Dehydration Leads to Hydrocarbon Gas Formation in Thermal Degradation of Gas-Phase Polyalcohols



Abstract
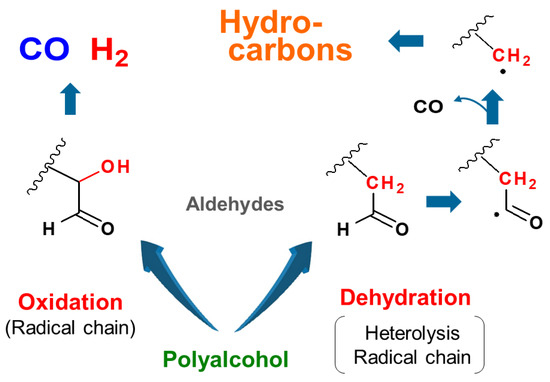
1. Introduction
2. Materials and Methods
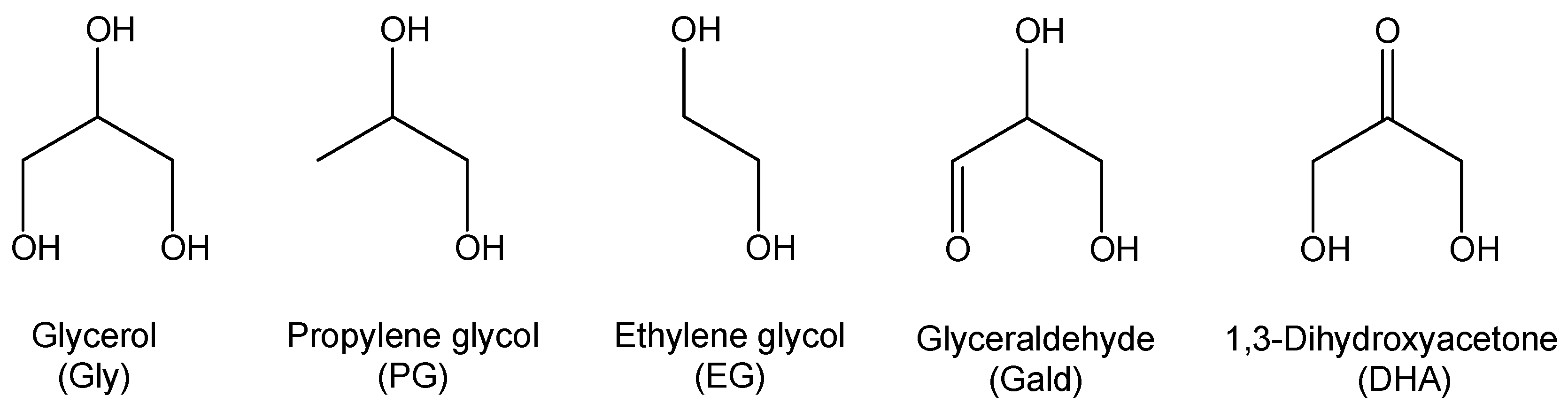
2.1. Materials
2.2. Pyrolysis
2.3. Product Analysis
2.4. Computational Methods
