, P.-Y.; Hsu, Y.-J. Photoelectrochemical cells for solar hydrogen production: Challenges and opportunities. Appl. Mater. 2019, 7, 080901. [Google Scholar] [CrossRef]
- Cheng, W.H.; Richter, M.H.; May, M.M.; Ohlmann, J.; Lackner, D.; Dimroth, F.; Hannappel, T.; Atwater, H.A.; Lewerenz, H.J. Monolithic photoelectrochemical device for direct water splitting with 19% efficiency. ACS Energy Lett. 2018, 3, 1795–1800. [Google Scholar] [CrossRef]
- Adán-Más, A.; Wei, D. Photoelectrochemical properties of graphene and its derivatives. Nanomaterials 2013, 3, 325–356. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, K.; Reddy, J.K.; Phanikrishna Sharma, M.V.; Kumari, V.D.; Subrahmanyam, M. Continuous hydrogen production activity over finely dispersed Ag2O/TiO2 catalysts from methanol: water mixtures under solar irradiation: A structure-activity correlation. Int. J. Hydrog. Energy 2010, 35, 3991–4001. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Baum, G.N.; Perez, J.; Baum, K.N. Technoeconomic Analysis of Photoelectrochemical (PEC) Hydrogen Production; Office of Energy Efficiency and Renewable Energy: Washington, DC, USA, 2009.
- Zhang, J.; Zhang, L.; Liu, H.; Sun, A.; Liu, R.S. Electrochemical Technologies for Energy Storage and Conversion; Wiley: Hoboken, NY, USA, 2012. [Google Scholar]
- Chen, Q.; Fan, G.; Fu, H.; Li, Z.; Zou, Z. Tandem photoelectrochemical cells for solar water splitting. Adv. Phys. X 2018, 3, 863–884. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I. A review and evaluation of photoelectrode coating materials and methods for photoelectrochemical hydrogen production. Int. J. Hydrog. Energy 2016, 41, 7950–7959. [Google Scholar] [CrossRef]
- Choudhary, S.; Upadhyay, S.; Kumar, P.; Singh, N.; Satsangi, V.R.; Shrivastav, R.; Dass, S. Nanostructured bilayered thin films in photoelectrochemical water splitting—A review. Int. J. Hydrog. Energy 2012, 37, 18713–18730. [Google Scholar] [CrossRef]
- Miller, E.L.; Gaillard, N.; Kaneshiro, J.; DeAngelis, A.; Garland, R. Progress in new semiconductor materials classes for solar photoelectrolysis. Int. J. Energy Res. 2010, 34, 1215–1222. [Google Scholar] [CrossRef]
- Gan, J.; Lu, X.; Rajeeva, B.B.; Menz, R.; Tong, Y.; Zheng, Y. Efficient photoelectrochemical water oxidation over hydrogen-reduced nanoporous BiVO4 with Ni-B electrocatalyst. ChemElectroChem 2015, 2, 1385–1395. [Google Scholar] [CrossRef]
- Mishra, M.; Chun, D.M. α-Fe2O3 as a photocatalytic material: A review. Appl. Catal. A Gen. 2015, 498, 126–141. [Google Scholar] [CrossRef]
- Khaselev, O.; Turner, J.A. A monolithic photovoltaic-photoelectrochemical device for hydrogen production via water splitting. Science 1998, 280, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wu, J.; Moniz, S.J.A.; Guo, D.; Tang, M.; Jiang, Q.; Chen, S.; Liu, H.; Wang, A.; Zhang, T.; et al. Stabilization of GaAs photoanodes by in situ deposition of nickel-borate surface catalysts as hole trapping sites. Sustain. Energy Fuels 2019, 3, 814–822. [Google Scholar] [CrossRef]
- Fang, M.; Dong, G.; Wei, R.; Ho, J.C. Hierarchical nanostructures: Design for sustainable water splitting. Adv. Energy Mater. 2017, 7, 1–25. [Google Scholar]
- Joy, J.; Mathew, J.; George, S.C. Nanomaterials for photoelectrochemical water splitting—Review. Int. J. Hydrog. Energy 2018, 43, 4804–4817. [Google Scholar] [CrossRef]
- Zeng, Z.; Chen, S.; Tan, T.T.Y.; Xiao, F.X. Graphene quantum dots (GQDs) and its derivatives for multifarious photocatalysis and photoelectrocatalysis. Catal. Today 2018, 315, 171–183. [Google Scholar] [CrossRef]
- Kandi, D.; Martha, S.; Parida, K.M. Quantum dots as enhancer in photocatalytic hydrogen evolution: A review. Int. J. Hydrog. Energy 2017, 42, 9467–9481. [Google Scholar] [CrossRef]
- Ye, M.Y.; Zhao, Z.H.; Hu, Z.F.; Liu, L.Q.; Ji, H.M.; Shen, Z.R.; Ma, T.Y. 0D/2D heterojunctions of vanadate quantum dots/graphitic carbon nitride nanosheets for enhanced visible-light-driven photocatalysis. Angew. Chem. Int. Ed. 2017, 56, 8407–8411. [Google Scholar] [CrossRef]
- Lou, Y.; Chen, J. Recent developments in one dimensional (1D) nanostructured TiO2 for photoelectrochemical water splitting. Nanosci. Nanotechnol. Lett. 2014, 6, 361–371. [Google Scholar] [CrossRef]
- Zheng, G.; Wang, J.; Liu, H.; Murugadoss, V.; Zu, G.; Che, H.; Lai, C.; Li, H.; Ding, T.; Gao, Q.; et al. Tungsten oxide nanostructures and nanocomposites for photoelectrochemical water splitting. Nanoscale 2019, 11, 18968–18994. [Google Scholar] [CrossRef]
- Babu, V.J.; Vempati, S.; Uyar, T.; Ramakrishna, S. Review of one-dimensional and two-dimensional nanostructured materials for hydrogen generation. Phys. Chem. Chem. Phys. 2015, 17, 2960–2986. [Google Scholar] [CrossRef]
- Chen, J.; Guo, L. Size effect of one-dimensional nanostructures on bubble nucleation in water splitting. Appl. Phys. Lett. 2019, 115, 101602. [Google Scholar] [CrossRef]
- Faraji, M.; Yousefi, M.; Yousefzadeh, S.; Zirak, M.; Naseri, N.; Jeon, T.H.; Choi, W.; Moshfegh, A.Z. Two-dimensional materials in semiconductor photoelectrocatalytic systems for water splitting. Energy Environ. Sci. 2019, 12, 59–95. [Google Scholar] [CrossRef]
- Kusior, A.; Wnuk, A.; Trenczek-Zajac, A.; Zakrzewska, K.; Radecka, M. TiO2 nanostructures for photoelectrochemical cells (PECs). Int. J. Hydrog. Energy 2015, 40, 4936–4944. [Google Scholar] [CrossRef]
- Cai, M.; Fan, P.; Long, J.; Han, J.; Lin, Y.; Zhang, H.; Zhong, M. Large-scale tunable 3D self-supporting WO3 micro-nano architectures as direct photoanodes for efficient photoelectrochemical water splitting. ACS Appl. Mater. Interfaces 2017, 9, 17856–17864. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Riboni, F.; Karlicky, F.; Kment, S.; Goswami, A.; Sudhagar, P.; Yoo, J.; Wang, L.; Tomanec, O.; Petr, M.; et al. α-Fe2O3/TiO2 3D hierarchical nanostructures for enhanced photoelectrochemical water splitting. Nanoscale 2017, 9, 134–142. [Google Scholar] [CrossRef]
- Wu, F.; Cao, F.; Liu, Q.; Lu, H.; Li, L. Enhancing photoelectrochemical activity with three-dimensional p-CuO/n-ZnO junction photocathodes. Sci. China Mater. 2016, 59, 825–832. [Google Scholar] [CrossRef]
- Yan, H.; Wang, X.; Yao, M.; Yao, X. Band structure design of semiconductors for enhanced photocatalytic activity: The case of TiO2. Prog. Nat. Sci. Mater. Int. 2013, 23, 402–407. [Google Scholar] [CrossRef]
- Park, H.S.; Kweon, K.E.; Ye, H.; Paek, E.; Hwang, G.S.; Bard, A.J. Factors in the metal doping of BiVO4 for improved photoelectrocatalytic activity as studied by scanning electrochemical microscopy and first-principles density-functional calculation. J. Phys. Chem. C 2011, 115, 17870–17879. [Google Scholar] [CrossRef]
- Yousefi, M.; Alimard, P. Synthesis of M-Nd doped Fe3O4 nanoparticles (M=Co,Ce,Cr, Ni) with tunable magnetic properties. Bull. Chem. Soc. Ethiop. 2013, 27, 49–56. [Google Scholar]
- Ikeda, S.; Kawaguchi, T.; Higuchi, Y.; Kawasaki, N.; Harada, T.; Remeika, M.; Islam, M.M.; Sakurai, T. Effects of zirconium doping into a monoclinic scheelite BiVO4 crystal on its structural, photocatalytic, and photoelectrochemical properties. Front. Chem. 2018, 6, 266. [Google Scholar] [CrossRef]
- Dass, S.; Chaudhary, Y.S.; AgjrawaP, M.; Shrivastav, A.; Shhvastav, R.; Saisangi-, V.R. Nanostructured Mn-doped and undoped CuO thin films-PEC studies. Indian J. Phys 2004, 78, 229–231. [Google Scholar]
- Xu, H.M.; Wang, H.; Shi, J.; Lin, Y.; Nan, C. Photoelectrochemical performance observed in Mn-doped BiFeO3 heterostructured thin films. Nanomaterials 2016, 6, 215. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Sheu, J.K.; Lin, Y.-C.; Tu, S.J.; Huang, F.W.; Lee, M.L.; Lai, W.C. Mn-doped GaN as photoelectrodes for the photoelectrolysis of water under visible light. Opt. Express 2012, 20, A678–A683. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, Z.; Chen, H.; Bai, Y.; Xiao, S.; Zheng, X.; Xue, Q.; Yang, S. Iron-doping-enhanced photoelectrochemical water splitting performance of nanostructured WO3: A combined experimental and theoretical study. Nanoscale 2015, 7, 2933–2940. [Google Scholar] [CrossRef]
- Kong, D.; Qi, J.; Liu, D.; Zhang, X.; Pan, L.; Zou, J. Ni-doped BiVO4 with V4+ species and oxygen vacancies for efficient photoelectrochemical water splitting. Trans. Tianjin Univ. 2019, 25, 340–347. [Google Scholar] [CrossRef]
- Jakub, Z.; Hulva, J.; Mirabella, F.; Kraushofer, F.; Meier, M.; Bliem, R.; Diebold, U.; Parkinson, G.S. Nickel doping enhances the reactivity of Fe3O4(001) to water. J. Phys. Chem.C 2019, 123, 15038–15045. [Google Scholar] [CrossRef]
- Tayebi, M.; Tayyebi, A.; Lee, B.K. Improved photoelectrochemical performance of molybdenum (Mo)-doped monoclinic bismuth vanadate with increasing donor concentration. Catal. Today 2019, 328, 35–42. [Google Scholar] [CrossRef]
- Kalanur, S.S.; Seo, H. Influence of molybdenum doping on the structural, optical and electronic properties of WO3 for improved solar water splitting. J. Colloid Interface Sci. 2018, 509, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tafen, D.N.; Lewis, J.P.; Hong, Z.; Manivannan, A.; Zhi, M.; Li, M.; Wu, N. Origin of photocatalytic activity of nitrogen-doped TiO2 nanobelts. J. Am. Chem. Soc. 2009, 131, 12290–12297. [Google Scholar] [CrossRef] [PubMed]
- Tayyebi, A.; Soltani, T.; Lee, B.K.; Outokesh, M.; Tayebi, M. Novel visible light photocatalytic and photoelectrochemical (PEC) activity of carbon-doped zinc oxide/reduced graphene oxide: Supercritical methanol synthesis with enhanced photocorrosion suppression. J. Alloys Compd. 2017, 723, 1001–1010. [Google Scholar] [CrossRef]
- Khan, A.; Ahmed, M.I.; Adam, A.; Azad, A.M.; Qamar, M. A novel fabrication methodology for sulfur-doped ZnO nanorods as an active photoanode for improved water oxidation in visible-light regime. Nanotechnology 2017, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Fang, Y.; Chen, T.; Qu, F.-L.; Liu, Z.; Du, G.; Asiri, A.M.; Gao, T.; Sun, X. Enhanced photoelectrochemical water oxidation performance of Fe2O3 nanorods array by S doping. ACS Sustain. Chem. Eng. 2017, 5, 7502–7506. [Google Scholar] [CrossRef]
- Sharma, A.; Chakraborty, M.; Anwar, Z.; Sahoo, P.; Thangavel, R. Structural, Optical and Photoelectrochemical Properties of Trivalent Impurity (B3+) Doped ZnO Nanorods Grown by Facile Hydrothermal Technique. In Proceedings of the International Conference on Energy, Communication, Data Analytics and Soft Computing, ICECDS 2017, Chennai, India, 1–2 August 2017; pp. 2491–2493. [Google Scholar]
- Kang, X.; Liu, S.; Dai, Z.; He, Y.; Song, X.; Tan, Z. Titanium Dioxide: From Engineering to Applications. Catalysts 2019, 9, 191. [Google Scholar] [CrossRef]
- Mao, S.S.; Shen, S.; Guo, L. Nanomaterials for renewable hydrogen production, storage and utilization. Prog. Nat. Sci. Mater. Int. 2012, 22, 522–534. [Google Scholar] [CrossRef]
- Wang, G.; Ling, Y.; Wang, H.; Xihong, L.; Li, Y. Chemically modified nanostructures for photoelectrochemical water splitting. J. Photochem. Photobiol. C Photochem. Rev. 2014, 18, 35–51. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Kang, Z. 3D branched ZnO nanowire arrays decorated with plasmonic Au nanoparticles for high-performance photoelectrochemical water splitting. Acs Appl. Mater. Interfaces 2014, 6, 4480–4489. [Google Scholar] [CrossRef]
- Wu, N. Plasmonic metal-semiconductor photocatalysts and photoelectrochemical cells: A review. Nanoscale 2018, 10, 2679–2696. [Google Scholar] [CrossRef]
- Xiao, F.X.; Liu, B. Plasmon-dictated photo-electrochemical water splitting for solar-to-chemical energy conversion: Current status and future perspectives. Adv. Mater. Interfaces 2018, 5, 1–21. [Google Scholar] [CrossRef]
- Yu, Z.; Li, F.; Sun, L. Recent advances in dye-sensitized photoelectrochemical cells for solar hydrogen production based on molecular components. Energy Environ. Sci. 2015, 8, 760–775. [Google Scholar] [CrossRef]
- Li, F.; Fan, K.; Xu, B.; Gabrielsson, E.; Daniel, Q.; Li, L.; Sun, L. Organic dye-sensitized tandem photoelectrochemical cell for light driven total water splitting. J. Am. Chem. Soc. 2015, 137, 9153–9159. [Google Scholar] [CrossRef]
- Swierk, J.R.; Mallouk, T.E. Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem. Soc. Rev. 2013, 42, 2357–2387. [Google Scholar] [CrossRef] [PubMed]
- Swierk, J.R.; Méndez-Hernández, D.D.; McCool, N.S.; Liddell, P.; Terazono, Y.; Pahk, I.; Tomlin, J.J.; Oster, N.V.; Moore, T.A.; Moore, A.L.; et al. Metal-free organic sensitizers for use in water-splitting dye-sensitized photoelectrochemical cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, J.W.; Lee, S.H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, Y.; Ding, X.; Yu, Z.; Sun, L. High-performance photoelectrochemical cells based on a binuclear ruthenium catalyst for visible-light-driven water oxidation. ChemSusChem 2014, 7, 2801–2804. [Google Scholar] [CrossRef]
- Li, X.; Liu, A.; Chu, D.; Zhang, C.; Du, Y.; Huang, J.; Yang, P. High performance of manganese porphyrin sensitized p-type CuFe2O4 photocathode for solar water splitting to produce hydrogen in a tandem photoelectrochemical cell. Catalysts 2018, 8, 108. [Google Scholar] [CrossRef]
- Thorne, J.E.; Zhao, Y.; He, D.; Fan, S.; Vanka, S.; Mi, Z.; Wang, D. Understanding the role of co-catalysts on silicon photocathodes using intensity modulated photocurrent spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 29653–29659. [Google Scholar] [CrossRef]
- Li, D.; Shi, J.; Li, C. Transition-metal-based electrocatalysts as cocatalysts for photoelectrochemical water splitting: A mini review. Small 2018, 14, 1–22. [Google Scholar] [CrossRef]
- Xu, X.-T.; Pan, L.; Zhang, X.; Wang, L.; Zou, J.-J. Rational design and construction of cocatalysts for semiconductor-based photo-electrochemical oxygen evolution: A comprehensive review. Adv. Sci. 2019, 6, 1801505. [Google Scholar] [CrossRef]
- Song, J.T.; Iwasaki, T.; Hatano, M. Pt co-catalyst effect on photoelectrochemical properties of 3C-SiC photo-anode. Jpn. J. Appl. Phys. 2014, 53, 2–6. [Google Scholar] [CrossRef]
- Haschke, S.; Zhuo, Y.; Schlicht, S.; Barr, M.K.S.; Kloth, R.; Dufond, M.E.; Santinacci, L.; Bachmann, J. Enhanced oxygen evolution reaction activity of nanoporous SnO2/Fe2O3/IrO2 thin film composite electrodes with ultralow noble metal loading. Adv. Mater. Interfaces 2019, 6, 1–8. [Google Scholar]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; An, H.J.; Kim, T.-W.; Lee, K.-Y.; Kim, H.J.; Kim, Y.; Chae, H.-J. Effect of structure-controlled ruthenium oxide by nanocasting in electrocatalytic oxygen and chlorine evolution reactions in acidic conditions. Catalysts 2019, 9, 549. [Google Scholar] [CrossRef]
- Peerakiatkhajohn, P.; Yun, J.-H.; Wang, S.; Wang, L. Review of recent progress in unassisted photoelectrochemical water splitting: From material modification to configuration design. J. Photonics Energy 2016, 7, 1–21. [Google Scholar] [CrossRef]
- Dong, G.; Hu, H.; Huang, X.; Zhang, Y.; Bi, Y. Rapid activation of Co3O4 cocatalysts with oxygen vacancies on TiO2 photoanodes for efficient water splitting. J. Mater. Chem. A 2018, 6, 21003–21009. [Google Scholar] [CrossRef]
- Wan, X.; Su, J.; Guo, L. Enhanced photoelectrochemical water oxidation on BiVO4 with mesoporous cobalt nitride sheets as oxygen-evolution cocatalysts. Eur. J. Inorg. Chem. 2018, 2018, 2557–2563. [Google Scholar] [CrossRef]
- Zhang, W.; Li, R.; Zhao, X.; Chen, Z.; Law, A.W.K.; Zhou, K. A cobalt-based metal–organic framework as cocatalyst on BiVO4 photoanode for enhanced photoelectrochemical water oxidation. ChemSusChem 2018, 11, 2710–2716. [Google Scholar] [CrossRef]
- Dang, K.; Wang, T.; Li, C.; Zhang, J.; Liu, S.; Gong, J. Improved oxygen evolution kinetics and surface states passivation of Ni-Bi co-catalyst for a hematite photoanode. Engineering 2017, 3, 285–289. [Google Scholar] [CrossRef]
- Xu, D.; Fu, Z.; Wang, D.; Lin, Y.; Sun, Y.; Meng, D.; Feng Xie, T. A Ni(OH)2-modified Ti-doped α-Fe2O3 photoanode for improved photoelectrochemical oxidation of urea: The role of Ni(OH)2 as a cocatalyst. Phys. Chem. Chem. Phys. 2015, 17, 23924–23930. [Google Scholar] [CrossRef]
- Rosser, T.E.; Gross, M.A.; Lai, Y.H.; Reisner, E. Precious-metal free photoelectrochemical water splitting with immobilised molecular Ni and Fe redox catalysts. Chem. Sci. 2016, 7, 4024–4035. [Google Scholar] [CrossRef]
- Guan, H.; Zhang, S.; Cai, X.; Gao, Q.; Yu, X.; Zhou, X.; Peng, F.; Fang, Y.; Yang, S. CdS@Ni3S2 core-shell nanorod arrays on nickel foam: A multifunctional catalyst for efficient electrochemical catalytic, photoelectrochemical and photocatalytic H2 production reaction. J. Mater. Chem. A 2019, 7, 2560–2574. [Google Scholar] [CrossRef]
- Fan, R.; Cheng, S.; Huang, G.; Wang, Y.; Zhang, Y.; Vanka, S.; Botton, G.A.; Mi, Z.; Shen, M. Unassisted solar water splitting with 9.8% efficiency and over 100 h stability based on Si solar cells and photoelectrodes catalyzed by bifunctional Ni-Mo/Ni. J. Mater. Chem. A 2019, 7, 2200–2209. [Google Scholar] [CrossRef]
- Van de Krol, R.; Grätzel, M. Photoelectrochemical Hydrogen Production; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Young, J.L.; Steiner, M.A.; Döscher, H.; France, R.M.; Turner, J.A.; Deutsch, T.G. Direct solar-to-hydrogen conversion via inverted metamorphic multi-junction semiconductor architectures. Nat. Energy 2017, 2, 17028. [Google Scholar] [CrossRef]
- Rocheleau, R.E.; Miller, E.L.; Misra, A. High-efficiency photoelectrochemical hydrogen production using multijunction amorphous silicon photoelectrodes. Energy Fuels 1998, 12, 3–10. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Mukerji, S.; Soga, T.; Umeno, M.; Tributsch, H. Over 18% solar energy conversion to generation of hydrogen fuel; theory and experiment for efficient solar water splitting. Int. J. Hydrog. Energy 2001, 26, 653–659. [Google Scholar] [CrossRef]
- Fountaine, K.T.; Lewerenz, H.J.; Atwater, H.A. Efficiency limits for photoelectrochemical water-splitting. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Ipek, B.; Uner, D. Artificial Photosynthesis from a Chemical Engineering Perspective. In Artificial Photosynthesis; IntechOpen: London, UK, 2012; pp. 13–36. [Google Scholar]
- Arifin, K.; Majlan, E.H.; Wan Daud, W.R.; Kassim, M.B. Bimetallic complexes in artificial photosynthesis for hydrogen production: A review. Int. J. Hydrog. Energy 2012, 37, 3066–3087. [Google Scholar] [CrossRef]
- Bard, A.J. Photoelectrochemistry and heterogeneous photo-catalysis at semiconductors. J. Photochem. 1979, 10, 59–75. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Domen, K. Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 2019, 48, 2109–2125. [Google Scholar] [CrossRef]
- Wang, Y.; Suzuki, H.; Xie, J.; Tomita, O.; Martin, D.J.; Higashi, M.; Kong, D.; Abe, R.; Tang, J. Mimicking natural photosynthesis: Solar to renewable H2 fuel synthesis by z-scheme water splitting systems. Chem. Rev. 2018, 118, 5201–5241. [Google Scholar] [CrossRef]
- Bard, A.J.; Fox, M.A. Artificial photosynthesis: Solar splitting of water to hydrogen and oxygen. Acc. Chem. Res. 1995, 28, 141–145. [Google Scholar] [CrossRef]
- Kato, H.; Sasaki, Y.; Shirakura, N.; Kudo, A. Synthesis of highly active rhodium-doped SrTiO3 powders in Z-scheme systems for visible-light-driven photocatalytic overall water splitting. J. Mater. Chem. A 2013, 1, 12327–12333. [Google Scholar] [CrossRef]
- Kato, H.; Hori, M.; Konta, R.; Shimodaira, Y.; Kudo, A. Construction of Z-Scheme type heterogeneous photocatalysis systems for water splitting into H2 and O2 under visible light irradiation. Chem. Lett. 2004, 33, 1348–1349. [Google Scholar] [CrossRef]
- Sasaki, Y.; Iwase, A.; Kato, H.; Kudo, A. The effect of co-catalyst for z-scheme photocatalysis systems with an Fe3+/Fe2+ electron mediator on overall water splitting under visible light irradiation. J. Catal. 2008, 259, 133–137. [Google Scholar] [CrossRef]
- Sayama, K.; Mukasa, K.; Abe, R.; Hironori, A. A new photocatalytic water splitting system under visible light irradiation mimicking a z-scheme mechanism in photosynthesis. J. Photochem. Photobiol. A Chem. 2002, 148, 71–77. [Google Scholar] [CrossRef]
- Sekizawa, K.; Maeda, K.; Domen, K.; Koike, K.; Ishitani, O. Artificial Z-scheme constructed with a supramolecular metal complex and semiconductor for the photocatalytic reduction of CO2. J. Am. Chem. Soc. 2013, 135, 4596–4599. [Google Scholar] [CrossRef]
- Wang, J.; Shen, H.; Dai, X.; Li, C.; Shi, W.; Yan, Y. Graphene oxide as solid-state electron mediator enhanced photocatalytic activities of GO-Ag3PO4/Bi2O3 Z-scheme photocatalyst efficiently by visible-light driven. Mater. Technol. 2018, 33, 421–432. [Google Scholar] [CrossRef]
- Iwase, A.; Ng, Y.H.; Ishiguro, Y.; Kudo, A.; Amal, R. Reduced graphene oxide as a solid-state electron mediator in Z-scheme photocatalytic water splitting under visible light. J. Am. Chem. Soc. 2011, 133, 11054–11057. [Google Scholar] [CrossRef]
- Xia, X.; Song, M.; Wang, H.; Zhang, X.; Sui, N.; Zhang, Q.; Colvin, V.L.; Yu, W.W. Latest progress in constructing solid-state Z scheme photocatalysts for water splitting. Nanoscale 2019, 11, 11071–11082. [Google Scholar] [CrossRef]
- Winkler, M.; Kawelke, S.; Happe, T. Light driven hydrogen production in protein based semi-artificial systems. Bioresour. Technol. 2011, 102, 8493–8500. [Google Scholar] [CrossRef]
- Kornienko, N.; Zhang, J.Z.; Sakimoto, K.K.; Yang, P.; Reisner, E. Interfacing nature’s catalytic machinery with synthetic materials for semi-artificial photosynthesis. Nat. Nanotechnol. 2018, 13, 890–899. [Google Scholar] [CrossRef]
- Lee, C.Y.; Zou, J.; Bullock, J.; Wallace, G.G. Emerging approach in semiconductor photocatalysis: Towards 3D architectures for efficient solar fuels generation in semi-artificial photosynthetic systems. J. Photochem. Photobiol. C Photochem. Rev. 2019, 39, 142–160. [Google Scholar] [CrossRef]
- Honda, Y.; Hagiwara, H.; Ida, S.; Ishihara, T. Application to photocatalytic H2 production of a whole-cell reaction by recombinant Escherichia coli cells expressing [FeFe]-hydrogenase and maturases genes. Angew. Chem. Int. Ed. 2016, 55, 8045–8048. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511–518. [Google Scholar] [CrossRef]
- IEA Hydrogen Project Database. Available online: ieahydrogen.org/Activities/National-Projects-%281%29/Netherlands/NL-Power-Plant-Delfzijl.aspx (accessed on 1 December 2019).