3. Results and Discussion
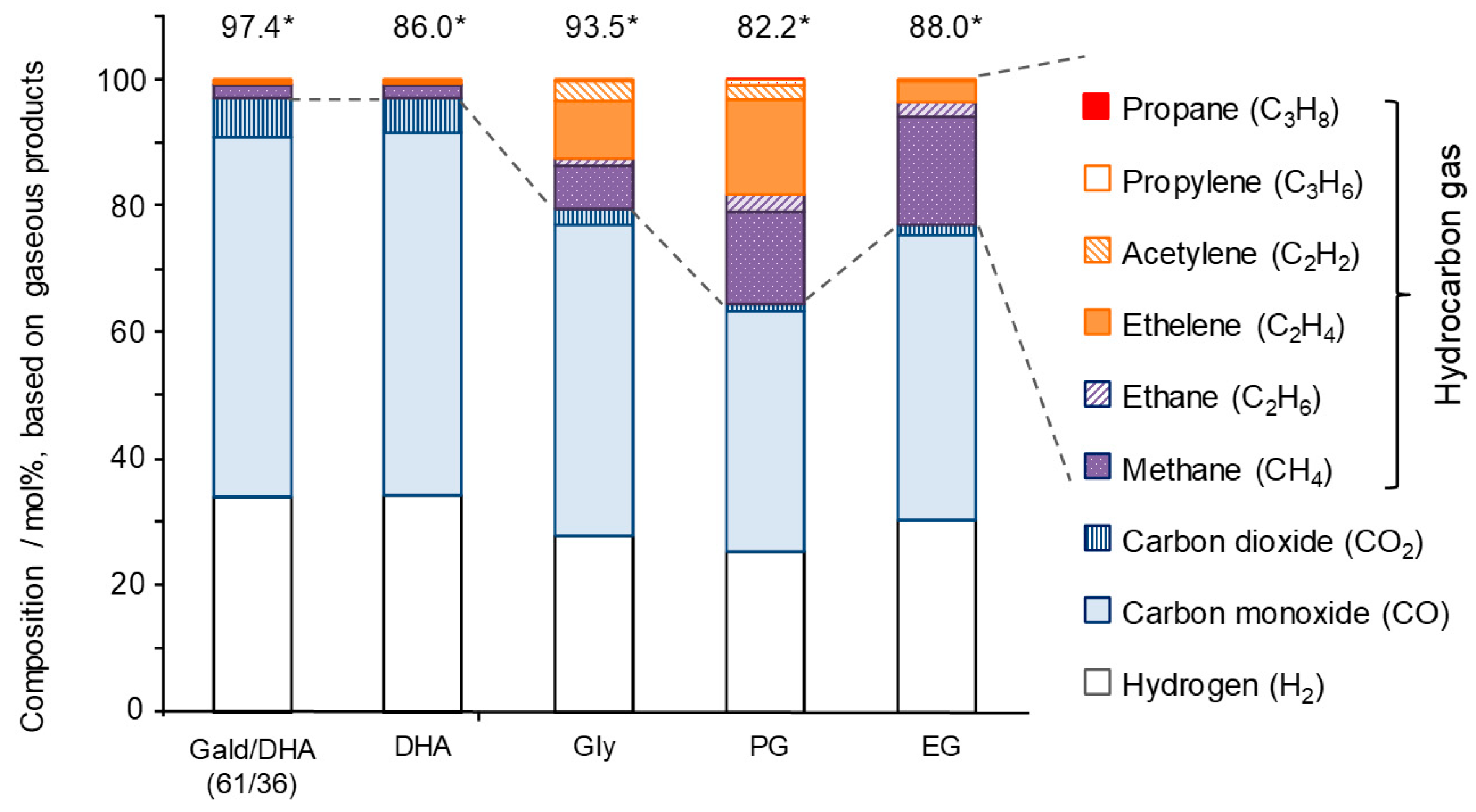
3.1. Experimental Study of Gas Formation via Fragmentation of Intermediates