- Krogsgaard, J.; Løkke, J.A. Regarding Potential Large Scale Energy Storage Project in Fredericia, Denmark; NEL: Oslo, Norway, 2017. [Google Scholar]
- Ipek, B.; Uner, D. Construction begins on Fukushima hydrogen energy research Fieldfuel. Cells Bull. 2018, 2018, 9. [Google Scholar]
- H2V. Projet H2V59-Usine de Production D’hydrogène Vert Dans les Hauts-de-France et Son Raccordement Électrique; H2V: Paris, France, 2019. [Google Scholar]
- Marcuello, P.; Embid, D.; Albertín, E.; García, G. Improvements to integrate high pressure alkaline electrolyzers for electricity/H2 production from renewable energies to balance the GRID—ELYGRID project. In Proceedings of the 20th World Hydrogen Energy Conference, WHEC 2014, Gwangju, Korea, 15–20 June 2014; pp. 13–18. [Google Scholar]
- Kolb, T.; Kleinschmidt, R.; Freytag, K. Windgas for the Energy Transition: Uniper Lays Cornerstone for Methanation Plant 2017. Available online: https://www.storeandgo.info/fileadmin/press_releases/20160705_PM_Grundstein_Falkenhagen_StoreGo_ENG.pdf (accessed on 1 December 2019).
- Arnone, D.; Arcaya, A.D.; Caldarola, C.G.; Guenoux, L.; Rossi, A.; Vaes, J. INGRID Project-High-Capacity Hydrogen-Based Green-Energy Storage Solutions for Grid Balancing; Deutscher Industrieverlag GmbH: Munich, Germany, 2016. [Google Scholar]
- Skøtt, A.T. Bakterier Kan Lave Brint og Kuldioxid om til Metangas 2016. Available online: http://biocat-project.com/wp-content/uploads/2016/09/Article-from-grand-opening.pdf (accessed on 1 December 2019).
- Hassl, M. Wasserstoff-Initiative bei MPREIS in Völs. 2019. Available online: https://www.green-energy-center.com/wp-content/uploads/2019/10/19-10-19-BEZIRKSBLATT-Wasserstoff-Initiative-bei-MPREIS-in-Völs-Westliches-Mittelgebirge.pdf (accessed on 1 December 2019).
- ZePros News. RTE et GRTgaz Accélèrent le Développement de Solutions Multi-Énergies Pour Les Territoires. 2017. Available online: http://www.grtgaz.com/medias/communiques-de-presse/rte-et-grtgaz-accelerent-le-developpement-de-solutions-multi-energies-pour-les-territoires.html (accessed on 1 December 2019).
- Power-To-Gas: McPhy delivers 4 MW of H2 Production Units to China’s Hebei Province and Strengthens its Position for Multi-MW International Projects. Available online: https://mcphy.com/en/press-releases/power_to_gas_mcphy_hebei_delivery/ (accessed on 1 December 2019).
- Hydrogenics selected for 2 MW Ontario energy storage facility. Fuel Cells Bull. 2014, 8, 9. [CrossRef]
- Canadian multi-MW P2G facility enters service. Fuel Cells Bull. 2018, 7, 1. [CrossRef]
- Hydrogenics links with StratosFuel on 2.5 MW California project. Fuel Cells Bull. 2016, 11, 7–8. [CrossRef]
- Liu, Z.; Kendall, K.; Yan, X. China progress on renewable energy vehicles: Fuel cells, hydrogen and battery hybrid vehicles. Energies 2019, 12, 54. [Google Scholar] [CrossRef]
- Kopp, M.; Coleman, D.; Stiller, C.; Scheffer, K.; Aichinger, J.; Scheppat, B. Energiepark Mainz: Technical and economic analysis of the worldwide largest Power-to-Gas plant with PEM electrolysis. Int. J. Hydrog. Energy 2017, 42, 13311–13320. [Google Scholar] [CrossRef]
- Sampson, J. H&R Inaugurates World’s Largest Dynamic Hydrogen Electrolysis Plant. Available online: https://www.gasworld.com/handr-inaugurates-hydrogen-electrolysis-plant-/2013848.article (accessed on 1 December 2019).
- Petersen, N.H.; Geitmann, S. H2 International Wind Power Boosts Hydrogen Transportation. Available online: https://www.h2-international.com/2019/03/11/wind-power-boosts-hydrogen-transportation/ (accessed on 1 December 2019).
- Voestalpine, A.G.; VERBUND. Siemens European Commission Funds H2FUTURE Project. 2017. Available online: https://www.h2future-project.eu/images/news/H2FUTURE-Press-Release-070217.pdf (accessed on 1 December 2019).
- Zeese, J. Launch of Refhyne, Word’s Largest Electrolysis Plant in Rhineland Refinery. Available online: https://www.fch.europa.eu/news/launch-refhyne-worlds-largest-electrolysis-plant-rhineland-refinery (accessed on 1 December 2019).
- Bellini, E. ITM Power Plans 100 MW Power-To-Gas Storage Project in the UK. Available online: https://www.pv-magazine.com/2018/10/01/itm-power-plans-100-mw-power-to-gas-storage-project-in-the-uk/ (accessed on 1 December 2019).
- Tichler, R.; Lehner, M.; Steinmüller, H.; Koppe, M. Power-To-Gas: Technology and Business Models; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Sunfire supplies Boeing with largest reversible solid oxide electrolyser/fuel cell System. Cells Bull. 2016, 2016, 1. [CrossRef]
- GrinHy project demos high-temp Electrolysis. fuel Cells Bull. 2016, 2016, 10. [CrossRef]
- First Commercial Plant for the Production of Blue Crude Planned in Norway. Available online: https://www.sunfire.de/en/company/news/detail/first-commercial-plant-for-the-production-of-blue-crude-planned-in-norway (accessed on 1 December 2019).
- Kim, K.N.; Lee, S.H.; Kim, H.; Park, Y.H.; In, S. Improved microbial electrolysis cell hydrogen production by hybridization with a TiO2 nanotube array photoanode. Energies 2018, 11, 3184. [Google Scholar] [CrossRef]
- Escapa, A.; Mateos, R.; Martínez, E.J.; Blanes, J. Microbial electrolysis cells: An emerging technology for wastewater treatment and energy recovery. From laboratory to pilot plant and beyond. Renew. Sustain. Energy Rev. 2016, 55, 942–956. [Google Scholar] [CrossRef]
- Rader, G.K.; Logan, B.E. Multi-electrode continuous flow microbial electrolysis cell for biogas production from acetate. Int. J. Hydrog. Energy 2010, 35, 8848–8854. [Google Scholar] [CrossRef]
- Cusick, R.D.; Bryan, B.; Parker, D.S.; Merrill, M.D.; Mehanna, M.; Kiely, P.D.; Liu, G.; Logan, B.E. Performance of a pilot-scale continuous flow microbial electrolysis cell fed winery wastewater. Appl. Microbiol. Biotechnol. 2011, 89, 2053–2063. [Google Scholar] [CrossRef]
- Heidrich, E.S.; Dolfing, J.; Scott, K.; Edwards, S.R.; Jones, C.; Curtis, T.P. Production of hydrogen from domestic wastewater in a pilot-scale microbial electrolysis cell. Appl. Microbiol. Biotechnol. 2013, 97, 6979–6989. [Google Scholar] [CrossRef]
- Heidrich, E.S.; Edwards, S.R.; Dolfing, J.; Cotterill, S.E.; Curtis, T.P. Performance of a pilot scale microbial electrolysis cell fed on domestic wastewater at ambient temperatures for a 12 month period. Bioresour. Technol. 2014, 173, 87–95. [Google Scholar] [CrossRef]
- Cotterill, S.E.; Dolfing, J.; Curtis, T.P.; Heidrich, E.S. Community assembly in wastewater-fed pilot-scale microbial electrolysis cells. Front. Energy Res. 2018, 6, 98. [Google Scholar] [CrossRef]
- Miller, E.L.; Garland, R.; Peterson, D. Photoelectrochemical Hydrogen Production: DOE PEC Working Group Overview; Hawaii Natural Energy Institute University of Hawaii at Manoa: Honolulu, HI, USA, 2011. [Google Scholar]
- Miller, E.L. IEA-HIA Task 26 Research and Development Progress in Renewable Hydrogen Production Through Photoelectrochemical Water Splitting. Energy Procedia 2012, 29, 438–444. [Google Scholar] [CrossRef]
- Rothschild, A.; Dotan, H. Beating the efficiency of photovoltaics-powered electrolysis with tandem cell photoelectrolysis. ACS Energy Lett. 2017, 2, 45–51. [Google Scholar] [CrossRef]
- Abdi, F.F.; Jang, J.; Ma, Y.; Ahmet, I.; van de Krol, R.; Stannowski, B.; Mayer, M.; Son, M.-K.; Rothschild, A.; Dotan, H.; et al. Photoelectrochemical Demonstrator Device for Solar Hydrogen Generation; Helmholtz-Zentrum Berlin: Berlin, Germany, 2013. [Google Scholar]
- Van De Krol, R. Project Final Report. 2016. Available online: https://cordis.europa.eu/project/id/621252/reporting (accessed on 1 December 2019).
- Goto, Y.; Hisatomi, T.; Wang, Q.; Higashi, T.; Ishikiriyama, K.; Maeda, T.; Sakata, Y.; Okunaka, S.; Tokudome, H.; Katayama, M.; et al. A particulate photocatalyst water-splitting panel for large-scale solar hydrogen generation. Joule 2018, 2, 509–520. [Google Scholar] [CrossRef]
- Yamada, T.; Domen, K. Development of sunlight driven water splitting devices towards future artificial photosynthetic industry. Chem. Eng. 2018, 2, 36. [Google Scholar] [CrossRef]
- Saladini, F.; Patrizi, N.; Pulselli, F.M.; Marchettini, N.; Bastianoni, S. Guidelines for emergy evaluation of first, second and third generation biofuels. Renew. Sustain. Energy Rev. 2016, 66, 221–227. [Google Scholar] [CrossRef]
- Johnson, E. Goodbye to carbon neutral: Getting biomass footprints right. Environ. Impact Assess. Rev. 2009, 29, 165–168. [Google Scholar] [CrossRef]
- Demirbas, A. Biofuels sources, biofuel policy, biofuel economy and global biofuel projections. Energy Convers. Manag. 2008, 49, 2106–2116. [Google Scholar] [CrossRef]
- Buceti, G. Sustainable power density in electricity generation. Manag. Environ. Qual. Int. J. 2014, 25, 5–18. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Vassileva, C.G.; Vassilev, V.S. Advantages and disadvantages of composition and properties of biomass in comparison with coal: An overview. Fuel 2015, 158, 330–350. [Google Scholar] [CrossRef]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Production of synthetic natural gas (SNG) from coal and dry biomass - A technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Basu, P. Gasification theory and modeling of gasifiers. In Biomass Gasification Design Handbook; Elsevier: Amsterdam, The Netherlands, 2010; pp. 117–165. [Google Scholar]
- Peter, M. Energy production from biomass (part 1): Overview of biomass. Bioresour. Technol. 2001, 83, 37–46. [Google Scholar]
- Svoboda, K.; Martinec, J.; Pohořelý, M.; Baxter, D. Integration of biomass drying with combustion/gasification technologies and minimization of emissions of organic compounds. Chem. Pap. 2009, 63, 15–25. [Google Scholar] [CrossRef]
- Hernández, J.J.; Aranda-Almansa, G.; Bula, A. Gasification of biomass wastes in an entrained flow gasifier: Effect of the particle size and the residence time. Fuel Process. Technol. 2010, 91, 681–692. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Li, Z.; Xiang, Y. Syngas composition study. Front. Energy Power Eng. China 2009, 3, 369–372. [Google Scholar] [CrossRef]
- Lv, P.M.; Xiong, Z.H.; Chang, J.; Wu, C.Z.; Chen, Y.; Zhu, J.X. An experimental study on biomass air-steam gasification in a fluidized bed. Bioresour. Technol. 2004, 95, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Vuthaluru, H.B.; Zhang, D.K.; Linjewile, T.M. Behaviour of inorganic constituents and ash characteristics during fluidized-bed combustion of several Australian low-rank coals. Fuel Process. Technol. 2000, 67, 165–176. [Google Scholar] [CrossRef]
- Arvelakis, S.; Koukios, E.G. Physicochemical upgrading of agroresidues as feedstocks for energy production via thermochemical conversion methods. Biomass Bioenergy 2002, 22, 331–348. [Google Scholar] [CrossRef]
- Arvelakis, S.; Gehrmann, H.; Beckmann, M.; Koukios, E.G. Preliminary results on the ash behavior of peach stones during fluidized bed gasification: Evaluation of fractionation and leaching as pre-treatments. Biomass Bioenergy 2005, 28, 331–338. [Google Scholar] [CrossRef]
- Arvelakis, S.; Gehrmann, H.; Beckmann, M.; Koukios, E.G. Agglomeration problems during fluidized bed gasification of olive-oil residue: Evaluation of fractionation and leaching as pre-treatments. Fuel 2003, 82, 1261–1270. [Google Scholar] [CrossRef]
- Arvelakis, S.; Vourliotis, P.; Kakaras, E.; Koukios, E.G. Effect of leaching on the ash behavior of wheat straw and olive residue during fluidized bed combustion. Biomass Bioenergy 2001, 20, 459–470. [Google Scholar] [CrossRef]
- Chen, W.H.; Peng, J.; Bi, X.T. A state-of-the-art review of biomass torrefaction, densification and applications. Renew. Sustain. Energy Rev. 2015, 44, 847–866. [Google Scholar] [CrossRef]
- Svoboda, K.; Pohořelý, M.; Hartman, M.; Martinec, J. Pretreatment and feeding of biomass for pressurized entrained flow gasification. Fuel Process. Technol. 2009, 90, 629–635. [Google Scholar] [CrossRef]
- Sokhansanj, S. Cost Benefit of Biomass Supply and Pre-Processing; BIOCAP Canada Foundation: Kingston, ON, Canada, 2006. [Google Scholar]
- Axelsson, L.; Franzén, M.; Ostwald, M.; Berndes, G.; Lakshmi, G.; Ravindranath, N.H. A review of biomass densification systems to develop uniform feedstock commodities for bioenergy application. Biofuels Bioprod. Biorefin. 2012, 6, 246–256. [Google Scholar] [CrossRef]
- Jouhara, H.; Ahmad, D.; van den Boogaert, I.; Katsou, E.; Simons, S.; Spencer, N. Pyrolysis of domestic based feedstock at temperatures up to 300 °C. Therm. Sci. Eng. Prog. 2018, 5, 117–143. [Google Scholar] [CrossRef]
- Zeng, K.; Gauthier, D.; Minh, D.P.; Weiss-Hortala, E.; Nzihou, A.; Flamant, G. Characterization of solar fuels obtained from beech wood solar pyrolysis. Fuel 2017, 188, 285–293. [Google Scholar] [CrossRef]
- Widyawati, M.; Church, T.L.; Florin, N.H.; Harris, A.T. Hydrogen synthesis from biomass pyrolysis with in situ carbon dioxide capture using calcium oxide. Int. J. Hydrog. Energy 2011, 36, 4800–4813. [Google Scholar] [CrossRef]
- Basu, P. Chapter 5—Pyrolysis. In Biomass Gasification, Pyrolysis and Torrefaction, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 147–176. [Google Scholar]
- Yadav, K.; Jagadevan, S. Influence of Process Parameters on Synthesis of Biochar by Pyrolysis of Biomass: An Alternative Source of Energy; IntechOpen: London, UK, 2019. [Google Scholar]
- Zhang, Z.; Bei, H.; Li, H.; Li, X.; Gao, X. Understanding the co-pyrolysis behavior of indonesian oil sands and corn straw. Energy Fuels 2017, 31, 2538–2547. [Google Scholar] [CrossRef]
- Hu, X.; Gholizadeh, M. Biomass pyrolysis: A review of the process development and challenges from initial researches up to the commercialisation stage. J. Energy Chem. 2019, 39, 109–143. [Google Scholar] [CrossRef]
- Bridgwater, T. Review biomass for energy. J. Sci. Food Agric. 2006, 86, 1755–1768. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Gupta, A.K.; De, A.; Aggarwal, S.K.; Kushari, A.; Runchal, A. Innovations in Sustainable Energy and Cleaner Environment; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Bridgwater, A.V. The production of biofuels and renewable chemicals by fast pyrolysis of biomass. Int. J. Glob. Energy Issues 2007, 27, 160–203. [Google Scholar] [CrossRef]
- Horne, P.A.; Williams, P.T. Influence of temperature on the products from the flash pyrolysis of biomass. Fuel 1996, 75, 1051–1059. [Google Scholar] [CrossRef]
- Jahirul, M.I.; Rasul, M.G.; Chowdhury, A.A.; Ashwath, N. Biofuels production through biomass pyrolysis-A technological review. Energies 2012, 5, 4952–5001. [Google Scholar] [CrossRef]
- Onay, O.; Kockar, O.M. Slow, fast and flash pyrolysis of rapeseed. Renew. Energy 2003, 28, 2417–2433. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic biomass pyrolysis: A review of product properties and effects of pyrolysis parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Mani, T.; Murugan, P.; Abedi, J.; Mahinpey, N. Pyrolysis of wheat straw in a thermogravimetric analyzer: Effect of particle size and heating rate on devolatilization and estimation of global kinetics. Chem. Eng. Res. Des. 2010, 88, 952–958. [Google Scholar] [CrossRef]
- Garcia-Nunez, J.A.; Pelaez-Samaniego, M.R.; Garcia-Perez, M.E.; Fonts, I.; Abrego, J.; Westerhof, R.J.M.; Garcia-Perez, M. Historical developments of pyrolysis reactors: A review. Energy Fuels 2017, 31, 5751–5775. [Google Scholar] [CrossRef]
- Lewandowski, W.M.; Januszewicz, K.; Kosakowski, W. Efficiency and proportions of waste tyre pyrolysis products depending on the reactor type—A review. J. Anal. Appl. Pyrol. 2019, 140, 25–53. [Google Scholar] [CrossRef]
- Park, D.K.; Kim, S.D.; Lee, S.H.; Lee, J.G. Co-pyrolysis characteristics of sawdust and coal blend in TGA and a fixed bed reactor. Bioresour. Technol. 2010, 101, 6151–6156. [Google Scholar] [CrossRef]
- Zhuo, Y.; Zhuo, Y.; Dugwell, D.R.; Dugwell, D.R.; Kandiyoti, R.; Kandiyoti, R. Co-pyrolysis and co-gasi cation of coal and biomass in bench-scale xed- bed and uidised bed reactors. Chem. Eng. 1999, 78, 667–679. [Google Scholar]
- Onay, O.; Koçkar, O.M. Fixed-bed pyrolysis of rapeseed (Brassica napus L.). Biomass Bioenergy 2004, 26, 289–299. [Google Scholar] [CrossRef]
- Isahak, W.N.R.W.; Hisham, M.W.M.; Yarmo, M.A.; Yun Hin, T.Y. A review on bio-oil production from biomass by using pyrolysis method. Renew. Sustain. Energy Rev. 2012, 16, 5910–5923. [Google Scholar] [CrossRef]
- Xu, R.; Ferrante, L.; Briens, C.; Berruti, F. Flash pyrolysis of grape residues into biofuel in a bubbling fluid bed. J. Anal. Appl. Pyrolysis 2009, 86, 58–65. [Google Scholar] [CrossRef]
- Lee, S.H.; Eom, M.S.; Yoo, K.S.; Kim, N.C.; Jeon, J.K.; Park, Y.K.; Song, B.H.; Lee, S.H. The yields and composition of bio-oil produced from Quercus Acutissima in a bubbling fluidized bed pyrolyzer. J. Anal. Appl. Pyrolysis 2008, 83, 110–114. [Google Scholar] [CrossRef]
- Dai, X.; Yin, X.; Wu, C.; Zhang, W.; Chen, Y. Pyrolysis of waste tires in a circulating fluidized-bed reactor. Energy 2001, 26, 385–399. [Google Scholar] [CrossRef]
- Lappas, A.A.; Samolada, M.C.; Iatridis, D.K.; Voutetakis, S.S.; Vasalos, I.A. Biomass pyrolysis in a circulating fluid bed reactor for the production of fuels and chemicals. Fuel 2002, 81, 2087–2095. [Google Scholar] [CrossRef]
- Sivell, A.; Beeckmans, J.M.; Webster, A.R. Ultrarapid pyrolysis of biomass using an electrical discharge. J. Anal. Appl. Pyrolysis 1984, 7, 185–191. [Google Scholar] [CrossRef]
- Luo, G.; Eng, R.J.; Jia, P.; Resende, F.L.P. Ablative pyrolysis of wood chips: Effect of operating conditions. Energy Technol. 2017, 5, 2128–2137. [Google Scholar] [CrossRef]
- Peacocke, G.V.C.; Bridgwater, A.V. Ablative plate pyrolysis of biomass for liquids. Biomass Bioenergy 1994, 7, 147–154. [Google Scholar] [CrossRef]
- Wagenaar, B.M.; Kuipers, J.A.M.; Prins, W.; van Swaaij, W.P.M. The Rotating Cone Flash Pyrolysis Reactor. In Advances in Thermochemical Biomass Conversion; Bridgwater, A.V., Ed.; Springer: Dordrecht, The Netherlands, 1993; pp. 1122–1133. [Google Scholar]
- Wagenaar, B.M.; Prins, W.; van Swaaij, W.P.M. Pyrolysis of biomass in the rotating cone reactor: Modelling and experimental justification. Chem. Eng. Sci. 1994, 49, 5109–5126. [Google Scholar] [CrossRef]
- Wentrup, C. Flash vacuum pyrolysis: Techniques and reactions. Angew. Chem. Int. Ed. 2017, 56, 14808–14835. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, T.; Ma, L.; Chang, J. Vacuum pyrolysis of waste tires with basic additives. Waste Manag. 2008, 28, 2301–2310. [Google Scholar] [CrossRef]
- Perkins, C.; Woodruff, B.; Andrews, L. Synthesis gas production by rapid solar thermal gasification of corn stover. In Proceedings of the 14th Biennial CSP SolarPACES Symposium, Las Vegas, NV, USA, 4–7 March 2008; pp. 4–7. [Google Scholar]
- Arribas, L.; Arconada, N.; González-Fernández, C.; Löhrl, C.; González-Aguilar, J.; Kaltschmitt, M.; Romero, M. Solar-driven pyrolysis and gasification of low-grade carbonaceous materials. Int. J. Hydrog. Energy 2017, 42, 13598–13606. [Google Scholar] [CrossRef]
- Chuayboon, S.; Abanades, S.; Rodat, S. Co-production of syngas and zinc via combined solar-driven biomass gasification and ZnO carbo-thermal reduction in a continuously-operated solar reactor. In Proceedings of the AIP Conference Proceedings, Maharashtra, India, 5–6 July 2018; p. 130004. [Google Scholar]
- Nzihou, A.; Flamant, G.; Stanmore, B.; Nzihou, A.; Flamant, G.; Stanmore, B.; Nzihou, A.; Flamant, G.; Stanmore, B. Synthetic fuels from biomass using concentrated solar energy—A review. Energy 2012, 42, 121–131. [Google Scholar] [CrossRef]
- Loutzenhiser, P.G.; Muroyama, A.P. A review of the state-of-the-art in solar-driven gasification processes with carbonaceous materials. Sol. Energy 2017, 156, 93–100. [Google Scholar] [CrossRef]
- Barrio, M.; Gbel, B.; Rimes, H.; Henriksen, U.; Hustad, J.E.; Srensen, L.H. Steam gasification of wood char and the effect of hydrogen inhibition on the chemical kinetics. In Progress in Thermochemical Biomass Conversion; Blackwell Science Ltd.: Oxford, UK, 2008; pp. 32–46. [Google Scholar]
- Mckendry, P. Energy production from biomass (part 3): Gasification technologies. Bioresour. Technol. 2002, 83, 55–63. [Google Scholar] [CrossRef]
- Kumar, A.; Jones, D.D.; Hanna, M.A. Thermochemical biomass gasification: A review of the current status of the technology. Energies 2009, 2, 556–581. [Google Scholar] [CrossRef]
- Piatkowski, N.; Wieckert, C.; Weimer, A.W.; Steinfeld, A. Solar-driven gasification of carbonaceous feedstock—A review. Energy Environ. Sci. 2011, 4, 73–82. [Google Scholar] [CrossRef]
- Sansaniwal, S.K.; Pal, K.; Rosen, M.A.; Tyagi, S.K. Recent advances in the development of biomass gasification technology: A comprehensive review. Renew. Sustain Energy Rev. 2017, 72, 363–384. [Google Scholar] [CrossRef]
- Ruiz, J.A.; Juárez, M.C.; Morales, M.P.; Muñoz, P.; Mendívil, M.A. Biomass gasification for electricity generation: Review of current technology barriers. Renew. Sustain. Energy Rev. 2013, 18, 174–183. [Google Scholar] [CrossRef]
- Villarini, M.; Marcantonio, V.; Colantoni, A.; Bocci, E. Sensitivity analysis of different parameters on the performance of a CHP internal combustion engine system fed by a biomass waste gasifier. Energies 2019, 12, 688. [Google Scholar] [CrossRef]
- Guo, F.; Dong, Y.; Dong, L.; Guo, C. Effect of design and operating parameters on the gasification process of biomass in a downdraft fixed bed: An experimental study. Int. J. Hydrog. Energy 2014, 39, 5625–5633. [Google Scholar] [CrossRef]
- Ghassemi, H.; Shahsavan-markadeh, R. Effects of various operational parameters on biomass gasification process; a modified equilibrium model. Energy Convers. Manag. 2014, 79, 18–24. [Google Scholar] [CrossRef]
- Kirsanovs, V.; Žandeckis, A.; Blumberga, D.; Veidenbergs, I. The influence of process temperature, equivalence ratio and fuel moisture content on gasification process: A review. In Proceedings of the 27th International Conference on Efficiency, Cost, Optimization, Simulation and Environmental Impact of Energy Systems, ECOS 2014, Turku, Finland, 15–19 June 2014. [Google Scholar]
- Parthasarathy, P.; Narayanan, K.S. Hydrogen production from steam gasification of biomass: Influence of process parameters on hydrogen yield-A review. Renew. Energy 2014, 66, 570–579. [Google Scholar] [CrossRef]
- Hernández, J.J.; Ballesteros, R.; Aranda, G. Characterisation of tars from biomass gasification: Effect of the operating conditions. Energy 2013, 50, 333–342. [Google Scholar] [CrossRef]
- Beenackers, A.A.C.M. Biomass gasification in moving beds, a review of European technologies. Renew. Energy 1999, 16, 1180–1186. [Google Scholar] [CrossRef]
- Martínez, D.J.; Mahkamov, K.; Andrade, R.V.; Lora, E.E.S. Syngas production in downdraft biomass gasi fi ers and its application using internal combustion engines. Renew. Energy 2012, 38, 1–9. [Google Scholar] [CrossRef]
- Warnecke, R. Gasification of biomass: Comparison of fixed bed and fluidized bed gasifier. Biomass Bioenergy 2000, 18, 489–497. [Google Scholar] [CrossRef]
- Basu, P. Chapter 8—Design of Biomass Gasifiers. In Biomass Gasification, Pyrolysis and Torrefaction, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 249–313. [Google Scholar]
- Njikam, F.; Shahbazi, A.; Shirley, V.; Lee, C.; Serre, S.; Lemieux, P. Optimizing Synthesis Gas Yield from the Cross Draft Gasification of Woody Biomass. In Proceedings of the Air and Waste Management Association’s Annual Conference and Exhibition, AWMA, New Orleans, LA, USA, 20–23 June 2006. [Google Scholar]
- Rüdisüli, M.; Schildhauer, T.J.; Biollaz, S.M.A.; Van Ommen, J.R. Scale-up of bubbling fluidized bed reactors—A review. Powder Technol. 2012, 217, 21–38. [Google Scholar] [CrossRef]
- Kunii, D.; Levenspiel, O. Circulating fluidized-bed reactors. Chem. Eng. Sci. 1997, 52, 2471–2482. [Google Scholar] [CrossRef]
- Couto, N.; Rouboa, A.; Silva, V.; Monteiro, E.; Bouziane, K. Influence of the biomass gasification processes on the final composition of syngas. Energy Procedia 2013, 36, 596–606. [Google Scholar] [CrossRef]
- Corella, J.; Toledo, J.M.; Molina, G. A review on dual fluidized-bed biomass gasifiers. Ind. Eng. Chem. Res. 2007, 46, 6831–6839. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, Q.; Zhao, H.; Cao, X.; Mei, Q.; Luo, Z.; Cen, K. Biomass—oxygen gasification in a high-temperature entrained-flow gasifier. Biotechnol. Adv. 2009, 27, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Kolb, T.; Krebs, L.; Santo, U.; Seifert, H.; Kuhn, D.; Wiemer, H.-J.; Pantouflas, E.; Zarzalis, N. Conversion of biomass based slurry in an entrained flow gasifier. Chem. Eng. Technol. 2007, 30, 967–969. [Google Scholar]
- Li, F.; Li, Z.; Huang, J.; Fang, Y. Understanding mineral behaviors during anthracite fluidized-bed gasification based on slag characteristics. Appl. Energy 2014, 131, 279–287. [Google Scholar] [CrossRef]
- Milne, T.; Evans, R.J. Biomass Gasifier “Tars”: Their Nature, Formation, and Conversion; National Renewable Energy Laboratory: Golden, CO, USA, 1998.
- Kiel, J.; Van Paasen, S.; Neeft, J. Primary Measures to Reduce tar Formation in Fluidised-Bed Biomass Gasifiers; Final SDE Project P1999-012; ECN: Putten, The Netherlands, 2004. [Google Scholar]
- Woolcock, P.J.; Brown, R.C. A review of cleaning technologies for biomass-derived syngas. Biomass Bioenergy 2013, 52, 54–84. [Google Scholar] [CrossRef]
- Wang, L.; Weller, C.L.; Jones, D.D.; Hanna, M.A. Contemporary issues in thermal gasification of biomass and its application to electricity and fuel production. Biomass Bioenergy 2008, 32, 573–581. [Google Scholar] [CrossRef]
- Han, J.; Kim, H. The reduction and control technology of tar during biomass gasification/pyrolysis: An overview. Renew. Sustain. Energy Rev. 2008, 12, 397–416. [Google Scholar] [CrossRef]
- Devi, L.; Ptasinski, K.J.; Janssen, F.J.J.G. A review of the primary measures for tar elimination in biomass gasification processes. Biomass Bioenergy 2003, 24, 125–140. [Google Scholar] [CrossRef]
- Abu El-Rub, Z.; Bramer, E.A.; Brem, G. Review of catalysts for tar elimination in biomass gasification processes. Ind. Eng. Chem. Res. 2004, 43, 6911–6919. [Google Scholar] [CrossRef]
- Sutton, D.; Kelleher, B.; Ross, J.R.H. Review of literature on catalysts for biomass gasification. Fuel Process. Technol. 2001, 73, 155–173. [Google Scholar] [CrossRef]
- Rueda, Y.G.; Helsen, L. The role of plasma in syngas tar cracking. Biomass Convers. Biorefinery 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Valderrama Rios, M.L.; González, A.M.; Lora, E.E.S.; Almazán del Olmo, O.A. Reduction of tar generated during biomass gasification: A review. Biomass Bioenergy 2018, 108, 345–370. [Google Scholar] [CrossRef]
- de Diego, L.F.; García-Labiano, F.; Gayán, P.; Abad, A.; Mendiara, T.; Adánez, J.; Nacken, M.; Heidenreich, S. Tar abatement for clean syngas production during biomass gasification in a dual fluidized bed. Fuel Process. Technol. 2016, 152, 116–123. [Google Scholar] [CrossRef]
- Anis, S.; Zainal, Z.A. Tar reduction in biomass producer gas via mechanical, catalytic and thermal methods: A review. Renew. Sustain. Energy Rev. 2011, 15, 2355–2377. [Google Scholar] [CrossRef]
- IEA Bioenergy Task 33. Available online: http://www.ieabioenergytask33.org/content/taks_description (accessed on 1 December 2019).
- NREL. National Renewable Energy Laboratory Institutional Plan: 2001-2005; DIANE Publishing: Collingdale, PA, USA, 2001. [Google Scholar]
- Lemieux, A.; Lavoie, J.-M. Conversion of Non-Homogeneous Biomass to Ultraclean Syngas and Catalytic Conversion to Ethanol. In Biofuel’s Engineering Process Technology; IntechOpen: London, UK, 2011. [Google Scholar]
- Van der Meijden, C.M.; Könemann, J.W.; Sierhuis, W.; van der Drift, A.; Rietveld, G. Wood to Bio-Methane demonstration project in the Netherlands; ECN Report Number ECN-M–13-009; Energy research Centre of the Netherlands ECN: Petten, The Netherlands, 2013; Volume 21, pp. 1–10. [Google Scholar]
- Gogreengas-First Project Progress Report 2016. Available online: https://gogreengas.com/wp-content/uploads/2015/11/BioSNG-Demonstration-Plant-June-2016-Project-Progress-Report.pdf (accessed on 1 December 2019).
- Gøbel, B.; Henriksen, U.B.; Ahrenfeldt, J.; Jensen, T.K.; Hindsgaul, C.; Bentzen, J.D.; Sørensen, L.H. Status—2000 hours of operation with the Viking gasifier. In Proceedings of the 2nd World Conference and Technology Exhibition on Biomass for Energy and Industry, Rome, Italy, 10–14 May 2004; pp. 1–5. [Google Scholar]
- Henriksen, U.; Ahrenfeldt, J.; Jensen, T.K.; Gøbel, B.; Bentzen, J.D.; Hindsgaul, C.; Sørensen, L.H. The design, construction and operation of a 75 kW two-stage gasifier. Energy 2006, 31, 1542–1553. [Google Scholar] [CrossRef]
- Gadsbøll, R.Ø.; Sárossy, Z.; Jørgensen, L.; Ahrenfeldt, J.; Henriksen, U.B. Oxygen-blown operation of the two stage Viking gasifier. Energy 2018, 158, 495–503. [Google Scholar] [CrossRef]
- Kuznecova, I.; Gusca, J. Property based ranking of CO and CO2 methanation catalysts. Energy Procedia 2017, 128, 255–260. [Google Scholar] [CrossRef]
- Wang, H.; Pei, Y.; Qiao, M.; Zong, B. Advances in methanation catalysis. Catalysis 2017, 29, 1–28. [Google Scholar]
- Sabatier, P.; Senderens, J. New synthesis of methane. Comptes Rendus Hebd. Des Séances De L’académie Des Sci. 1902, 134, 514–516. [Google Scholar]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Mills, G.A.; Steffgen, F.W. Catalytic methanation. Catal. Rev. Sci. Eng. 1974, 8, 159–210. [Google Scholar] [CrossRef]
- Wang, Z. Sabatier-Senderens reduction. In Comprehensive Organic Name Reactions and Reagents; American Cancer Society: Atlanta, GA, USA, 2010; pp. 2454–2457. [Google Scholar]
- Rajendran, S.; Naushad, M.; Raju, K.; Boukherroub, R. Emerging Nanostructured Materials for Energy and Environmental Science, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P.L. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Martínez, J.; Hernández, E.; Alfaro, S.; Medina, R.L.; Aguilar, G.V.; Albiter, E.; Valenzuela, M.A. High selectivity and stability of nickel catalysts for CO2 methanation: Support effects. Catalysts 2019, 9, 24. [Google Scholar] [CrossRef]
- Iglesias, G.M.; De Vries, C.; Claeys, M.; Schaub, G. Chemical energy storage in gaseous hydrocarbons via iron Fischer-Tropsch synthesis from H2/CO2—Kinetics, selectivity and process considerations. Catal. Today 2015, 242, 184–192. [Google Scholar] [CrossRef]
- Saito, M.; Anderson, R.B. The activity of several molybdenum compounds for the methanation of CO. J. Catal. 1980, 63, 438–446. [Google Scholar] [CrossRef]
- Tapia, J.; Acelas, N.Y.; López, D.; Moreno, A. NiMo-sulfide supported on activated carbon to produce renewable diesel. Univ. Sci. 2017, 22, 71–85. [Google Scholar] [CrossRef]
- Chang, F.W.; Kuo, M.S.; Tsay, M.T.; Hsieh, M.C. Hydrogenation of CO2 over nickel catalysts on rice husk ash-alumina prepared by incipient wetness impregnation. Appl. Catal. A Gen. 2003, 247, 309–320. [Google Scholar] [CrossRef]
- Jiajian, G.; Gu, F.; Zhang, M. Effect of nickel nanoparticle size in Ni/α-Al2O3 on CO methanation reaction for the production of synthetic natural gas. Catal. Sci. Technol. 2013, 3, 2009–2015. [Google Scholar]
- Wang, W.; Gong, J. Methanation of carbon dioxide: An overview. Front. Chem. Eng. China 2011, 5, 2–10. [Google Scholar]
- Delmelle, R.; Duarte, R.B.; Franken, T.; Burnat, D.; Holzer, L.; Borgschulte, A.; Heel, A. Development of improved nickel catalysts for sorption enhanced CO2 methanation. Int. J. Hydrog. Energy 2016, 41, 20185–20191. [Google Scholar] [CrossRef]
- Lee, G.D.; Moon, M.J.; Park, J.H.; Park, S.S.; Hong, S.S. Raney ni catalysts derived from different alloy precursors part II. CO and CO2 methanation activity. Korean J. Chem. Eng. 2005, 22, 541–546. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.; Wang, E.; Lin, C.; Shang, Y.; Ding, G.; Ma, X.; Qin, S.; Sun, Q. Effect of composite supports on the methanation activity of Co-Mo-based sulphur-resistant catalysts. J. Nat. Gas Chem. 2012, 21, 767–773. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Willauer, H.D. Heterogeneous catalytic CO2 conversion to value-added hydrocarbons. Energy Environ. Sci. 2010, 3, 884–890. [Google Scholar] [CrossRef]
- Barrientos, J.; Gonzalez, N.; Boutonnet, M.; Järås, S. Deactivation of Ni/Γ-Al2O3 catalysts in CO methanation: Effect of Zr, Mg, Ba and Ca oxide promoters. Top. Catal. 2017, 60, 1276–1284. [Google Scholar] [CrossRef]
- Daza, C.E.; Gamba, O.A.; Hernández, Y.; Centeno, M.A.; Mondragón, F.; Moreno, S.; Molina, R. High-stable mesoporous Ni-Ce/clay catalysts for syngas production. Catal. Lett. 2011, 141, 1037–1046. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Bebiano, S.S.; Lopes, J.M.; Henriques, C. Magnesium as promoter of CO2 methanation on Ni-based USY zeolites. Energy Fuels 2017, 31, 9776–9789. [Google Scholar] [CrossRef]
- Liu, Q.; Gu, F.; Lu, X.; Liu, Y.; Li, H.; Zhong, Z.; Xu, G.; Su, F. Enhanced catalytic performances of Ni/Al2O3 catalyst via addition of V2O3 for CO methanation. Appl. Catal. A Gen. 2014, 488, 37–47. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Cavattoni, T.; Finocchio, E.; Riani, P.; Flytzani-Stephanopoulos, M.; Busca, G. A study of Ni/La-Al2O3 catalysts: A competitive system for CO2 methanation. Appl. Catal. B Environ. 2019, 248, 286–297. [Google Scholar] [CrossRef]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent advancements, fundamental challenges, and opportunities in catalytic methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Gumerov, F.; Le Neindre, B. Regeneration of spent catalyst and impregnation of catalyst by supercritical fluid. Int. J. Anal. Mass Spectrom. Chromatogr. 2016, 4, 51–65. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Weatherbee, G.D.; Jarvi, G.A. Sulfur poisoning of nickel methanation catalysts. I. in situ deactivation by H2S of nickel and nickel bimetallics. J. Catal. 1979, 60, 257–269. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Shen, W.M.; Dumesic, J.A.; Hill, C.G. Criteria for stable Ni particle size under methanation reaction conditions: Nickel transport and particle size growth via nickel carbonyl. J. Catal. 1981, 68, 152–165. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Wissing, L.; Skjøth-Rasmussen, M.S. High temperature methanation: Catalyst considerations. Catal. Today 2013, 215, 233–238. [Google Scholar] [CrossRef]
- Wanke, S.E.; Flynn, P.C. The sintering of supported metal catalysts. Catal. Rev. 1975, 12, 93–135. [Google Scholar] [CrossRef]
- Bai, X.; Wang, S.; Sun, T.; Wang, S. The sintering of Ni/Al2O3 methanation catalyst for substitute natural gas production. React. Kinet. Mech. Catal. 2014, 112, 437–451. [Google Scholar] [CrossRef]
- Sehested, J. Four challenges for nickel steam-Reforming catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.; Pedersen, K.; Sehested, J. High temperature methanation. Sintering and structure sensitivity. Appl. Catal. A Gen. 2007, 330, 134–138. [Google Scholar] [CrossRef]
- Liu, Q.; Gu, F.; Zhong, Z.; Xu, G.; Su, F. Anti-sintering ZrO2-modified Ni/α-Al2O3 catalyst for CO methanation. RSC Adv. 2016, 6, 20979–20986. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; Van Helden, J.W.A.; Van Hoof, A.J.F.; Friedrich, H.; Groeneveld, E.; Pidko, E.A.; Hensen, E.J.M. Tunable colloidal Ni nanoparticles confined and redistributed in mesoporous silica for CO2 methanation. Catal. Sci. Technol. 2019, 9, 2578–2591. [Google Scholar] [CrossRef]
- Du, J.; Gao, J.; Gu, F.; Zhuang, J.; Lu, B.; Jia, L.; Xu, G.; Liu, Q.; Su, F. A strategy to regenerate coked and sintered Ni/Al2O3 catalyst for methanation reaction. Int. J. Hydrog. Energy 2018, 43, 20661–20670. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Bemrose, C.R.; Bridgwater, J. A review of attrition and attrition test methods. Powder Technol. 1987, 49, 97–126. [Google Scholar] [CrossRef]
- Kalman, H.; Goder, D. Design criteria for particle attrition. Adv. Powder Technol. 1998, 9, 153–167. [Google Scholar] [CrossRef]
- Seemann, M.; Thunman, H. Methane synthesis. In Substitute Natural Gas from Waste; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 221–243. [Google Scholar]
- Porubova, J.; Klemm, M.; Kiendl, I.; Valters, K.; Markova, D.; Repele, M.; Bazbauers, G. Influence of temperature and pressure change on adiabatic and isothermal methanation processes. Environ. Clim. Technol. 2012, 9, 22–27. [Google Scholar] [CrossRef]
- Kang, W.R.; Lee, K.B. Effect of operating parameters on methanation reaction for the production of synthetic natural gas. Korean J. Chem. Eng. 2013, 30, 1386–1394. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RCS Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Catalysis reviews: Science and reforming and methanation carbon deposition in steam reforming and methanation. Catal. Rev. Sci. Eng. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Eckle, S. Investigations of the Kinetics and Mechanism of the Selective Methanation of CO in CO2 and H2-Rich Reformates Over Ru Supported Catalysts. Ph.D. Thesis, Universität Ulm, Ulm, Germany, 2012. [Google Scholar]
- Er-rbib, H.; Bouallou, C. Modelling and simulation of methanation catalytic reactor for renewable electricity storage. Chem. Eng. Trans. 2013, 35, 541–546. [Google Scholar]
- Lazdans, A.; Dace, E.; Gusca, J. Development of the experimental scheme for methanation process. Energy Procedia 2016, 95, 540–545. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.Y.; Li, H.; Yu, Z. Active and stable Ni based catalysts and processes for biogas upgrading: The effect of temperature and initial methane concentration on CO2 methanation. Appl. Energy 2018, 227, 206–212. [Google Scholar] [CrossRef]
- Kiewidt, L.; Thöming, J. Predicting optimal temperature profiles in single-stage fixed-bed reactors for CO2-methanation. Chem. Eng. Sci. 2015, 132, 59–71. [Google Scholar] [CrossRef]
- Richardson, R.C. Design of Fixed Bed Catalytic Reactors. 1963. Available online: https://lib.dr.iastate.edu/cgi/viewcontent.cgi?article=3554&context=rtd (accessed on 1 December 2019).
- Danaci, S. Optimisation and Integration of Catalytic Porous Structures into Structured Reactors for CO2 Conversion to Methane. Ph.D. Thesis, Université Grenoble Alpes, Grenoble, France, 2017. [Google Scholar]
- Heyne, S.; Seemann, M.C.; Harvey, S. Integration study for alternative methanation technologies for the production of synthetic natural gas from gasified biomass. Chem. Eng. Trans. 2010, 21, 409–414. [Google Scholar]
- Fries, L.; Antonyuk, S.; Heinrich, S.; Palzer, S. Performance comparison of syngas methanation on fluidized and fixed bed reactors. In Proceedings of the 13th International Conference on Fluidization—New Paradigm in Fluidization Engineering, Gyeong-ju, Korea, 16–21 May 2010; pp. 1–9. [Google Scholar]
- Sudiro, M.; Bertucco, A. Synthetic Natural Gas (SNG) from Coal and Biomass: A Survey of Existing Process Technologies, Open Issues and Perspectives. In Natural Gas; IntechOpen: London, UK, 2010; pp. 105–126. [Google Scholar]
- Götz, M.; Koch, A.M.; Graf, F. State of the art and perspectives of CO2 methanation process concepts for power-to-gas applications. Int. Gas Res. Conf. Proc. 2014, 1, 314–327. [Google Scholar]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Methanation in a fluidized bed reactor with high initial CO partial pressure: Part I-Experimental investigation of hydrodynamics, mass transfer effects, and carbon deposition. Chem. Eng. Sci. 2011, 66, 924–934. [Google Scholar] [CrossRef]
- Lefebvre, J.; Götz, M.; Bajohr, S.; Reimert, R.; Kolb, T. Improvement of three-phase methanation reactor performance for steady-state and transient operation. Fuel Process. Technol. 2015, 132, 83–90. [Google Scholar] [CrossRef]
- Moulijn, J.A.; Kapteijn, F. Monolithic reactors in catalysis: Excellent control. Curr. Opin. Chem. Eng. 2013, 2, 346–353. [Google Scholar] [CrossRef]
- Pérez, S.; Aragón, J.J.; Peciña, I.; Garcia-Suarez, E.J. Enhanced CO2 methanation by new microstructured reactor concept and design. Top. Catal. 2019, 62, 518–523. [Google Scholar] [CrossRef]
- Brooks, K.P.; Hu, J.; Zhu, H.; Kee, R.J. Methanation of carbon dioxide by hydrogen reduction using the Sabatier process in microchannel reactors. Chem. Eng. Sci. 2007, 62, 1161–1170. [Google Scholar] [CrossRef]
- Liu, Z.; Chu, B.; Zhai, X.; Jin, Y.; Cheng, Y. Total methanation of syngas to synthetic natural gas over Ni catalyst in a micro-channel reactor. Fuel 2012, 95, 599–605. [Google Scholar] [CrossRef]
- Augustyn, A.; Gawlik, L.; Pepłowska, M. Power to gas—An innovative energy conversion and storage solution. IOP Conf. Ser. Earth Environ. Sci. 2019, 214, 1. [Google Scholar] [CrossRef]
- Bailera, M.; Lisbona, P.; Romeo, L.M.; Espatolero, S. Power to gas projects review: Lab, pilot and demo plants for storing renewable energy and CO2. Renew. Sustain. Energy Rev. 2017, 69, 292–312. [Google Scholar] [CrossRef]
- Rieke, S. Solar Fuel GmbH—Solar Fuels and Power-To-Gas Technologies; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Specht, M.; Zuberbühler, U.; Bandi, A. Experimental Data on Renewable Methanol/Methane Generation from Atmospheric CO2 in Pilot Plants. 2011. Available online: https://www.iass-potsdam.de/sites/default/files/files/specht_zsw_p2g.pdf (accessed on 1 December 2019).
- Otten, R.; Forster, S.; Block, T.; Gosda, H.; Dieminger, F. Wirkungsgrad-Optimierung von Methanisierungs-und Biogasanlagen-Technologie im Rahmen Eines EE-Speicherungs-Pilotprojekts. 2016. Available online: https://www.tib.eu/suchen/id/TIBKAT:88124936X/ (accessed on 1 December 2019).
- GreenHydrogen.dk; Elplatek A/S; Lemvig Biogasanlæg A.m.b.A; DTU Mekanik; AU-Herning MeGa-stoRE Final Report. 2015. Available online: https://www.lemvigbiogas.com/MeGa-stoREfinalreport.pdf (accessed on 1 December 2019).
- Stähr, D.; Rasmusson, H.; Gerstein, D. STORE&GO: Das größte power-to-gas-forschungsprojekt in Europa befindet sich auf der zielgeraden. Energie 2019, 11, 62–65. [Google Scholar]
- Larsson, A.; Gunnarsson, I.; Tengberg, F. The GoBiGas Project—Demonstration of the Production of Biomethane from Biomass via Gasification. 2018. Available online: https://research.chalmers.se/en/publication/509030 (accessed on 1 December 2019).
- Bünger, U.; Landinger, H.; Pschorr-Schoberer, E.; Schmidt, P.; Weindorf, W.; Jöhrens, J.; Lambrecht, U.; Naumann, K.; Lischke, A. Power-to-Gas (PtG) in Transport Status Quo and Perspectives for Development. Energy Procedia 2015, 73, 182–189. [Google Scholar]
- Bach, C. Renewable Energy in the Future Energy System. 2015. Available online: http://www.ccem.ch/renerg2.html (accessed on 1 December 2019).
- Kwak, M. Attero Power(ed) to (by) Gas—The Waste to Power to Gas W2P2G Project 2014. Available online: https://docplayer.net/19872136-Attero-power-ed-to-by-gas-the-waste-to-power-to-gas-w2p2g-project-marco-kwak-project-business-development.html (accessed on 1 December 2019).
- GL Noble Denton; Pöyry Management Consulting. Gas Quality Harmonisation-Cost Benefit Analysis; European Commission: Brussels, Belgium, 2012. [Google Scholar]
- Gondal, I.A.; Sahir, M.H. Prospects of natural gas pipeline infrastructure in hydrogen transportation. Int. J. Energy Res. 2011, 36, 1338–1345. [Google Scholar] [CrossRef]
- Thema, M.; Weidlich, T.; Hörl, M.; Bellack, A.; Mörs, F.; Hackl, F.; Kohlmayer, M.; Gleich, J.; Stabenau, C.; Trabold, T.; et al. Biological CO2-methanation: An approach to standardization. Energies 2019, 12, 1670. [Google Scholar] [CrossRef]
- Tractebel; Engie; Hinicio. Study on Early Business Cases for H2 in Energy Storage and More Broadly Power to H2 Applications; FCH JU: Bruxelles, Belgium, 2017. [Google Scholar]
- EASEE-Gas. Common Business Practice 2005-001/02 2005. Available online: https://easee-gas.eu/download_file/DownloadFile/4/cbp-2005-001-02-harmonisation-of-natural-gas-quality/ (accessed on 1 December 2019).
- Marcogaz. Impact of Hydrogen in Natural Gas on End-Use Applications 2017. Available online: https://www.marcogaz.org/app/download/7928117663/UTIL-GQ-17-29.pdf?t=1541666775 (accessed on 1 December 2019).
- NATURALHY. Using the Existing Natural Gas System for Hydrogen 2009. Available online: https://www.fwg-gross-bieberau.de/fileadmin/user_upload/Erneuerbare_Energie/Naturalhy_Brochure.pdf (accessed on 1 December 2019).
- NGC; Interchangeability Work Group. White Paper on Natural Gas Interchangeability and Non-Combustion End Use; NGC: Sarasota, FL, USA, 2005. [Google Scholar]
- CIMAC WG17 “Gas Engines”. Impact of Gas Quality on Gas Engine Performance 2015. Available online: https://www.cimac.com/cms/upload/workinggroups/WG17/CIMAC_WG17_Position_Paper_Impact_Gas_Quality_on_Gas_Engine_Performance_2015_Jul.pdf (accessed on 1 December 2019).
- Gas Infrastructure Europe. GIE Position Paper on Impact of Including Methane Number in the European Standard for Natural Gas; Gas Infrastructure Europe: Bruxelles, Belgium, 2012. [Google Scholar]
- Höcher, T.; Sterner, M.; Mlaker, H.; Köppel, W.; Müller-Syring, G.; Henel, M. Entwicklung von Modularen Konzepten Zur Erzeugung, Speicherung und Einspeisung von Wasserstoff und Methan ins Erdgasnetz 2013. Available online: https://www.dvgw.de/medien/dvgw/forschung/berichte/g1_07_10.pdf (accessed on 1 December 2019).
- Melaina, M.; Penev, M.; Steward, D.; Antonia, O.; Bush, B.; Daniel, B.; Heimiller, D.; Melius, J. Hydrogen Infrastructure Cost Estimates and Blending Hydrogen into Natural Gas Pipelines 2012. Available online: https://www.hydrogen.energy.gov/pdfs/htac_nov12_3_melaina.pdf (accessed on 1 December 2019).
- PG&E gas R&D and innovation. Pipeline Hydrogen 2018. Available online: https://www.pge.com/pge_global/common/pdfs/for-our-business-partners/interconnection-renewables/interconnections-renewables/Whitepaper_PipelineHydrogenAnalysis.pdf (accessed on 1 December 2019).
- Melaina, M.; Antonia, O.; Penev, M. Blending Hydrogen into Natural Gas Pipeline Networks: A Review of Key Issues; National Renewable Energy Laboratory: Golden, CO, USA, 2013.
- Saffers, J.B.; Molkov, V.V. Hydrogen safety engineering framework and elementary design safety tools. Int. J. Hydrog. Energy 2014, 39, 6268–6285. [Google Scholar] [CrossRef]
- Saffers, J.B.; Molkov, V.V. Towards hydrogen safety engineering for reacting and non-reacting hydrogen releases. J. Loss Prev. Process Ind. 2013, 26, 344–350. [Google Scholar] [CrossRef]
- Ogden, J.; Jaffe, A.M.; Scheitrum, D.; McDonald, Z.; Miller, M. Natural gas as a bridge to hydrogen transportation fuel: Insights from the literature. Energy Policy 2018, 115, 317–329. [Google Scholar] [CrossRef]
- Holm, T. Aspects of the mechanism of the flame ionization detector. J. Chromatogr. A 1999, 842, 221–227. [Google Scholar] [CrossRef]
- Ongkiehong, L. The Hydrogen Flame Ionization Detector. Ph.D. Thesis, Technische Hogeschool Eindhoven, Eindhoven, The Netherlands, 1960. [Google Scholar]
- Toonen, A.; van Loon, R. Hydrogen Detection with a TCD Using Mixed Carrier Gas on the Agilent Micro GC; Agilent Technologies, Inc.: Santa Clara, CA, USA, 2013. [Google Scholar]
- Emerson. Electrochemical vs. Semiconductor Gas Detection—A Critical Choice; Patent and Trademark Office: Washington, DC, USA, 2019.
- Trinchi, A.; Kandasamy, S.; Wlodarski, W. High temperature field effect hydrogen and hydrocarbon gas sensors based on SiC MOS devices. Sens. Actuators B Chem. 2008, 133, 705–716. [Google Scholar] [CrossRef]
- Park, N.H.; Akamatsu, T.; Itoh, T.; Izu, N.; Shin, W. Calorimetric thermoelectric gas sensor for the detection of hydrogen, methane and mixed gases. Sensors 2014, 14, 8350–8362. [Google Scholar] [CrossRef] [PubMed]
- Rego, R.; Caetano, N.; Mendes, A. Hydrogen/methane and hydrogen/nitrogen sensor based on the permselectivity of polymeric membranes. Sens. Actuators B Chem 2005, 112, 150–159. [Google Scholar] [CrossRef]
- Altfeld, K.; Pinchbeck, D. Admissible hydrogen concentrations in natural gas systems. Gas Energy 2013, 2103, 1–16. [Google Scholar]
- Leicher, J.; Nowakowski, T.; Giese, A.; Görner, K. Power-to-gas and the consequences: Impact of higher hydrogen concentrations in natural gas on industrial combustion processes. Energy Procedia 2017, 120, 96–103. [Google Scholar] [CrossRef]
- Graf, F.; Götz, M.; Bajohr, S. Injection of biogas, SNG and hydrogen. Gwf-Gas ErdgasInt. Issue 2011, 2, 30–40. [Google Scholar]
- Haeseldonckx, D.; D’haeseleer, W. The use of the natural-gas pipeline infrastructure for hydrogen transport in a changing market structure. Int. J. Hydrog. Energy 2007, 32, 1381–1386. [Google Scholar] [CrossRef]
- IEA. Reduction of CO2 Emission by Adding Hydrogen to Natural Gas; IEA: Paris, France, 2003. [Google Scholar]
- Dodds, P.E.; Demoullin, S. Conversion of the UK gas system to transport hydrogen. Int. J. Hydrog. Energy 2013, 38, 7189–7200. [Google Scholar] [CrossRef]
- Branan, C.R. Metallurgy. In Rules of Thumb for Chemical Engineers, 4th ed.; Branan, C.R., Ed.; Gulf Professional Publishing: Burlington, NY, USA, 2005; pp. 277–299. [Google Scholar]
- PG&E gas R&D and innovation. Hydrogen Technical Analysis 2018. Available online: https://www.pge.com/pge_global/common/pdfs/for-our-business-partners/interconnection-renewables/interconnections-renewables/Hydrogen_TechnicalAnalysis.pdf (accessed on 1 December 2019).
- Revie, R.W. Uhlig’s Corrosion Handbook; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Robertson, I.M.; Sofronis, P.; Nagao, A.; Martin, M.L.; Wang, S.; Gross, D.W.; Nygren, K.E. Hydrogen embrittlement understood. Metall. Mater. Trans. B 2015, 46, 1085–1103. [Google Scholar] [CrossRef]
- Cabrini, M.; Lorenzi, S.; Marcassoli, P.; Pastore, T. Hydrogen embrittlement behaviour of HSLA linepipe steels under cathodic protection. Corros. Rev. 2011, 29, 261–274. [Google Scholar] [CrossRef]
- Zabrzeski, L.; Janusz, P.; Liszka, K.; Łaciak, M.; Szurlej, A. Hydrogen-Natural Gas mixture compression in case of transporting through high-pressure gas pipelines. Iop Conf. Ser. Earth Environ. Sci. 2019, 214, 012137. [Google Scholar] [CrossRef]
- Haeseldonckx, D. Concrete Transition Issues Towards a Fully-Fledged Use of Hydrogen as an Energy Carrier. Ph.D. Thesis, KU Leuven, Leuven, Belgium, 2009. [Google Scholar]
- Mcdonell, V.; Colorado, A. Effect of Variable Fuel Composition on Emissions and Lean Blowoff Stability Performance Analysis of Nine Industrial Combustion Applications; California Energy Commission: Sacramento, CA, USA, 2016.
- U.S. Department of Energy. Blending of Hydrogen in Natural Gas Distribution Systems; U.S. Department of Energy: Washington, DC, USA, 1978.
- Scholten, F.L.; Wolters, M. Hydrogen diffusion throught plastic pipes. In Proceedings of the 17th International Plastic Fuel-Gas Pipe Symposium 2002, San Francisco, CA, USA, 20–23 October 2002; pp. 280–284. [Google Scholar]
- UNI EN ISO 14532:2017—“Natural gas- Vocabulary” 2017. Available online: http://store.uni.com/catalogo/index.php/catalog/product/view/id/265256/s/en-iso-14532-2017/category/6693/?___store=en&josso_back_to=http%3A%2F%2Fstore.uni.com%2Fjosso-security-check.php&josso_cmd=login_optional&josso_partnerapp_host=store.uni.com&___from_store=it (accessed on 1 December 2019).
- Kuczynski, S.; Łaciak, M.; Olijnyk, A.; Szurlej, A.; Włodek, T. Thermodynamic and technical issues of hydrogen and methane-hydrogen mixtures pipeline transmission. Energies 2019, 12, 569. [Google Scholar] [CrossRef]
- Kurdyumov, V.N.; Fernández, E.; Liñán, A. Flame flashback and propagation of premixed flames near a wall. In Proceedings of the Combustion Institute, Edinburgh, UK, 30 July–4 August 2000; pp. 1883–1889. [Google Scholar]
- BP; IGU. Guidebook to Gas Interchangeability and Gas Quality; BP: London, UK, 2011. [Google Scholar]
- Youcai, Z. Municipal solid waste incineration process and generation of bottom ash and fly ash. In Pollution Control and Resource Recovery: Municipal Solid Wastes Incineration; Youcai, Z., Ed.; Butterworth-Heinemann: Oxford, UK, 2017; pp. 1–59. [Google Scholar]
- Sterlepper, J.; Orrzewalla, J. Recovery of Commercially Valuable Products from Used Rubber. Tires. Patent EP0643264A1, 27 February 1995. [Google Scholar]
- Nitschke-kowsky, P. Impact of Hydrogen Admixture on Installed Gas Appliances 2012. Available online: http://members.igu.org/old/IGU%20Events/wgc/wgc-2012/wgc-2012-proceedings/working-committee-papers/working-committee-woc5/expert-forum-5.b/impact-of-hydrogen-admixture-on-installed-gas-appliances/view/++widget++form.widgets.download/@@download/Petra-Nitschke-Kowsky-%25E2%2580%2593-Werner-We%25C3%259Fing.pdf. (accessed on 1 December 2019).
- Huth, M.; Heilos, A. Fuel flexibility in gas turbine systems: Impact on burner design and performance. In Modern Gas Turbine Systems; Jansohn, P., Ed.; Woodhead Publishing: Cambridge, UK, 2013; pp. 635–684. [Google Scholar]
- De Vries, H.; Mokhov, A.V.; Levinsky, H.B. The impact of natural gas/hydrogen mixtures on the performance of end-use equipment: Interchangeability analysis for domestic appliances. Appl. Energy 2017, 208, 1007–1019. [Google Scholar] [CrossRef]
- Khatir, N.; Abdelkrim, L. Effect of hydrogen addition to methane on ICE emissions and performances: A brief review. J. Sci. Res. Rev. 2013, 2, 75–80. [Google Scholar]
- Staffell, I.; Scamman, D.; Velazquez Abad, A.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef]
- Benesch, R.; Jacksier, T. Hydrogen and material quality issues for PEM fuel cells. In Proceedings of the 2005 IEEE Vehicle Power and Propulsion Conference, Chicaco, IL, USA, 7–9 September 2005; pp. 646–651. [Google Scholar]
- U.S. Department of Energy; Ohi, J.M.; Vanderborgh, N.; Voecks, G. Hydrogen Fuel Quality Specifications for Polymer Electrolyte Fuel Cells in Road Vehicles; Office of Energy Efficiency and Renewable Energy: Washington, DC, USA, 2016.
- ISO 14687-2-“Hydrogen fuel—Product specification” 2012. Available online: https://www.iso.org/standard/55083.html (accessed on 1 December 2019).
- Haque, M.A.; Sulong, A.B.; Loh, K.S.; Majlan, E.H.; Husaini, T.; Rosli, R.E. Acid doped polybenzimidazoles based membrane electrode assembly for high temperature proton exchange membrane fuel cell: A review. Int. J. Hydrog. Energy 2017, 42, 9156–9179. [Google Scholar] [CrossRef]
- Ianniello, R.; Schmidt, V.M.; Stimming, U.; Stumper, J.; Wallau, A. CO adsorption and oxidation on Pt and Pt/Ru alloys: Dependence on substrate composition. Electrochim. Acta 1994, 39, 1863–1869. [Google Scholar] [CrossRef]
- Baschuk, J.J.; Li, X. Carbon monoxide poisoning of proton exchange membrane fuel cells. Int. J. Energy Res. 2001, 25, 695–713. [Google Scholar] [CrossRef]
- Sethuraman, V.A.; Weidner, J.W. Analysis of sulfur poisoning on a PEM fuel cell electrode. Electrochim. Acta 2010, 55, 5683–5694. [Google Scholar] [CrossRef]
- Hongsirikarn, K.; Jr, J.; Greenway, S.; Creager, S. Influence of ammonia on the conductivity of Nafion membranes. J. Power Sources 2010, 195, 30–38. [Google Scholar] [CrossRef]
- Besancon, B.M.; Hasanov, V.; Imbault-Lastapis, R.; Benesch, R.; Barrio, M.; Mølnvik, M.J. Hydrogen quality from decarbonized fossil fuels to fuel cells. Int. J. Hydrog. Energy 2009, 34, 2350–2360. [Google Scholar] [CrossRef]
- Díaz, M.A.; Iranzo, A.; Rosa, F.; Isorna, F.; López, E.; Bolivar, J.P. Effect of carbon dioxide on the contamination of low temperature and high temperature PEM (polymer electrolyte membrane) fuel cells. Influence of temperature, relative humidity and analysis of regeneration processes. Energy 2015, 90, 299–309. [Google Scholar]
- Bruijn, F.; Papageorgopoulos, D.C.; Sitters, E.; Janssen, G. The influence of carbon dioxide on PEM fuel cell anodes. J. Power Sources 2002, 110, 117–124. [Google Scholar] [CrossRef]
- Lanzini, A.; Madi, H.; Chiodo, V.; Papurello, D.; Maisano, S.; Santarelli, M.; Van herle, J. Dealing with fuel contaminants in biogas-fed solid oxide fuel cell (SOFC) and molten carbonate fuel cell (MCFC) plants: Degradation of catalytic and electro-catalytic active surfaces and related gas purification methods. Prog. Energy Combust. Sci. 2017, 61, 150–188. [Google Scholar] [CrossRef]
- Dayton, D.C.; Ratcliff, M.; Bain, R. Fuel Cell Integraton—A Study of the Impacts of Gas Quality and Impurities; NREL Milestone Report: Golden, CO, USA, 2001.
- Cavalli, A.; Bernardini, R.; Del Carlo, T.; Aravind, P.V. Effect of H2S and HCl on solid oxide fuel cells fed with simulated biosyngas containing primary tar. Energy Sci. Eng. 2019, 7, 2456–2468. [Google Scholar] [CrossRef]
- Sigot, L.; Ducom, G.; Benadda, B.; Labouré, C. Comparison of adsorbents for H2S and D4 removal for biogas conversion in a solid oxide fuel cell. Environ. Technol. 2016, 37, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Weinlaender, C.; Neubauer, R.; Hochenauer, C. Low-temperature H2S removal for solid oxide fuel cell application with metal oxide adsorbents. Adsorp. Sci. Technol. 2017, 35, 120–136. [Google Scholar] [CrossRef]
- Elwell, A.C.; Elsayed, N.H.; Kuhn, J.N.; Joseph, B. Design and analysis of siloxanes removal by adsorption from landfill gas for waste-to-energy processes. Waste Manag. 2018, 73, 189–196. [Google Scholar] [CrossRef]
- Ryckebosch, E.; Drouillon, M.; Vervaeren, H. Techniques for transformation of biogas to biomethane. Biomass Bioenergy 2011, 35, 1633–1645. [Google Scholar] [CrossRef]
- Boldrin, P.; Millan-Agorio, M.; Brandon, N.P. Effect of sulfur- and tar-contaminated syngas on solid oxide fuel cell anode materials. Energy Fuels 2015, 29, 442–446. [Google Scholar] [CrossRef]
- Vecchione, L.; Cossio, F.; Longo, L. Comparison of different systems for tar removal for renewable energy derivation from biomass gasification. Contemp. Eng. Sci. 2016, 9, 413–423. [Google Scholar] [CrossRef]
- Mcphail, S.J.; Moreno, A.; Belella, E.; Abate, L.; Cigolotti, V. The New Fuel Chain: From Renewables to Fuel Cells; ENEA: Kista, Sweden, 2016. [Google Scholar]
- Blanco, H.; Faaij, A. A review at the role of storage in energy systems with a focus on Power to Gas and long-term storage. Renew. Sustain. Energy Rev. 2018, 81, 1049–1086. [Google Scholar] [CrossRef]
- Ma, J.; Li, Q.; Kühn, M.; Nakaten, N. Power-to-gas based subsurface energy storage: A review. Renew. Sustain. Energy Rev. 2018, 97, 478–496. [Google Scholar] [CrossRef]
- Bang, H.J.; Stockar, S.; Muratori, M.; Rizzoni, G. Modeling and analysis of a CNG residential refueling system. In Proceedings of the ASME 2014 Dynamic Systems and Control Conference, DSCC 2014, San Antonio, TX, USA, 22–24 October 2014; pp. 1–9. [Google Scholar]
- Graf, F.; Riedl, J.; Kröger, K.; Reimert, R.; Meyer, J. Monitoring CNG quality in Germany. In Proceedings of the International Gas Union World Gas Conference Papers 2009, Buenos Aires, Argentina, 5–9 October 2009; pp. 3588–3597. [Google Scholar]
- Thomas, S.; Dawe, R.A. Review of ways to transport natural gas energy from countries which do not need the gas for domestic use. Energy 2003, 28, 1461–1477. [Google Scholar] [CrossRef]
- Hosseini, M.; Dincer, I.; Naterer, G.F.; Rosen, M.A. Thermodynamic analysis of filling compressed gaseous hydrogen storage tanks. Int. J. Hydrog. Energy 2012, 37, 5063–5071. [Google Scholar] [CrossRef]
- He, C.; Yu, R.; Sun, H.; Chen, Z. Lightweight multilayer composite structure for hydrogen storage tank. Int. J. Hydrog. Energy 2016, 41, 15812–15816. [Google Scholar] [CrossRef]
- Mokhatab, S.; Mak, J.Y.; Valappil, J.V.; Wood, D.A. Handbook of Liquefied Natural Gas; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Germanischer Lloyd. Rules for Classification and Construction: Ship Technology-Bridge Arrangement and Equipment on Seagoing Ships; Germanischer Lloyd: Hamborg, Germany, 2008. [Google Scholar]
- Aasadnia, M.; Mehrpooya, M. Large-scale liquid hydrogen production methods and approaches: A review. Appl. Energy 2018, 212, 57–83. [Google Scholar] [CrossRef]
- Makridis, S.S. Hydrogen storage and compression. In Methane and Hydrogen for Energy Storage; IET: London, UK, 2016; pp. 1–28. [Google Scholar]
- Sun, Z.G.; Wang, R.; Ma, R.; Guo, K.; Fan, S. Natural gas storage in hydrates with the presence of promoters. Energy Convers. Manag. 2003, 44, 2733–2742. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.; Zhu, Y. Porous carbon-based materials for hydrogen storage: Advancement and challenges. J. Mater. Chem. A 2013, 1, 9365–9381. [Google Scholar] [CrossRef]
- Poomisitiporn, K.; Rangsunvigit, P.; Kitiyanan, B.; Kulprathipanja, S. Competitive adsorption of methane and carbon dioxide on different activated carbons. Chem. Eng. Trans. 2016, 52, 121–126. [Google Scholar]
- Feroldi, M.; Neves, A.C.; Bach, V.R.; Alves, H.J. Adsorption technology for the storage of natural gas and biomethane from biogas. Int. J. Energy Res. 2016, 40, 1890–1900. [Google Scholar] [CrossRef]
- Nie, Z.; Lin, Y.; Jin, X. Research on the theory and application of adsorbed natural gas used in new energy vehicles: A review. Front. Mech. Eng. 2016, 11, 258–274. [Google Scholar] [CrossRef]
- Park, J.E.; Lee, G.B.; Hwang, S.Y.; Kim, J.H.; Hong, B.U.; Kim, H.; Kim, S. The effects of methane storage capacity using upgraded activated carbon by KOH. Appl. Sci. 2018, 8, 1596. [Google Scholar] [CrossRef]
- Mota, J.P.B.; Rodrigues, A.E.; Saatdjian, E.; Tondeur, D. Dynamics of natural gas adsorption storage systems employing activated carbon. Carbon 1997, 35, 1259–1270. [Google Scholar] [CrossRef]
- Mota, J.P.B. Impact of gas composition on natural gas storage by adsorption. Aiche J. 1999, 45, 986–996. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, C.; Su, W.; Zhou, Y.; Zhou, L. Principles of methane adsorption and natural gas storage. Adsorption 2009, 15, 133–137. [Google Scholar] [CrossRef]
- Taheri, Z.; Shabani, M.R.; Nazari, K.; Mehdizaheh, A. Natural gas transportation and storage by hydrate technology: Iran case study. J. Nat. Gas Sci. Eng. 2014, 21, 846–849. [Google Scholar] [CrossRef]
- Rogers, R.E.; Toghiani, R.K. Gas-hydrate storage of natural gas. In Proceedings of the ASEE Annual Conference Proceedings, Albuquerque, Mexico, 24 June 2001; pp. 5195–5201. [Google Scholar]
- Sloan, E.D. Clathrate hydrates: The other common solid water phase. Ind. Eng. Chem. Res. 2000, 39, 3123–3129. [Google Scholar] [CrossRef]
- Dawe, R.A.; Thomas, S.; Kromah, M. Hydrate technology for transporting natural gas. Eng. J. Univ. Qatar 2004, 16, 11–18. [Google Scholar]
- Chernov, A.A.; Pil’Nik, A.A.; Elistratov, D.S.; Mezentsev, I.V.; Meleshkin, A.V.; Bartashevich, M.V.; Vlasenko, M.G. New hydrate formation methods in a liquid-gas medium. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Khokhar, A.A.; Gudmundsson, J.S.; Sloan, E.D. Gas storage in structure H hydrates. Fluid Phase Equilibria 1998, 150, 383–392. [Google Scholar] [CrossRef]
- Czakkel, O.; Nagy, B.; Dobos, G.; Fouquet, P.; Bahn, E.; László, K. Static and dynamic studies of hydrogen adsorption on nanoporous carbon gels. Int. J. Hydrog. Energy 2019, 44, 18169–18178. [Google Scholar] [CrossRef]
- Terranova, M.L.; Orlanducci, S.; Rossi, M. Carbon Nanomaterials for Gas Adsorption; Jenny Stanford Publishing: Boca Raton, FL, USA, 2012. [Google Scholar]
- Eckert, J.; Cheetham, A.K. Hydrogen Storage Materials with Binding Intermediate between Physisorption and Chemisorption 2010. Available online: https://www.osti.gov/servlets/purl/1009133 (accessed on 1 December 2019).
- Bastos-Neto, M.; Patzschke, C.; Lange, M.; Möllmer, J.; Möller, A.; Fichtner, S.; Schrage, C.; Lässig, D.; Lincke, J.; Staudt, R.; et al. Assessment of hydrogen storage by physisorption in porous materials. Energy Environ. Sci. 2012, 5, 8294–8303. [Google Scholar] [CrossRef]
- Burress, J.; Kraus, M.; Beckner, M.; Cepel, R.; Suppes, G.; Wexler, C.; Pfeifer, P. Hydrogen storage in engineered carbon nanospaces. Nanotechnology 2009, 20, 204026. [Google Scholar] [CrossRef] [PubMed]
- Reyhani, A.; Mortazavi, S.Z.; Mirershadi, S.; Moshfegh, A.Z.; Parvin, P.; Golikand, A.N. Hydrogen storage in decorated multiwalled carbon nanotubes by Ca, Co, Fe, Ni, and Pd nanoparticles under ambient conditions. J. Phys. Chem. C 2011, 115, 6994–7001. [Google Scholar] [CrossRef]
- Brandon, N.N.P.; Thompsett, D. Fuel Cells Compendium.; Elsevier Science: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Lototskyy, M.V.; Davids, M.W.; Tolj, I.; Klochko, Y.V.; Sekhar, B.S.; Chidziva, S.; Smith, F.; Swanepoel, D.; Pollet, B.G. Metal hydride systems for hydrogen storage and supply for stationary and automotive low temperature PEM fuel cell power modules. Int. J. Hydrog. Energy 2015, 40, 11491–11497. [Google Scholar] [CrossRef]
- Westerwaal, R.J.; Haije, W.G. Evaluation solid-state hydrogen storage systems. ECN Hydrog. Clean Fossil Fuels 2008, 43, 1–75. [Google Scholar]
- Kiehne, H.A. Battery Technology Handbook; Taylor & Francis: Abingdon, UK, 2003. [Google Scholar]
- Orimo, S.; Nakamori, Y.; Eliseo, J.R.; Züttel, A.; Jensen, C.M. Complex hydrides for hydrogen storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef]
- Züttel, A. Materials for hydrogen storage. Mater. Today 2003, 6, 24–33. [Google Scholar] [CrossRef]
- Millet, P. Hydrogen storage in hydride-forming materials. In Advances in Hydrogen Production, Storage and Distribution; Woodhead Publishing Limited: Cambridge, UK, 2014; pp. 341–467. [Google Scholar]
- Evans, D.J. A review of underground fuel storage events and putting risk into perspective with other areas of the energy supply chain. Geol. Soc. Spec. Publ. 2009, 313, 173–216. [Google Scholar] [CrossRef]
- Sunjay, S.; Singh, V.K. P-28 Geological storage: Underground gas storage. In Proceedings of the 8th Biennal International Conference & Exposition on Petroleum Geophysics, SPG Hyderabad, India, 1–3 February 2010; pp. 1–8. [Google Scholar]
- Berest, P.; Brouard, B.; Durup, J.G. Tightness tests in salt-cavern wells. Oil Gas Sci. Technol. 2001, 56, 451–469. [Google Scholar] [CrossRef]
- Wang, T.; Yan, X.; Yang, H.; Yang, X.; Jiang, T.; Zhao, S. A new shape design method of salt cavern used as underground gas storage. Appl. Energy 2013, 104, 50–61. [Google Scholar] [CrossRef]
- Bérest, P.; Bergues, J.; Brouard, B. Review of static and dynamic compressibility issues relating to deep underground salt caverns. Int. J. Rock Mech. Min. Sci. 1999, 36, 1031–1049. [Google Scholar] [CrossRef]
- Plaat, H. Underground gas storage: Why and how. In Underground Gas Storage—Worldwide Experiences and Future Development in the UK and Europe; Geological Society Special Publication: London, UK, 2009; pp. 25–37. [Google Scholar]
- Tooseh, E.K.; Jafari, A.; Teymouri, A. Experimental investigation of injection pressure effect on the natural gas storage in aquifers. Int. J. Chem. Eng. Appl. 2017, 8, 351–354. [Google Scholar] [CrossRef]
- Shi, L.; Wang, J.M.; Liao, G.Z.; Xiong, W.; Gao, S.S. Mechanism of gas-water flow at pore-level in aquifer gas storage. J. Cent. South Univ. 2013, 20, 3620–3626. [Google Scholar] [CrossRef]
- Golghanddashti, H.; Saadat, M.; Abbasi, S.; Shahrabadi, A. Experimental investigation of salt precipitation during gas injection into a depleted gas reservoir. In Proceedings of the International Petroleum Technology Conference, Bangkok, Thailand, 15–17 November 2011; p. 8. [Google Scholar]
- Tooseh, E.K.; Jafari, A.; Teymouri, A. Gas-water-rock interactions and factors affecting gas storage capacity during natural gas storage in a low permeability aquifer. Pet. Explor. Dev. 2018, 45, 1123–1128. [Google Scholar] [CrossRef]
- Verga, F. What’s conventional and what’s special in a reservoir study for underground gas storage. Energies 2018, 11, 1245. [Google Scholar] [CrossRef]
- Anyadiegwu, C.I.C. Optimizing the benefits of conversion of depleted oil reservoirs for underground natural gas storage in Nigeria. Int. J. Sci. Technol. 2012, 1, 17–43. [Google Scholar]
- Anyadiegwu, C.I.C.; Anyanwu, E.E.; Obah, B.; Ogueke, N.V. Performance analysis of depleted oil reservoirs for underground gas storage. Asia Pac. J. Multidiscip. Res. 2014, 2, 99–114. [Google Scholar]
- Sandu, M.; Ernecic, B.; Radu, G.; Iskhakov, A. Study on Underground Gas Storage in Europe and Central Asia; Technical Report; United Nations Economic Commission for Europe: Geneva, Switzerland, 2012. [Google Scholar]
- Mazarei, M.; Davarpanah, A.; Ebadati, A.; Mirshekari, B. The feasibility analysis of underground gas storage during an integration of improved condensate recovery processes. J. Pet. Explor. Prod. Technol. 2019, 9, 397–408. [Google Scholar] [CrossRef]
- Teatini, P.; Castelletto, N.; Ferronato, M.; Gambolati, G.; Janna, C.; Cairo, E.; Marzorati, D.; Colombo, D.; Ferretti, A.; Bagliani, A.; et al. Geomechanical response to seasonal gas storage in depleted reservoirs: A case study in the Po River basin, Italy. J. Geophys. Res. Earth Surf. 2011, 116, 1–21. [Google Scholar] [CrossRef]
- Tarkowski, R. Underground hydrogen storage: Characteristics and prospects. Renew. Sustain. Energy Rev. 2019, 105, 86–94. [Google Scholar] [CrossRef]
- Ozarslan, A. Large-scale hydrogen energy storage in salt caverns. Int. J. Hydrog. Energy 2012, 37, 14265–14277. [Google Scholar] [CrossRef]
- Gregory, S.P.; Barnett, M.J.; Field, L.P.; Milodowski, A.E. Subsurface microbial hydrogen cycling: Natural occurrence and implications for industry. Microorganisms 2019, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- RAG. Underground Sun Storage—Storing Energy from Wind and Solar Power Below Ground 2016. Available online: https://www.underground-sun-storage.at/en/project/project-description.html (accessed on 1 December 2019).
- Aktiengesellschaft, R.-A.; Axiom angewandte Prozesstechnik GesmbH; Verbund AG; Montanuniversitat Leoben; Uuniversitat für Bodenkultur Wien; Energieinstitut an der Johannes Kepler Universität Linz. Publizierbarer Endbericht—Underground Sun Conversion; RAG: Vienna, Austria, 2013. [Google Scholar]
- RAG. Underground Sun Storage—Storing Sunshine 2016. Available online: https://www.rag-austria.at/fileadmin/bilder/0_NEU_RAG_Austria_AG/Unternehmen/sunconversion_broschuere_engl_180907_fin.pdf (accessed on 1 December 2019).
- Kirsanovs, V.; Blumberga, D.; Veidenbergs, I.; Rochas, C.; Vigants, E.; Vigants, G. Experimental investigation of downdraft gasifier at various conditions. Energy Procedia 2017, 128, 332–338. [Google Scholar] [CrossRef]
- Sarkar, S.; Kumar, A. Biohydrogen production from forest and agricultural residues for upgrading of bitumen from oil sands. Energy 2010, 35, 582–591. [Google Scholar] [CrossRef]
- Dascomb, J.; Krothapalli, A.; Fakhrai, R. Thermal conversion efficiency of producing hydrogen enriched syngas from biomass steam gasification. Int. J. Hydrog. Energy 2013, 38, 11790–11798. [Google Scholar] [CrossRef]
- Umeki, K.; Yamamoto, K.; Namioka, T.; Yoshikawa, K. High temperature steam-only gasification of woody biomass. Appl. Energy 2010, 87, 791–798. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Bhattacharya, A.; Datta, A. Modeling of hydrogen production process from biomass using oxygen blown gasification. Int. J. Hydrog. Energy 2012, 37, 18782–18790. [Google Scholar] [CrossRef]
- Solar Hydrogen Generation Research 2011. Available online: https://www.osti.gov/servlets/purl/1025597 (accessed on 1 December 2019).
- Kromer, M.; Roth, K.; Takata, R.; Chin, P. Support for Cost Analyses on Solar-Driven High Temperature Thermochemical Water-Splitting Cycles; TIAX: Lexington, MA, USA, 2011. [Google Scholar]
- Yang, W.; Prabhakar, R.R.; Tan, J.; Tilley, S.D.; Moon, J. Strategies for enhancing the photocurrent, photovoltage, and stability of photoelectrodes for photoelectrochemical water splitting. Chem. Soc. Rev. 2019, 48, 4979–5015. [Google Scholar] [CrossRef]
- Verkehrswende, A.; Energiewende, A.; Frontier, E. Die Zukünftigen Kosten Strombasierter Synthetischer Brennstoffe; Agora Energiewende: Berlin, Germany, 2018. [Google Scholar]
- Ochs, D.; Ahrer, W. Life cycle analysis of hydrogen production from biomass fermentation. Chem. Eng. Trans. 2010, 21, 1159–1164. [Google Scholar]
- Gorre, J.; Ortloff, F.; van Leeuwen, C. Production costs for synthetic methane in 2030 and 2050 of an optimized Power-to-Gas plant with intermediate hydrogen storage. Appl. Energy 2019, 253, 113594. [Google Scholar] [CrossRef]



























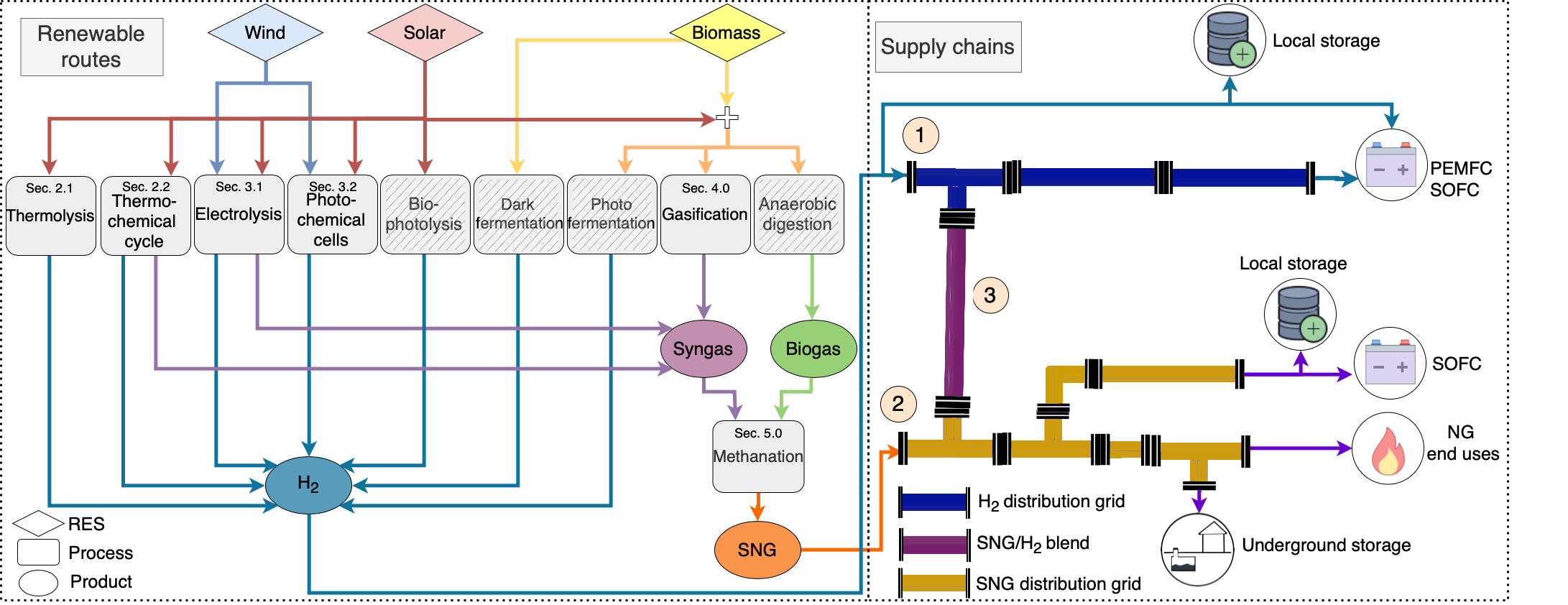{kind=link}
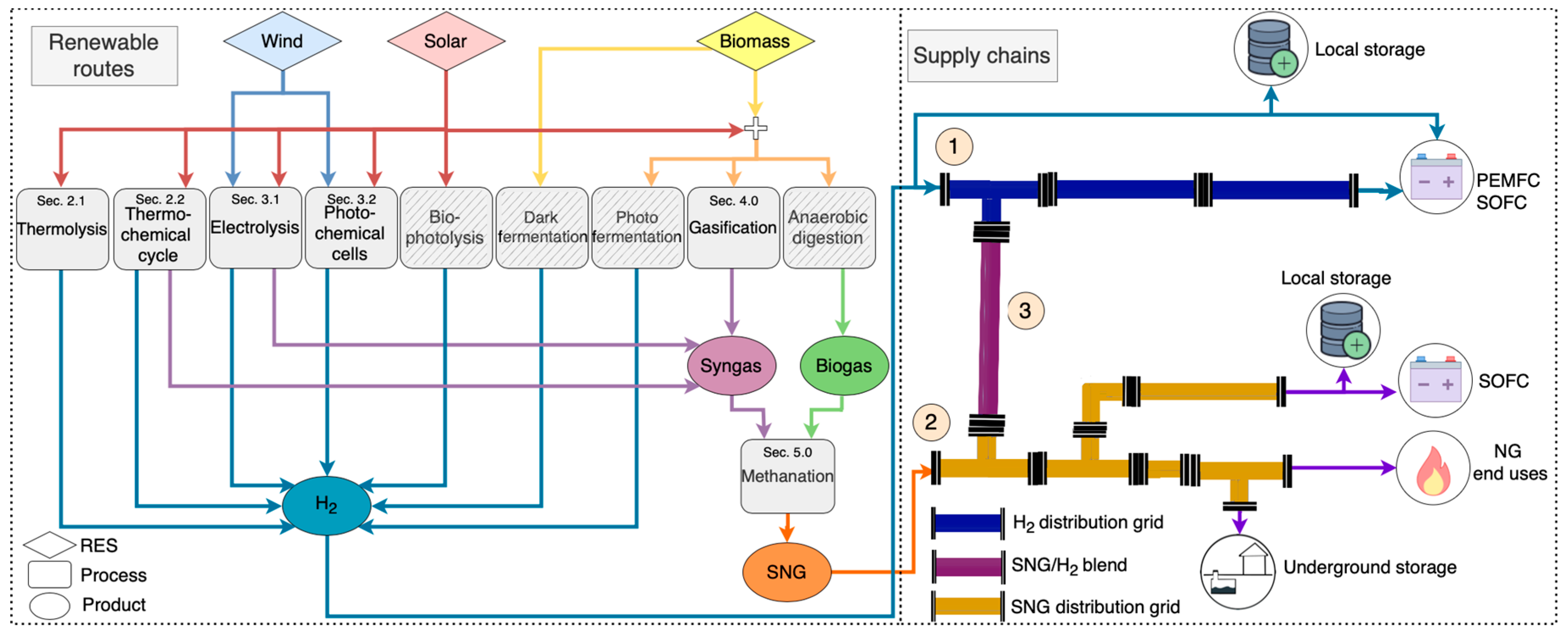{kind=link}
{kind=link}
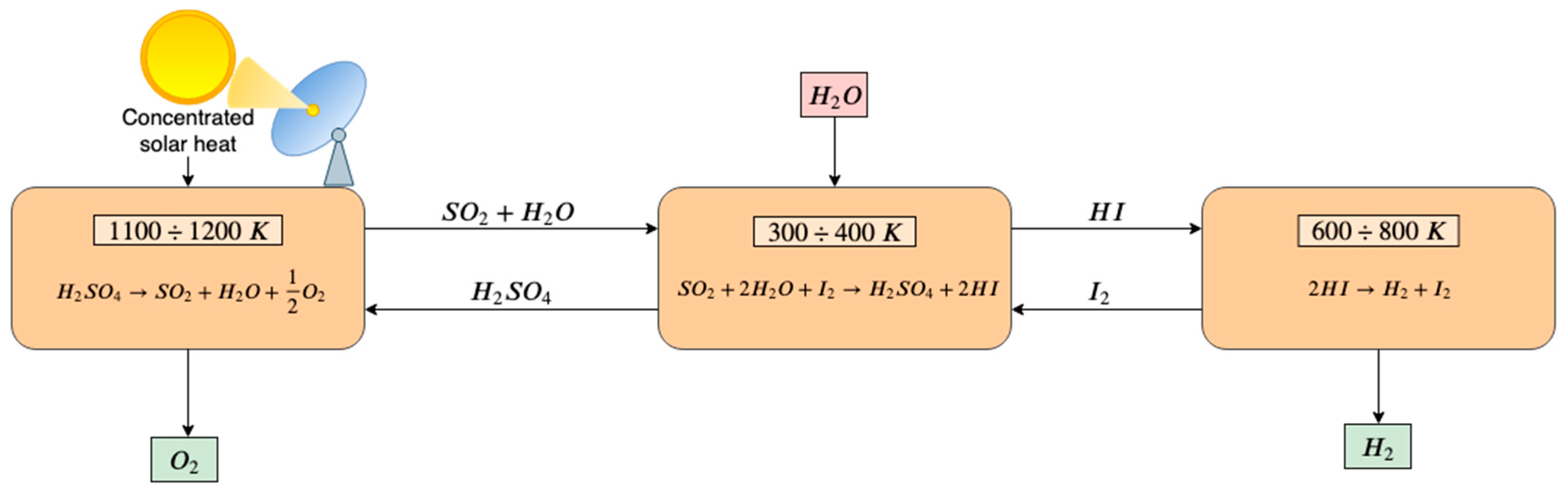{kind=link}
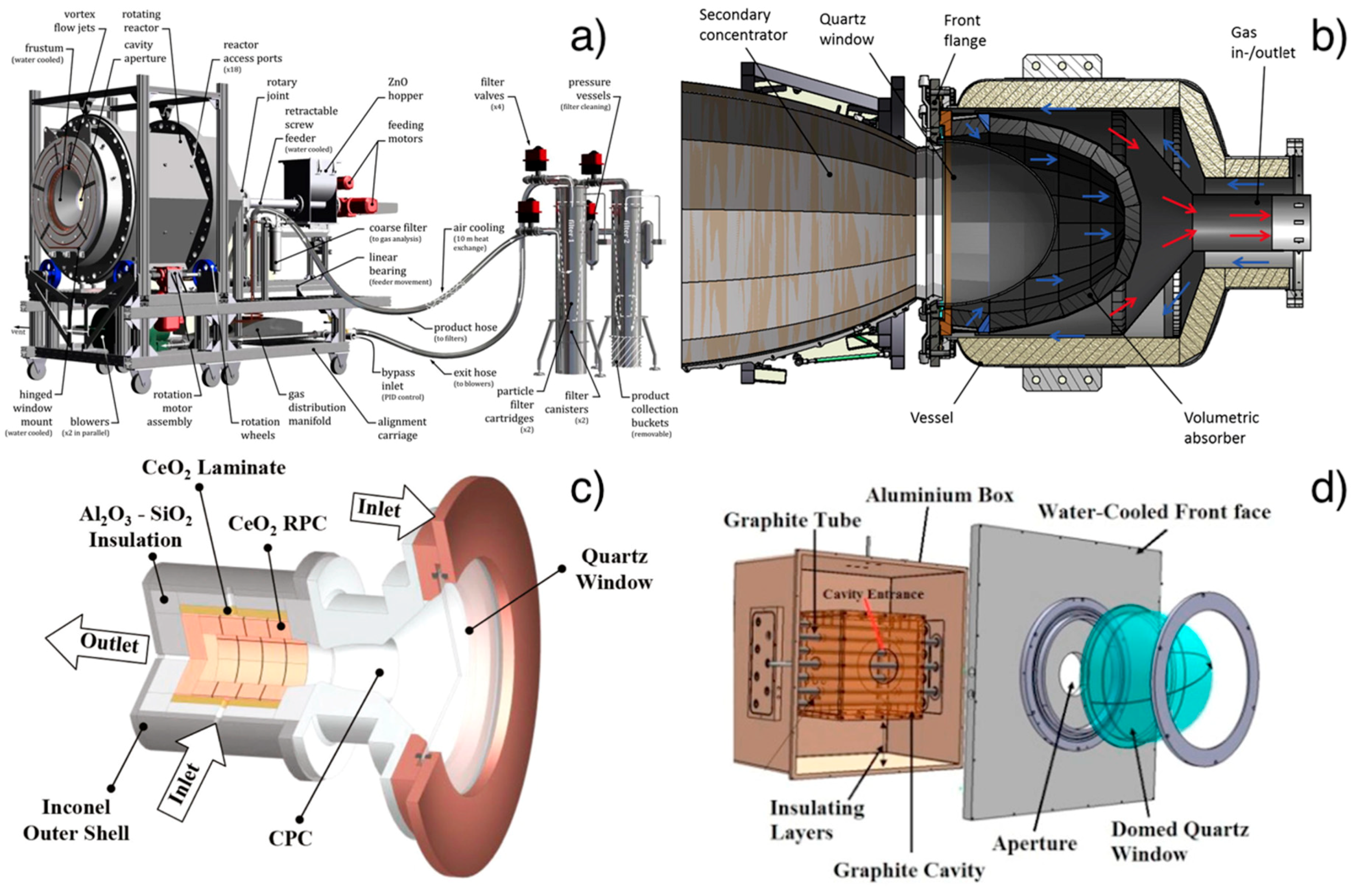{kind=link}
{kind=link}
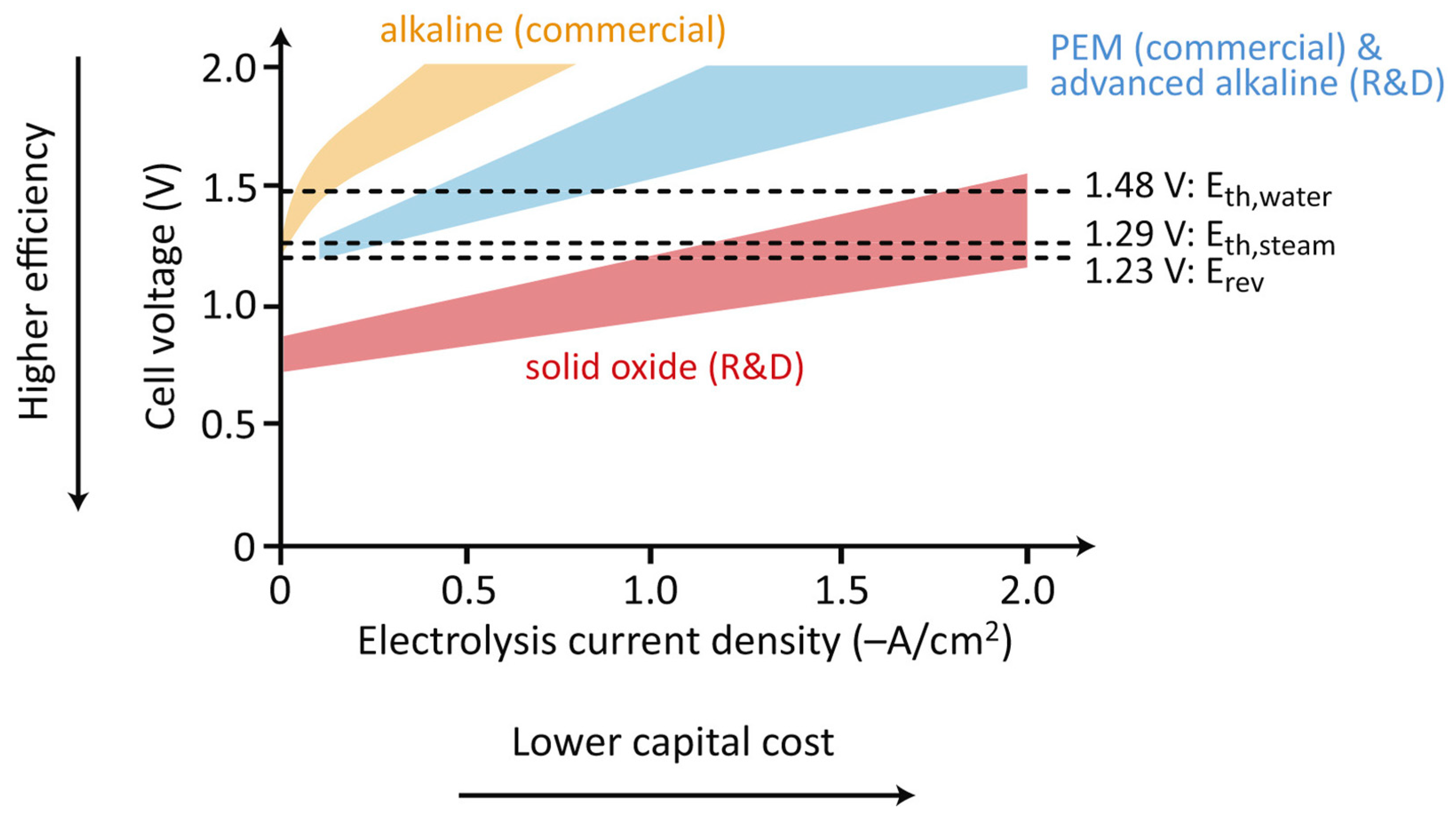{kind=link}
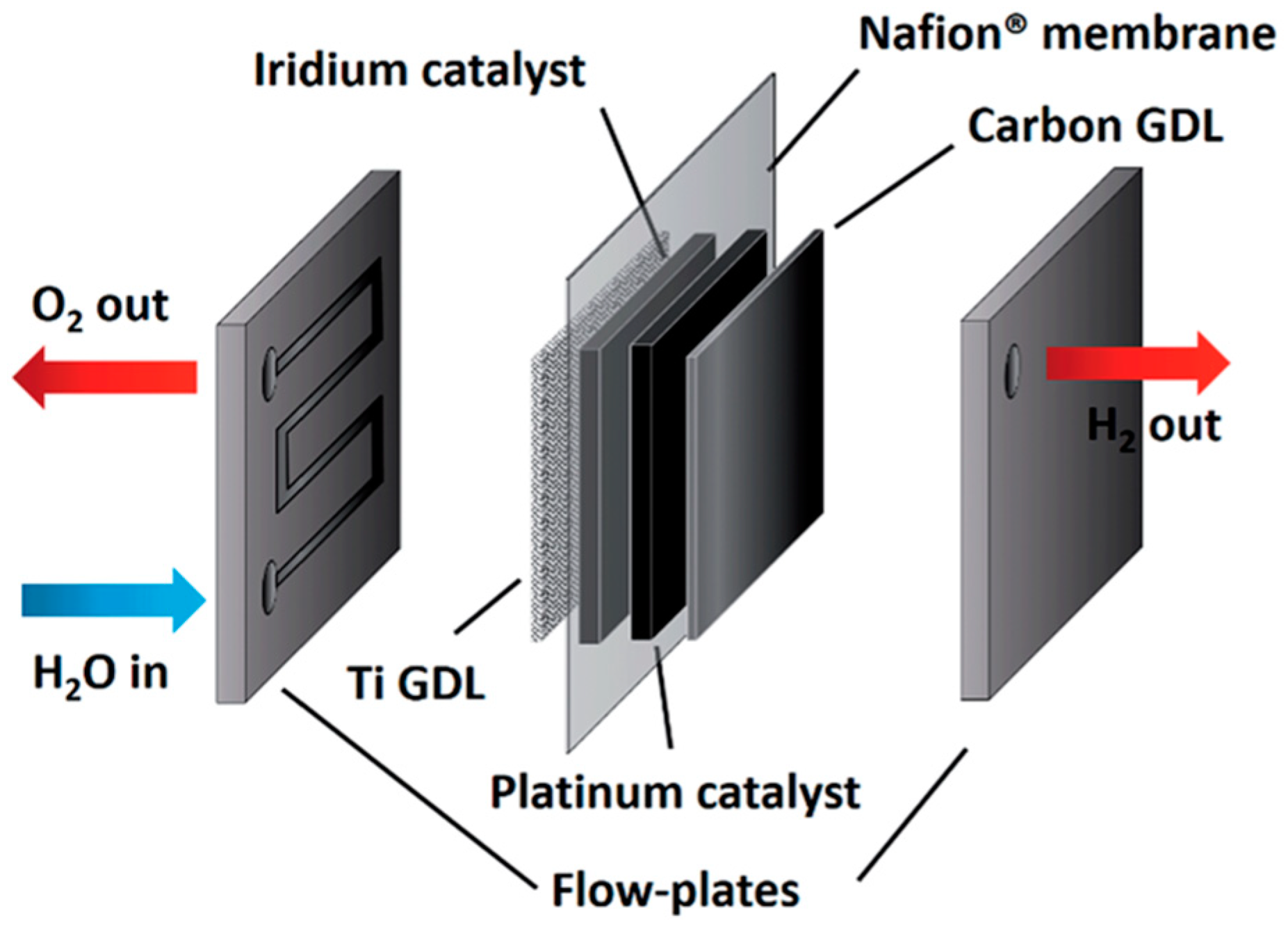{kind=link}
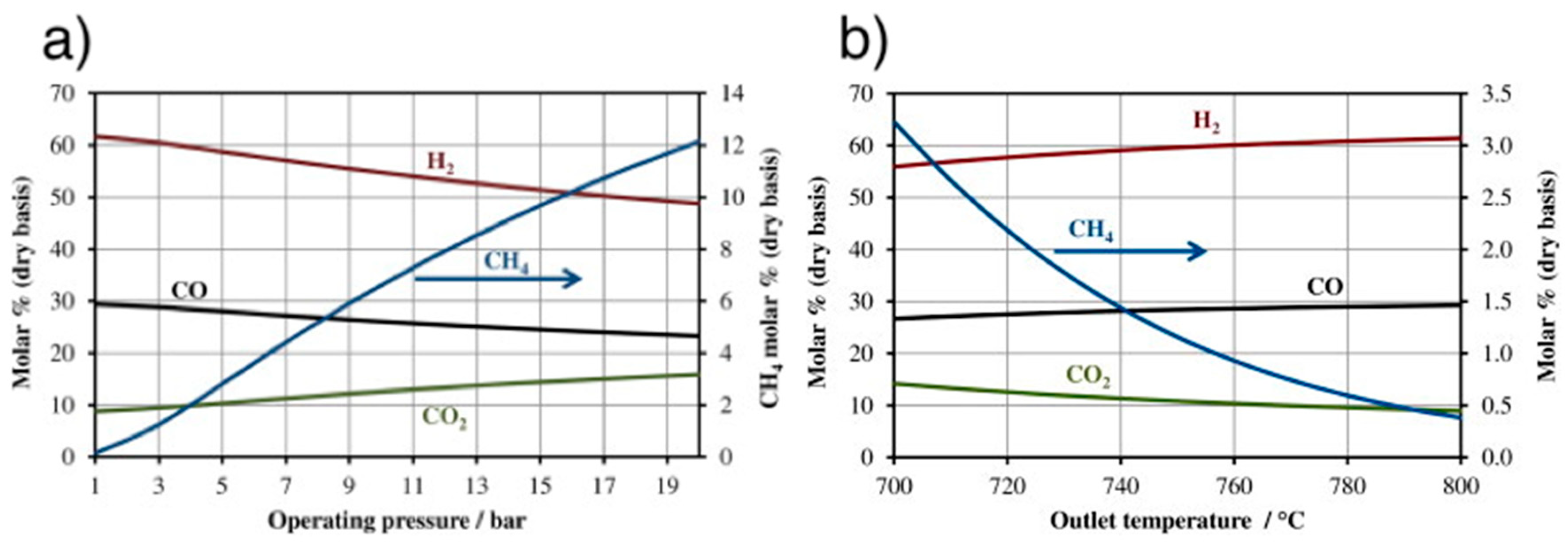{kind=link}
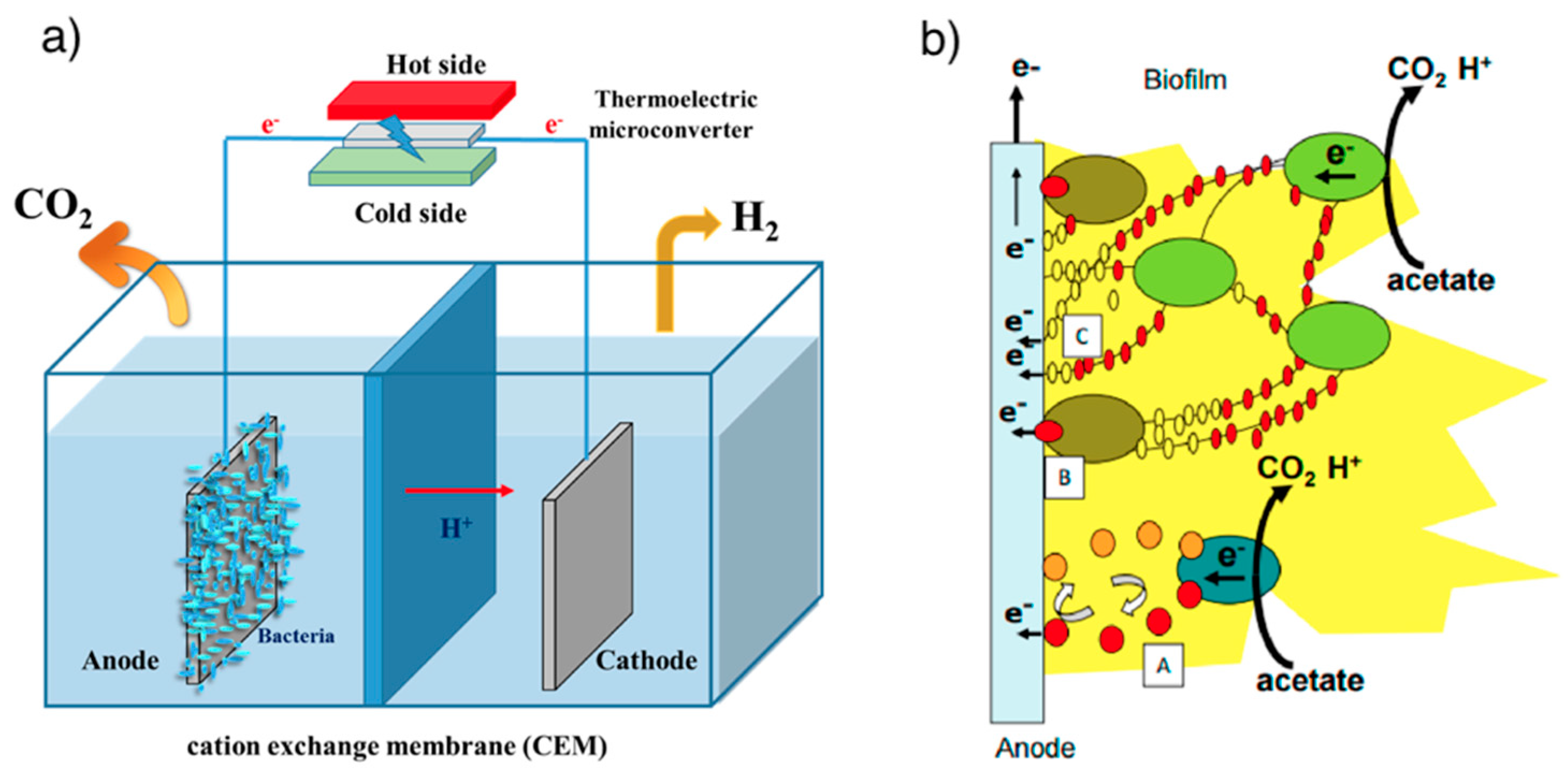{kind=link}
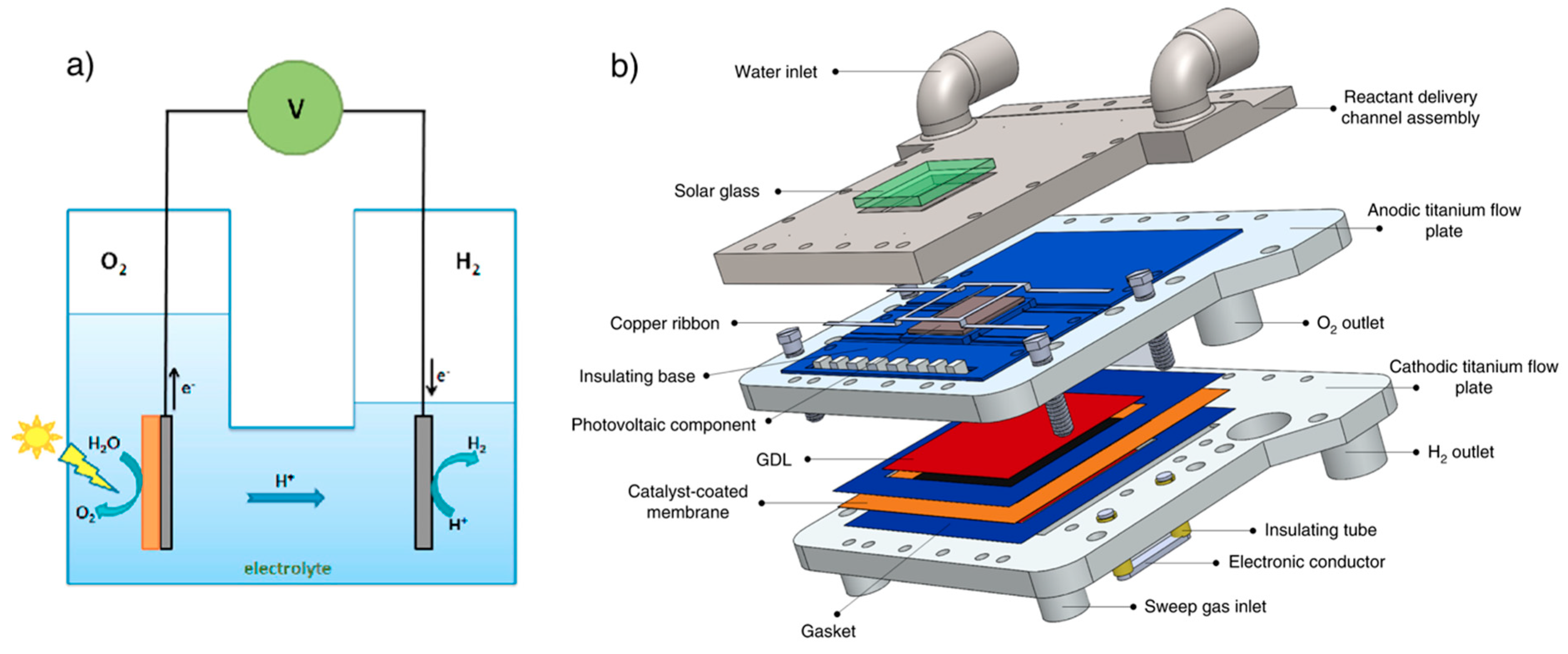{kind=link}
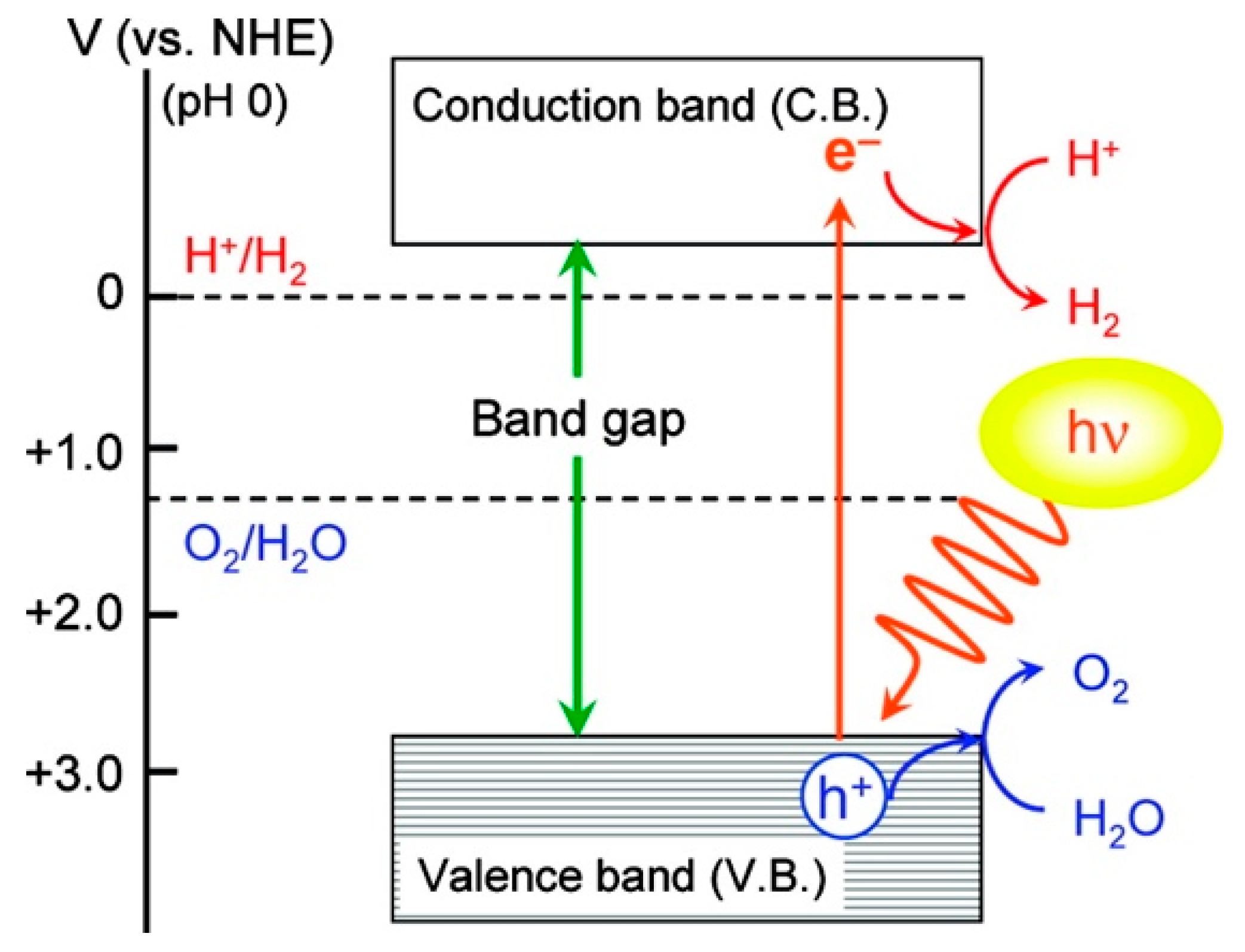{kind=link}
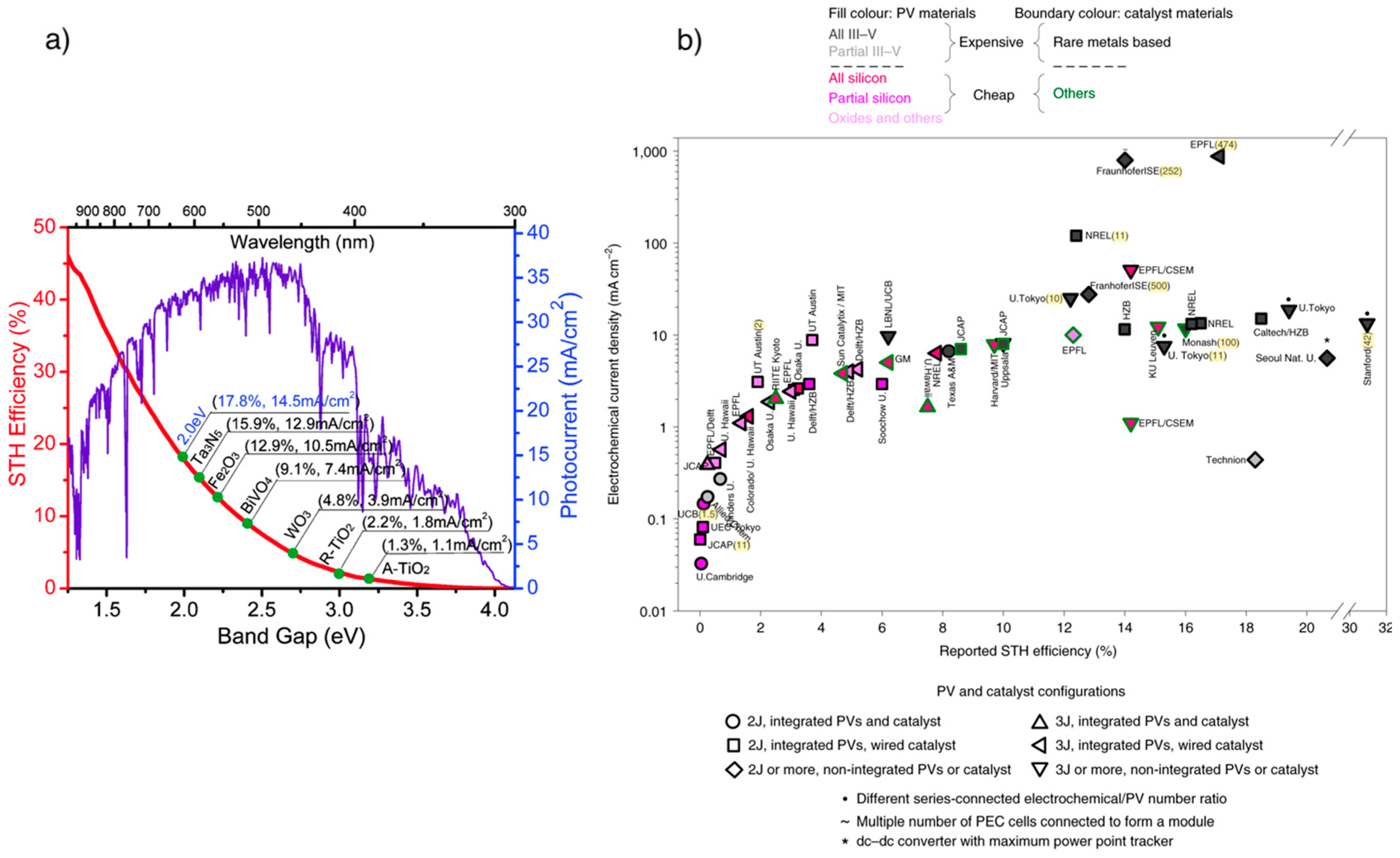{kind=link}
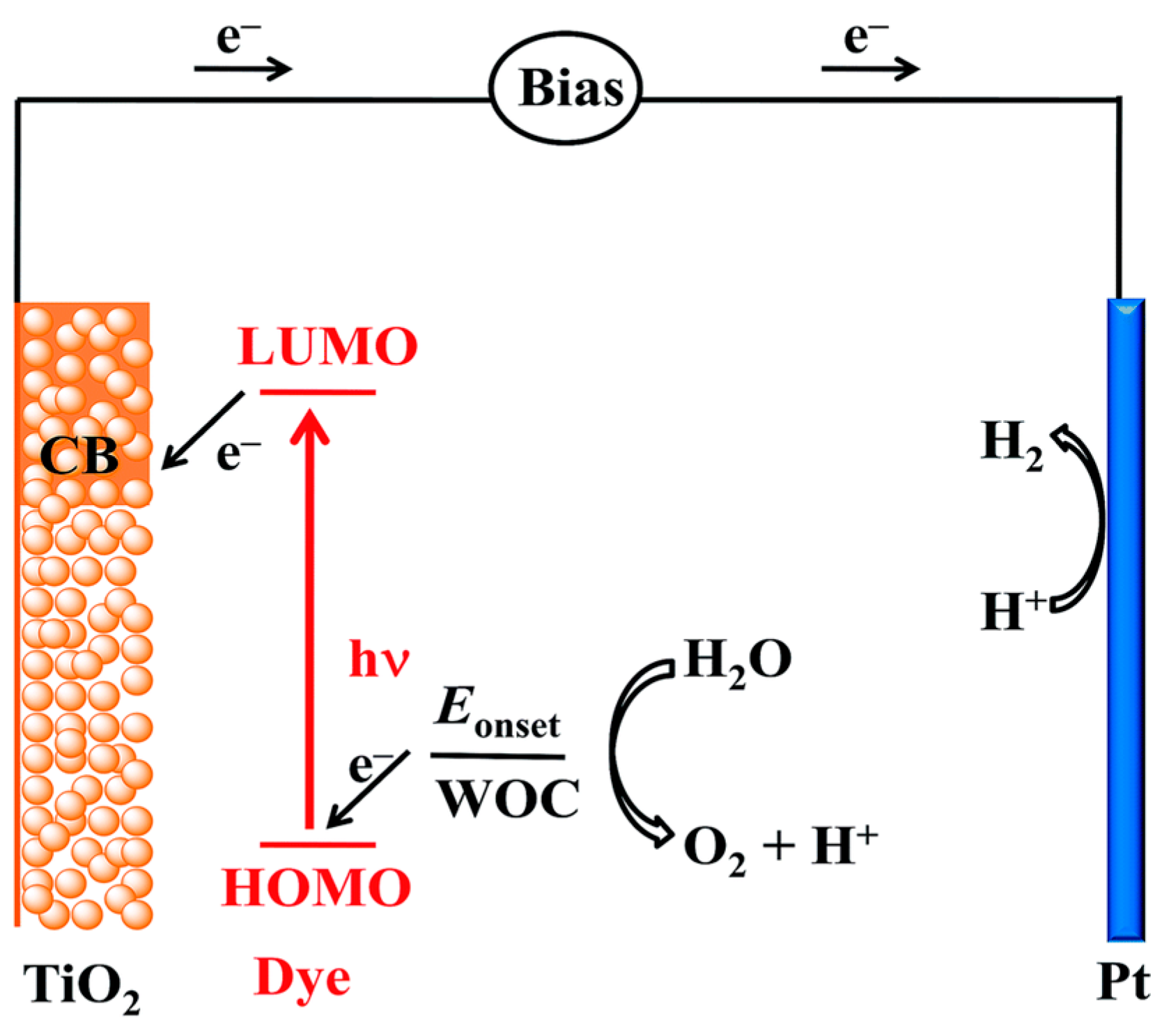{kind=link}
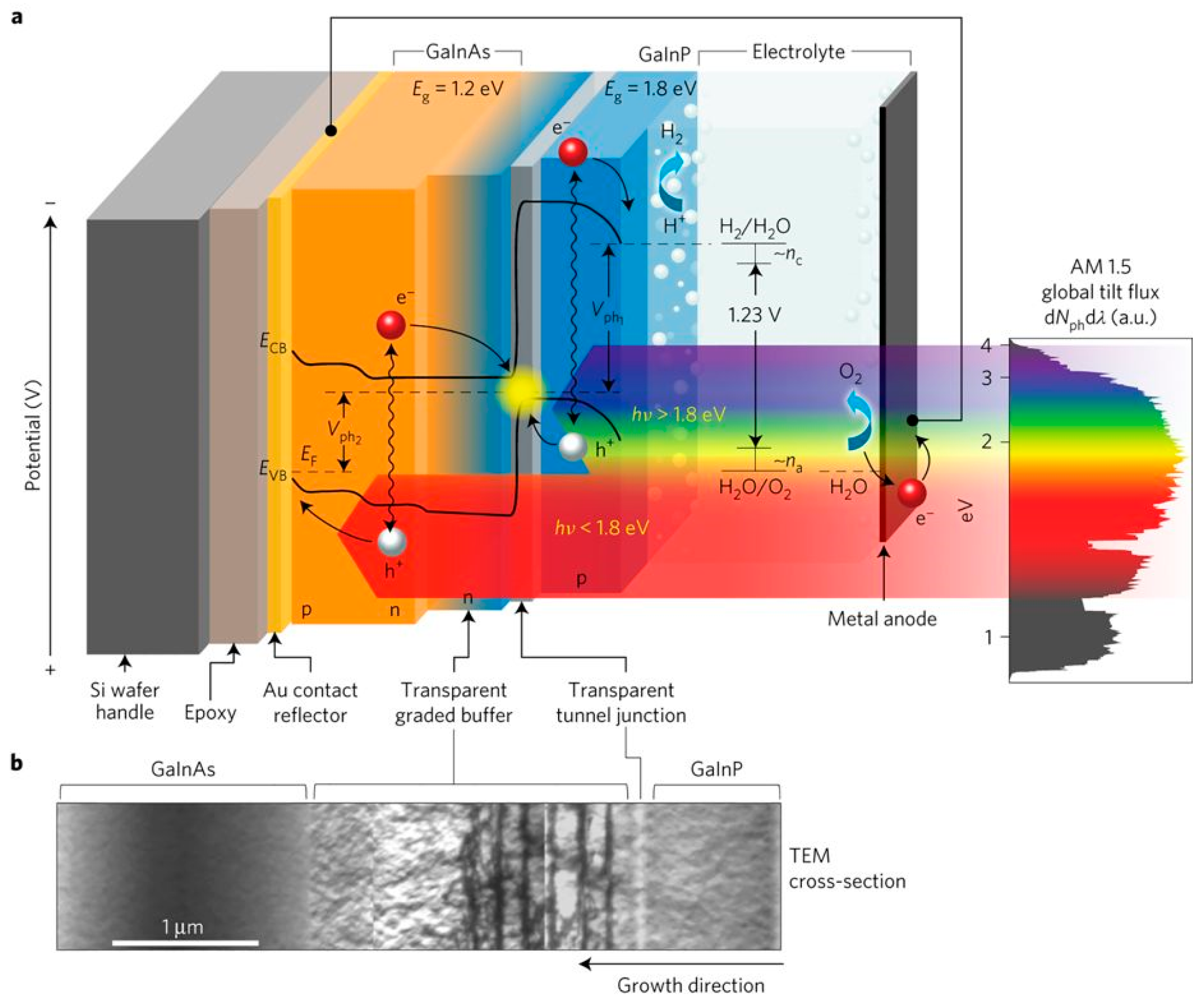{kind=link}
{kind=link}
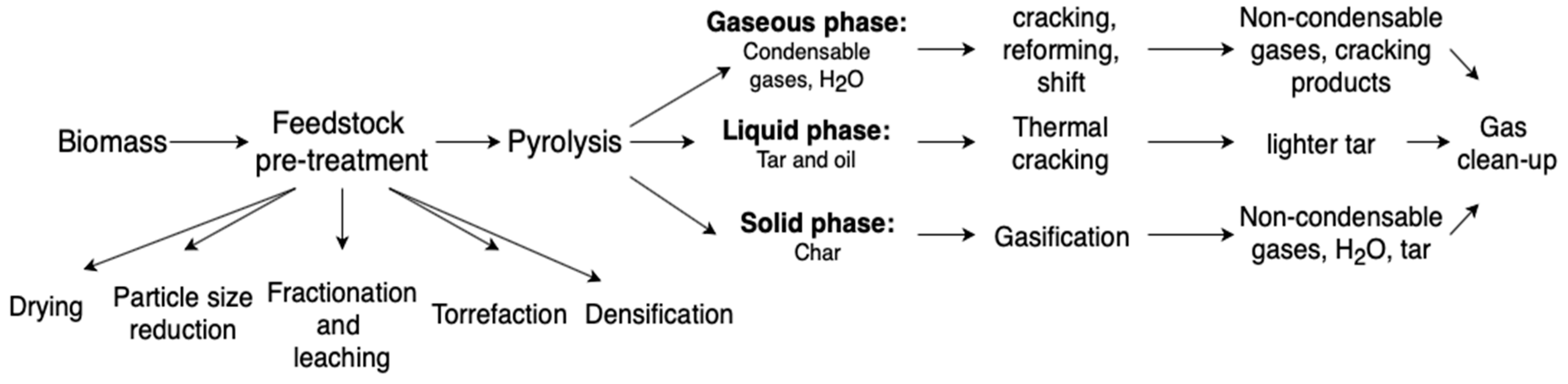{kind=link}
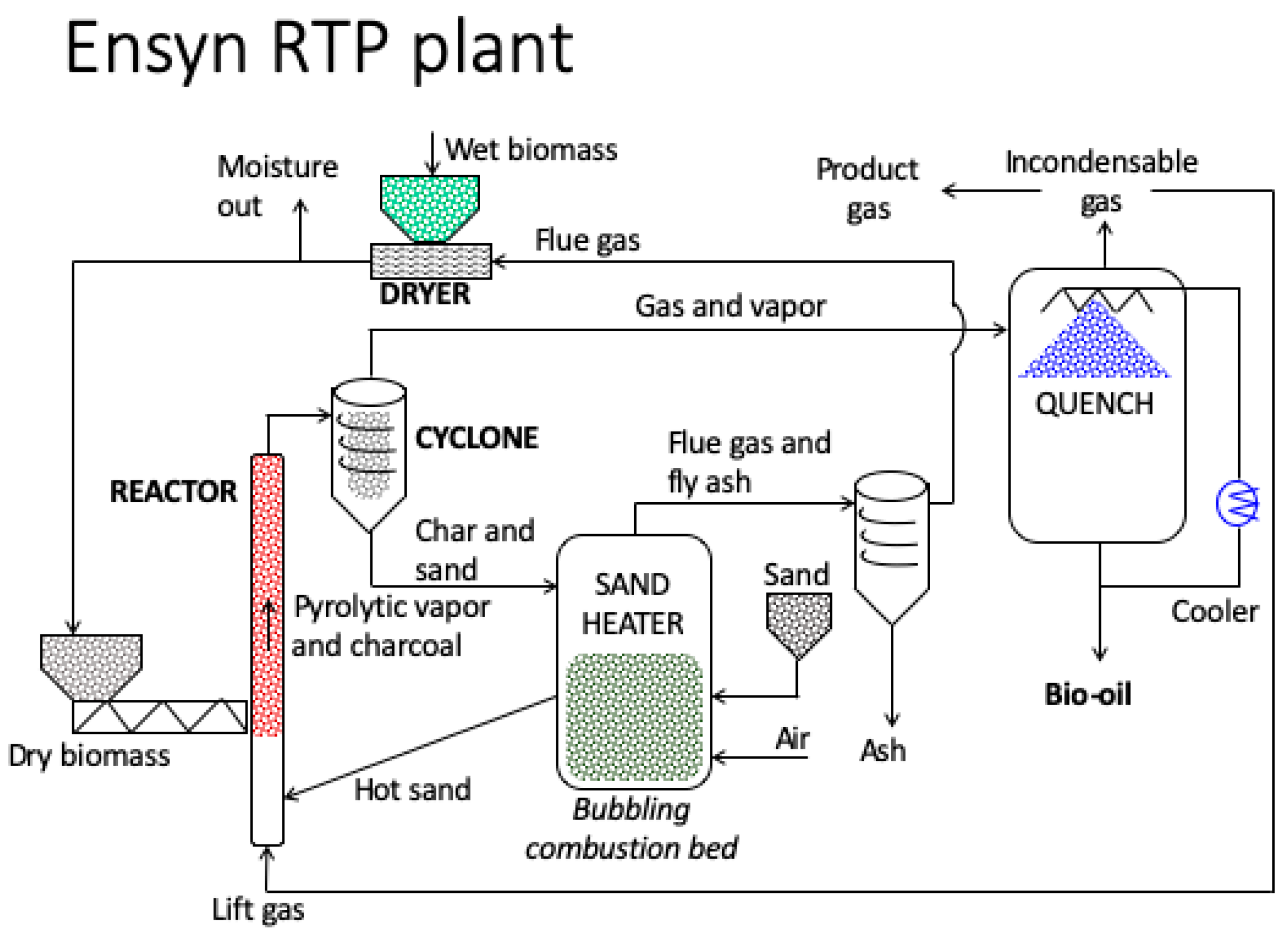{kind=link}
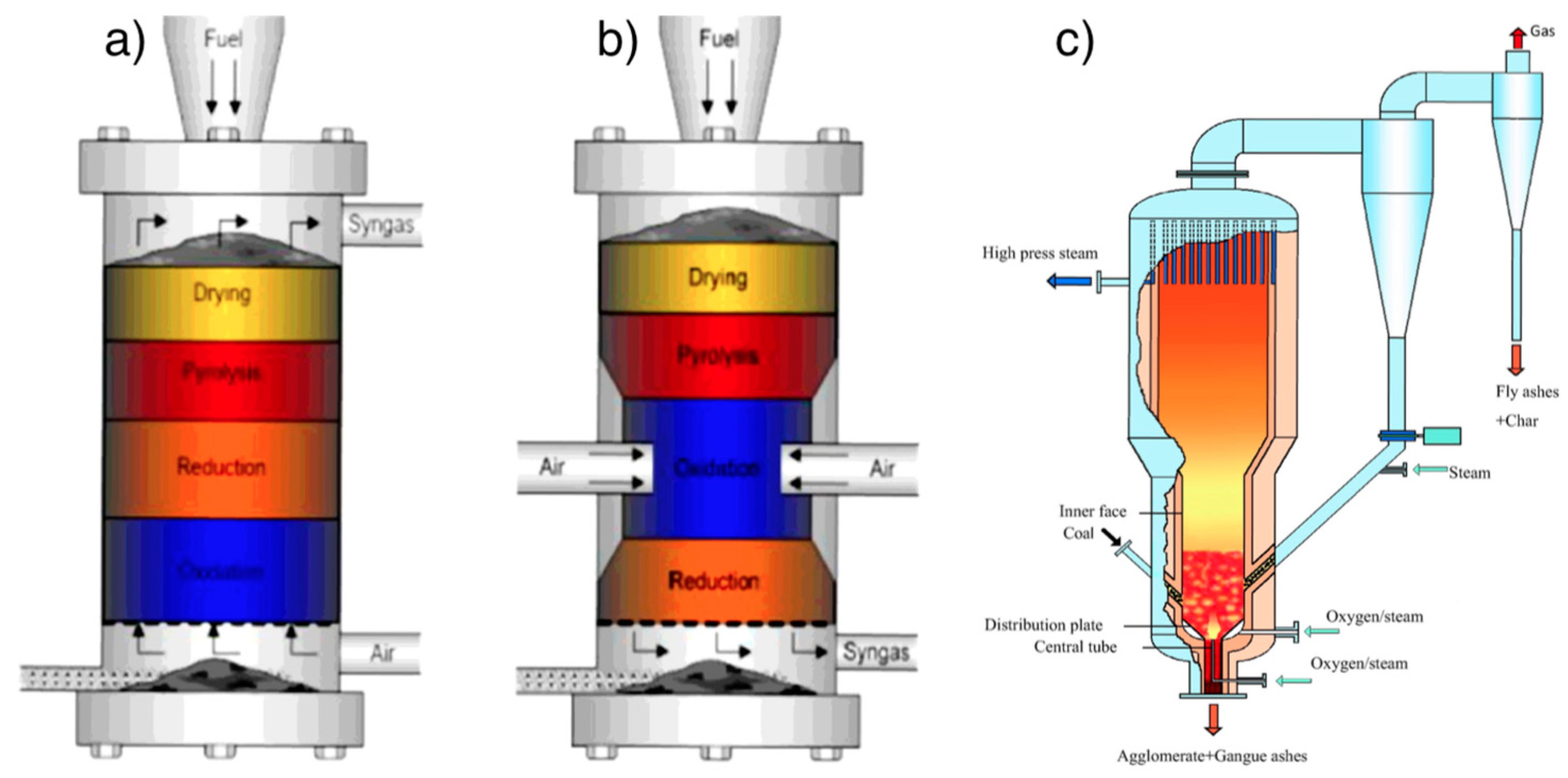{kind=link}
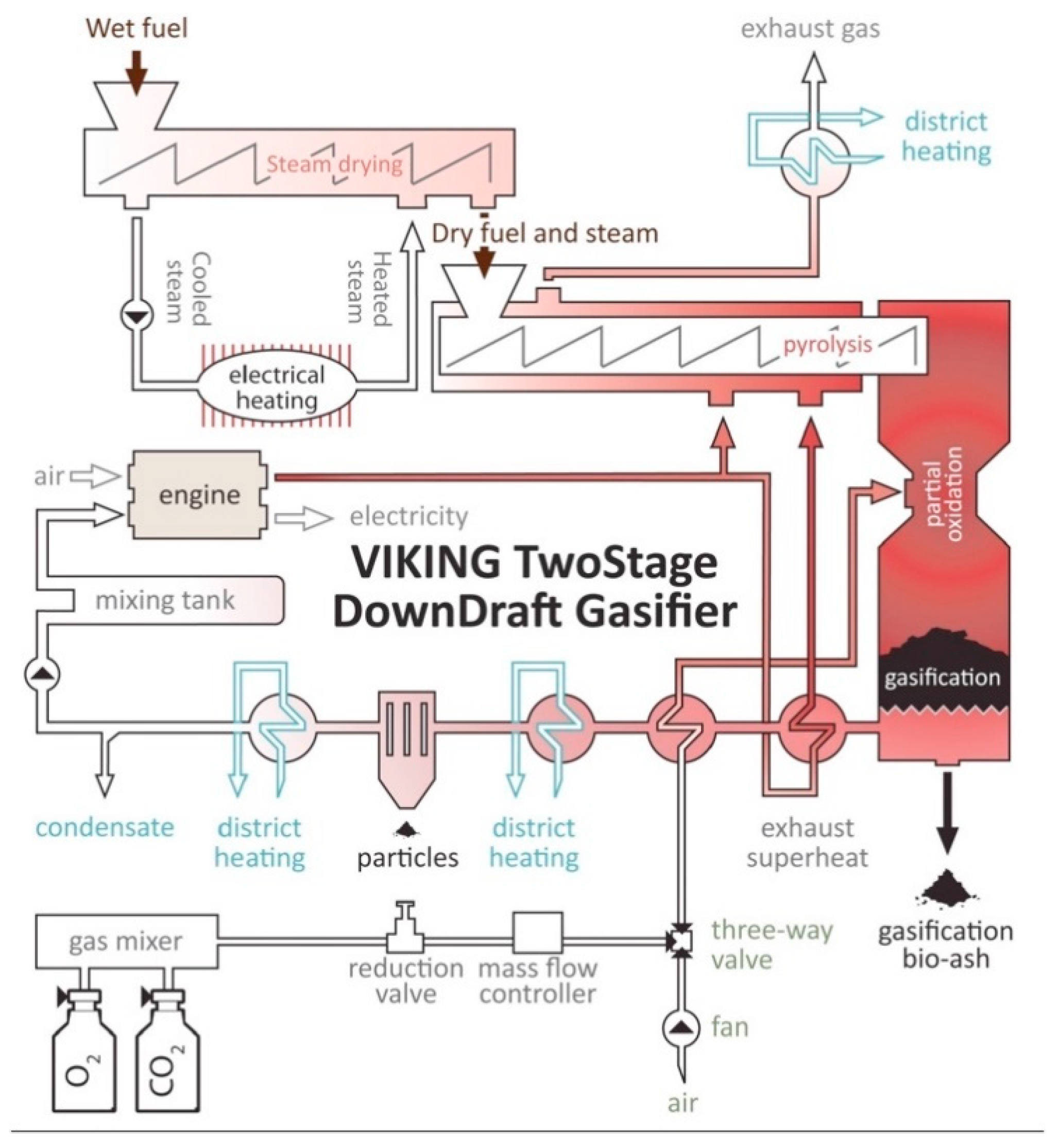{kind=link}
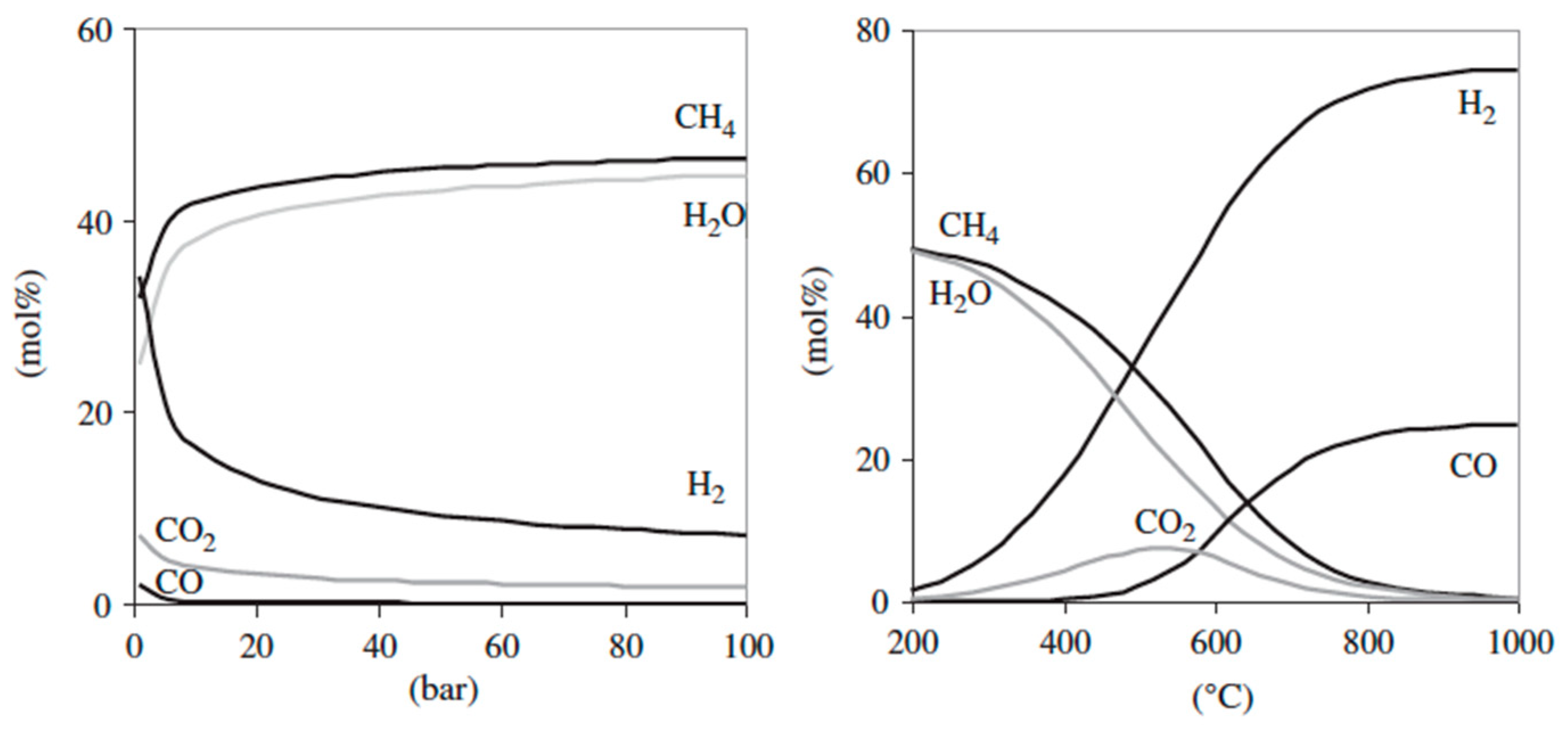{kind=link}
{kind=link}
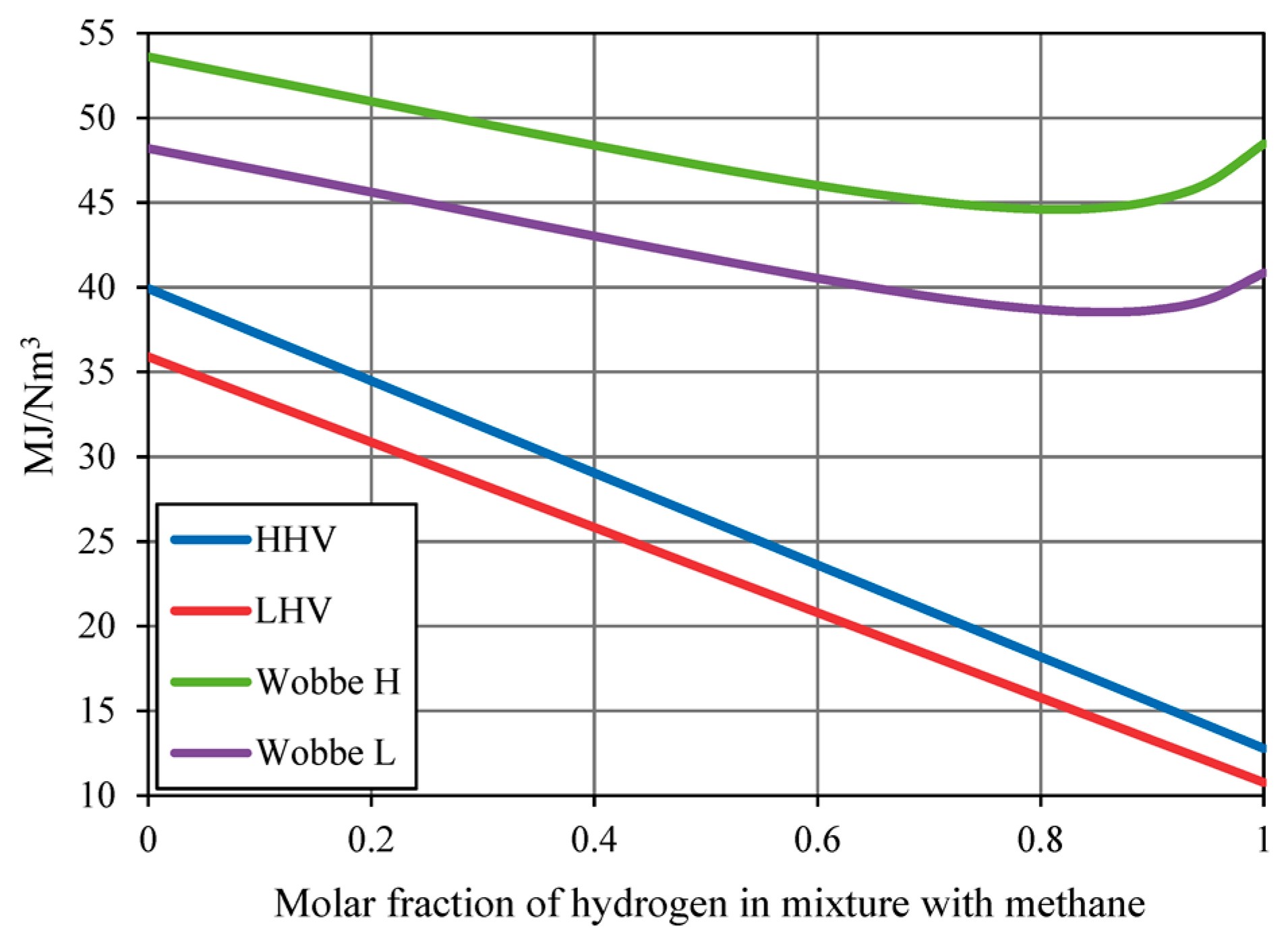{kind=link}
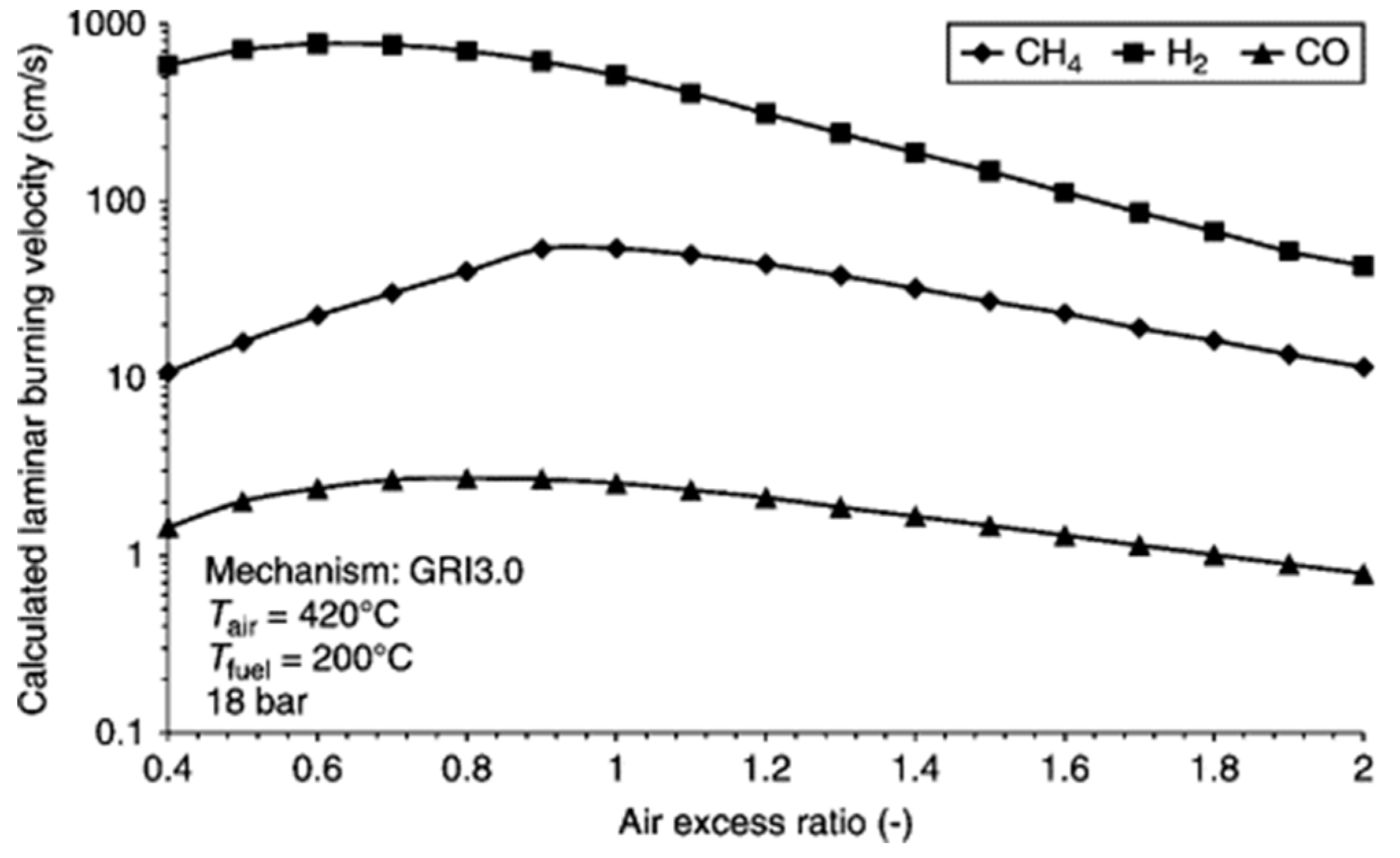{kind=link}
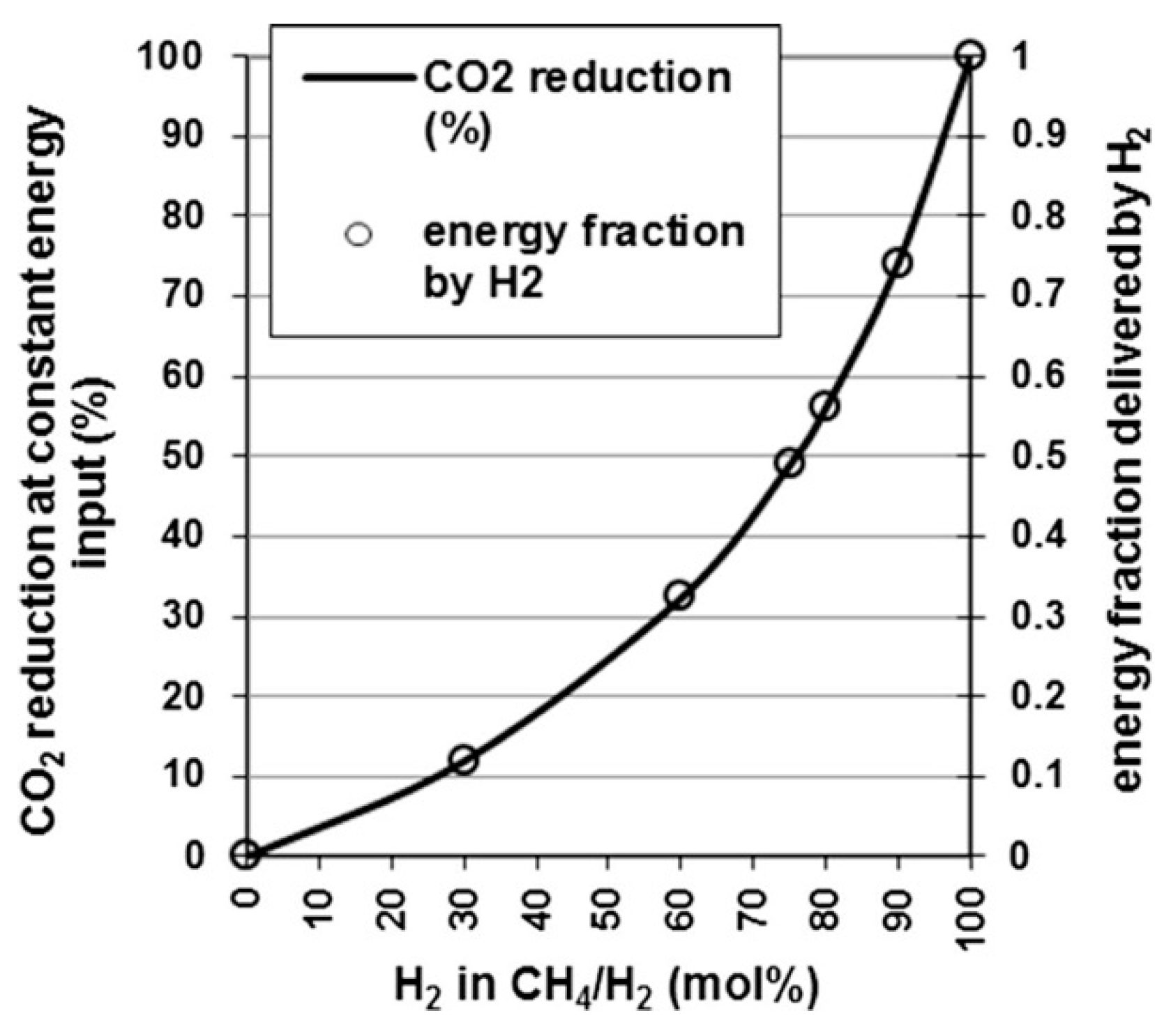{kind=link}
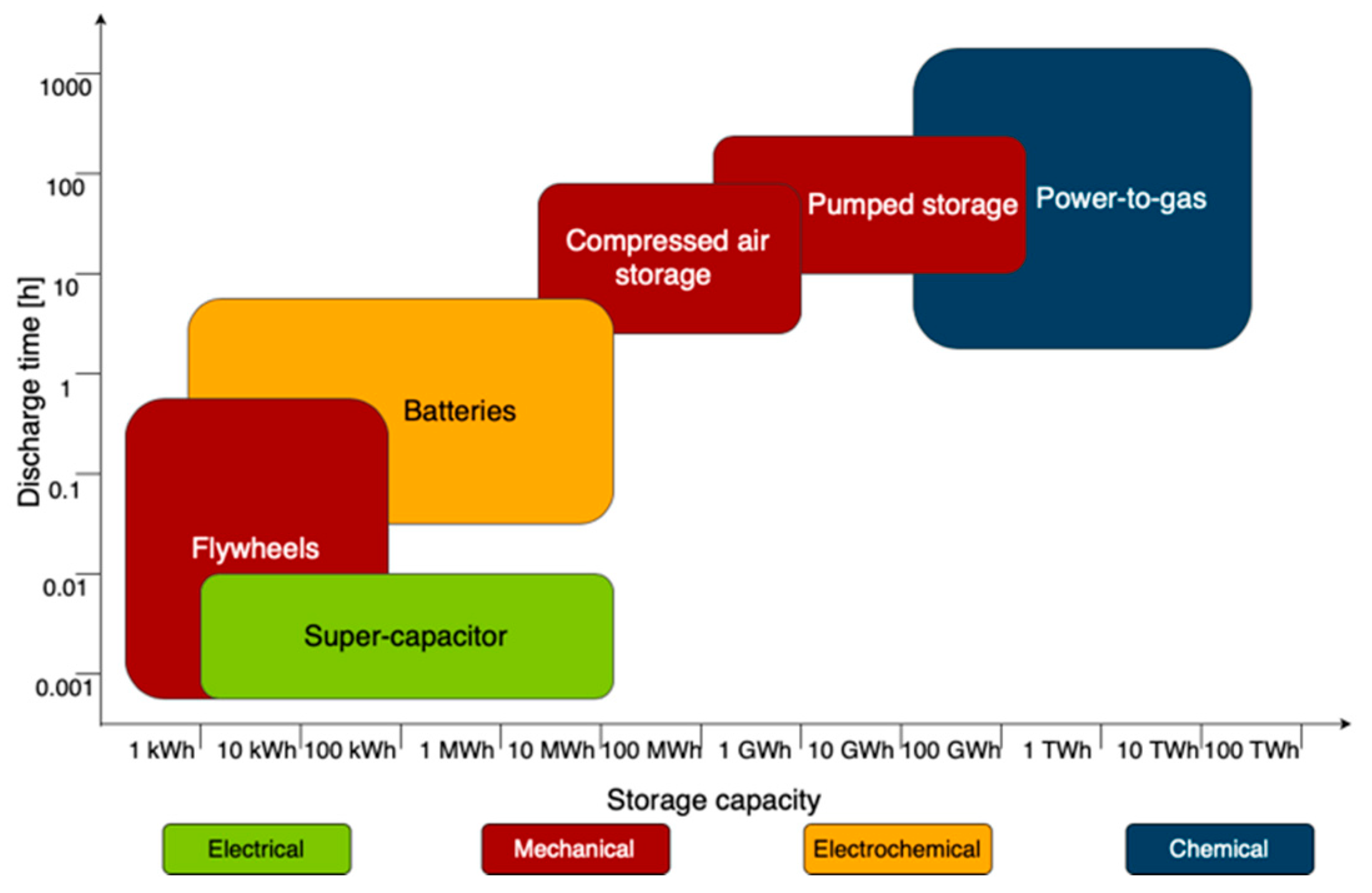{kind=link}
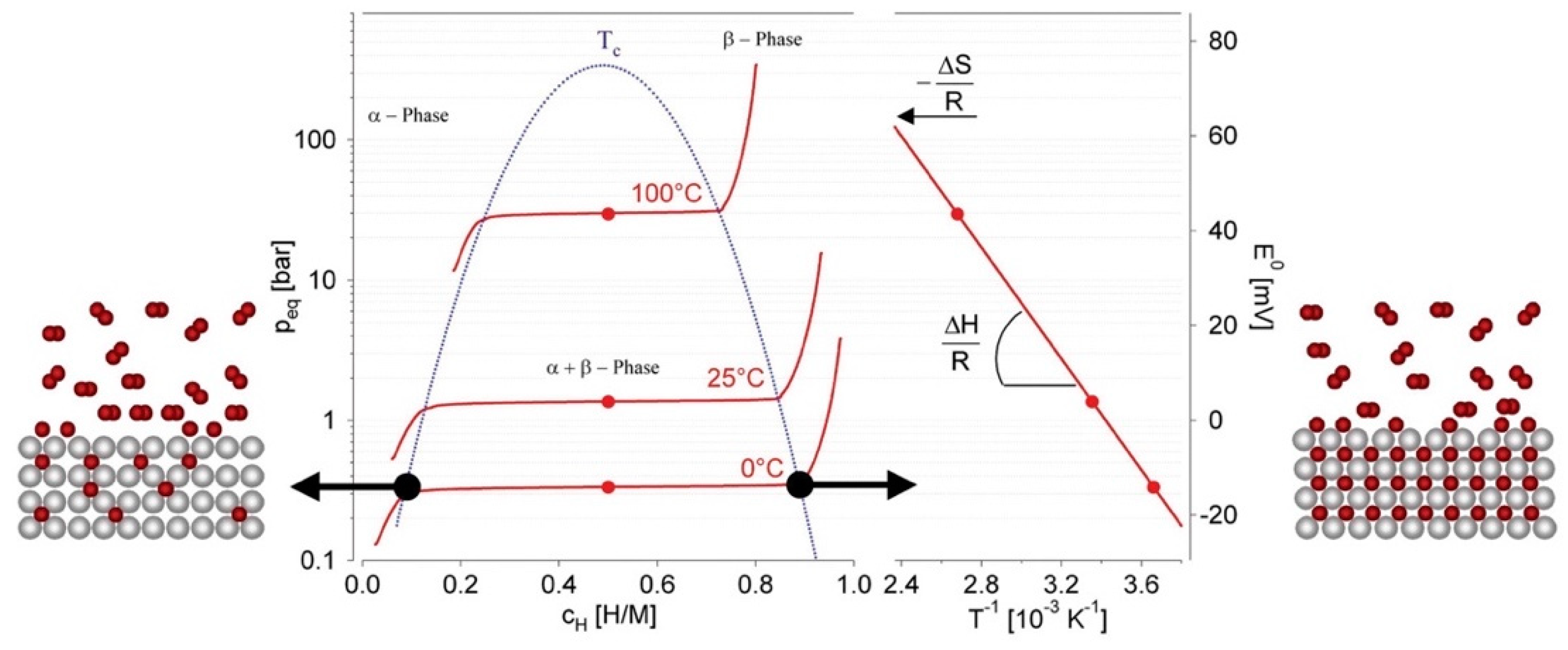{kind=link}
| Parameter | Unit | Value |
|---|---|---|
| Wobbe index | MJ/m3 | 48.96 ÷ 56.92 |
| Relative density | m3/m3 | 0.555 ÷ 0.700 |
| Total sulfur | mg/m3 | 30 |
| H2S and COS | mg/m3 | 5 |
| RSH | mg/m3 | 6 |
| O2 | mol % | 0.001 |
| CO2 | mol % | 2.5 |
| Water dew point | °C at 70 bar | −8 |
| Hydrocarbon dew point | °C at 1–70 bar | −2 |
| Parameter | CH4 | H2 |
|---|---|---|
| Higher heating value [MJ/m3] | 39.82 | 12.75 |
| Relative density [m3/m3] | 0.5548 | 0.0695 |
| Wobbe index [MJ/m3] | 53.54 | 48.37 |
| Stoichiometric air requirement [mol/mol] | 9.55 | 2.39 |
| Laminar flame velocity [cm/s] | 36.7 | 275 |
| Adiabatic flame temperature [K] | 1950 | 2210 |
| Flammability limits in air by volume [%] | 5.3 ÷ 15 | 4.1 ÷ 74 |
| Diffusion coefficient [cm2/s] | 0.21 | 0.63 |
| TRL | TRL Description | Energy Conversion Efficiency [%] | Production Cost [$/kgfuel] | CO2 Emission [kgCO2/kgfuel] | Note | ||||
|---|---|---|---|---|---|---|---|---|---|
| Biomass gasification | Air | 4–9 |
| 45–60 (cold gas efficiency of energy conversion to syngas) [757] | 1.17 (2000 dry tons/day plant using forest residues)–1.33 (2000 dry tons/day plant, straw-based biohydrogen) [758] | 1.10–1.61 [758] | |||
| Steam | 60–70 [759,760] | ||||||||
| Oxygen | 65–80 [528,761] | ||||||||
| Thermolysis | 1–3 | Laboratory scale | 10 [68] | 7.98–8.40 [34] | - | No pilot plant, more attention for two-step thermochemical cycles | |||
| Two-step Thermochemical cycle (ZnO/Zn) | 4–7 |
| 17(CY 2015 case assuming 70% ZnO dissociation and heliostats cost of 126.5$/m2)–21 (CY 2025 case assuming 85% ZnO dissociation and heliostat cost of 90$/m2) [762] | 6.07–4.18 [763] | - | Further research on reactor materials, quenching of products, separation of reactants and solar concentration technologies | |||
| Three-step thermochemical cycle (S–I) | 19 [149] | 5.01–4.68 [763] | - | ||||||
| Photoelectrochemical (PEC) cell | 1–4 | Scale-up of PEC reactors | 3–5 (solar to fuel efficiency demonstrated in single-junction devices based on low cost thin-film materials)12–16 (solar to fuel efficiency demonstrated in tandem devices based on high-quality crystalline semiconductors) [441] | 11.4 [315,764] | - | Pilot plants planned for next few year | |||
| Electrolysis | PV | Wind | PV | Wind | |||||
| Alkaline electrolyzer | 7–9 |
| 10–12 | 20–24 (photovoltaic efficiency of 20% and the wind power efficiency of 40% multiplied for the electrolyzer efficiency: AEC 50%–60%, PEMEC 55%–70%, SOEC 40%–60%, MEC 78% [178]) | 1.8 (forecasted production cost of hydrogen in 2050 through water electrolysis using electricity from PV or offshore wind)–4.3 (hydrogen production cost in 2020 through water electrolysis using electricity from PV or offshore wind) [765] | 3–6.7 [765] | - | Three planned first-of-a-kind commercial demonstration plants (10-20-700 MWel) | |
| PEM electrolyzer | 4–8 |
| 11–14 | 22–28 | - | Three planned first-of-a-kind commercial demonstration plants | |||
| Solid oxide electrolyzer | 3–5 |
| 8–12 | 16–24 | - | Planned the first commercial plant (20 MWel) | |||
| Microbial electrolyzer | 1–4 |
| 15.5 | 31 | 11.5 (NREL TEA calculation assuming 90% hydrolysis efficiency) [766] | Pilot plants are mostly fed by domestic wastewater | |||
| CO/CO2 methanation | 5–7 |
| 74–82 [606] | 2.83–4.17 [767] | - (No CO2 emissions if the CO2 conversion efficiency is 100%) | Two planned demonstration plants of 10 MW (MeGa-StoRE project) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozzi, E.; Minuto, F.D.; Lanzini, A.; Leone, P. Green Synthetic Fuels: Renewable Routes for the Conversion of Non-Fossil Feedstocks into Gaseous Fuels and Their End Uses. Energies 2020, 13, 420. https://doi.org/10.3390/en13020420
Rozzi E, Minuto FD, Lanzini A, Leone P. Green Synthetic Fuels: Renewable Routes for the Conversion of Non-Fossil Feedstocks into Gaseous Fuels and Their End Uses. Energies. 2020; 13(2):420. https://doi.org/10.3390/en13020420
Chicago/Turabian StyleRozzi, Elena, Francesco Demetrio Minuto, Andrea Lanzini, and Pierluigi Leone. 2020. "Green Synthetic Fuels: Renewable Routes for the Conversion of Non-Fossil Feedstocks into Gaseous Fuels and Their End Uses" Energies 13, no. 2: 420. https://doi.org/10.3390/en13020420
APA StyleRozzi, E., Minuto, F. D., Lanzini, A., & Leone, P. (2020). Green Synthetic Fuels: Renewable Routes for the Conversion of Non-Fossil Feedstocks into Gaseous Fuels and Their End Uses. Energies, 13(2), 420. https://doi.org/10.3390/en13020420