3.2. Theoretical Study of Intermediate Formation
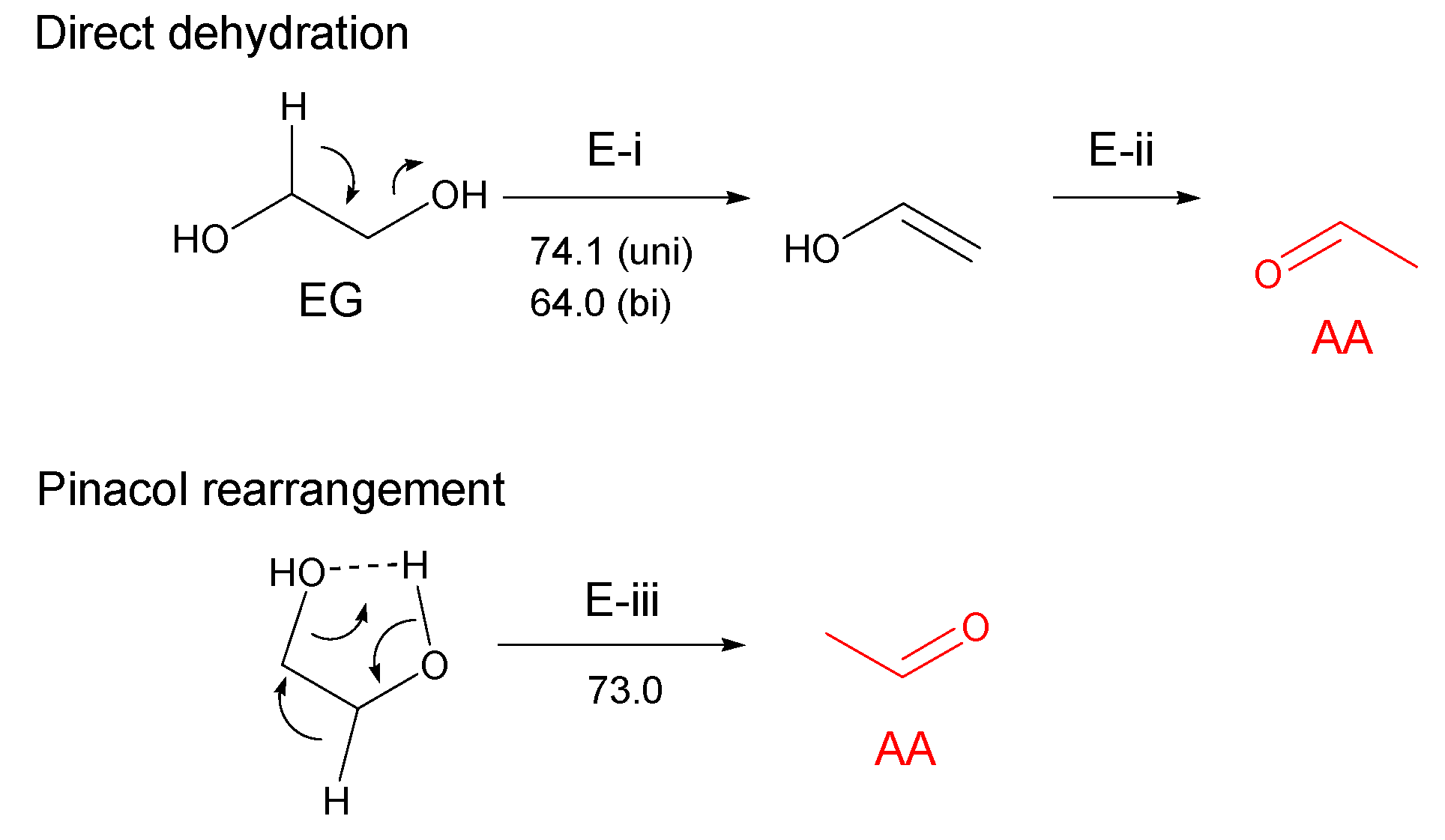
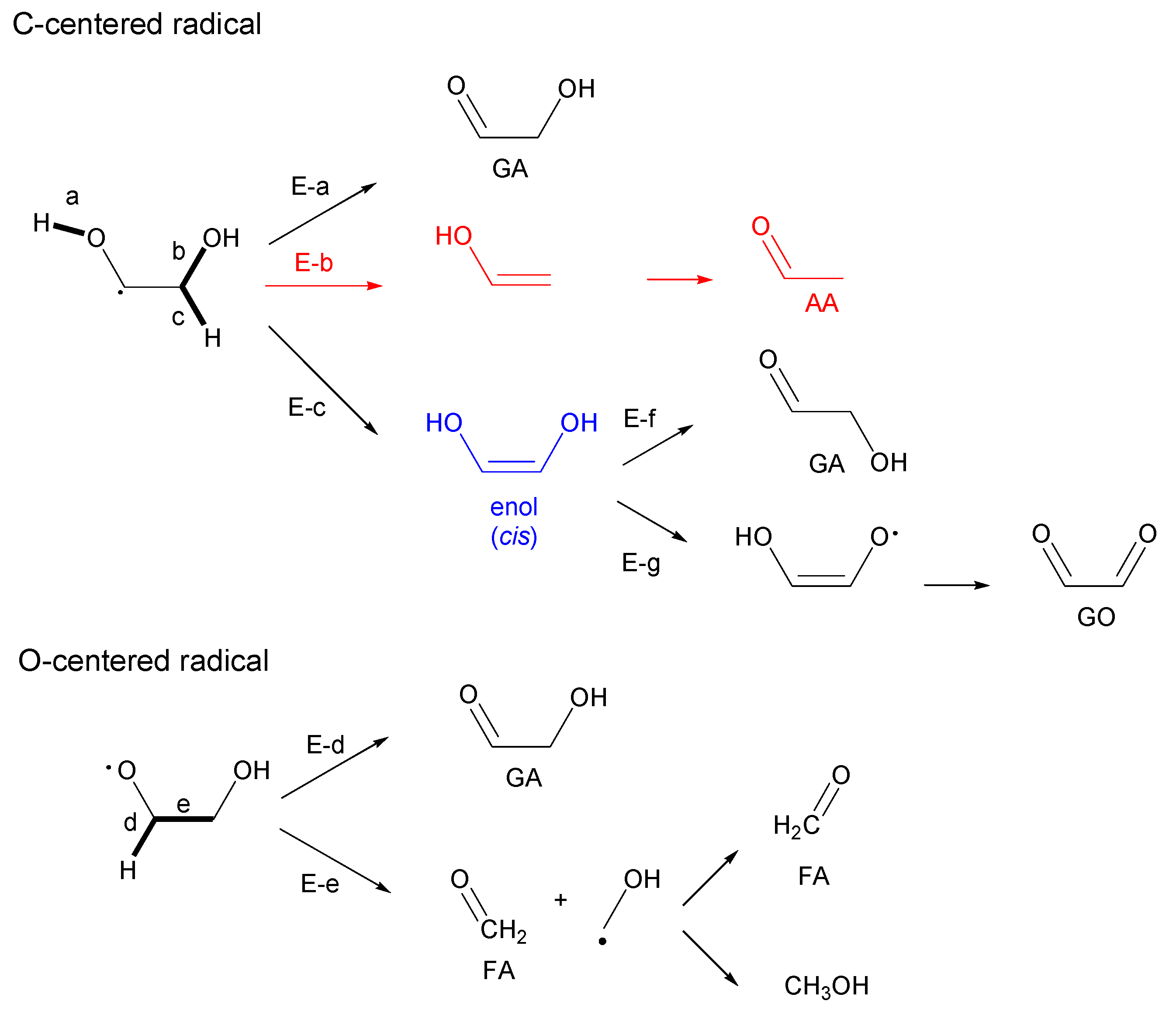
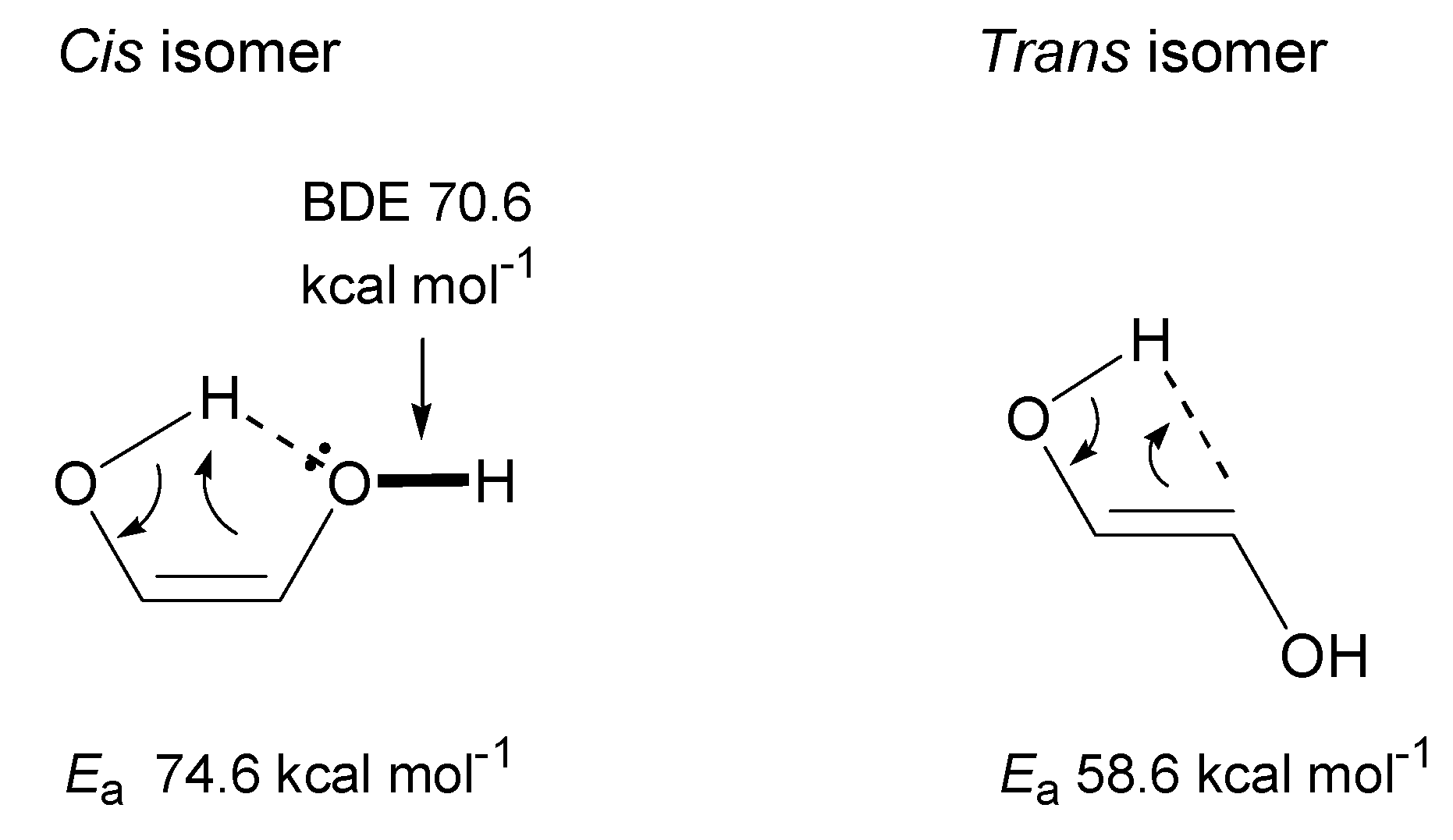
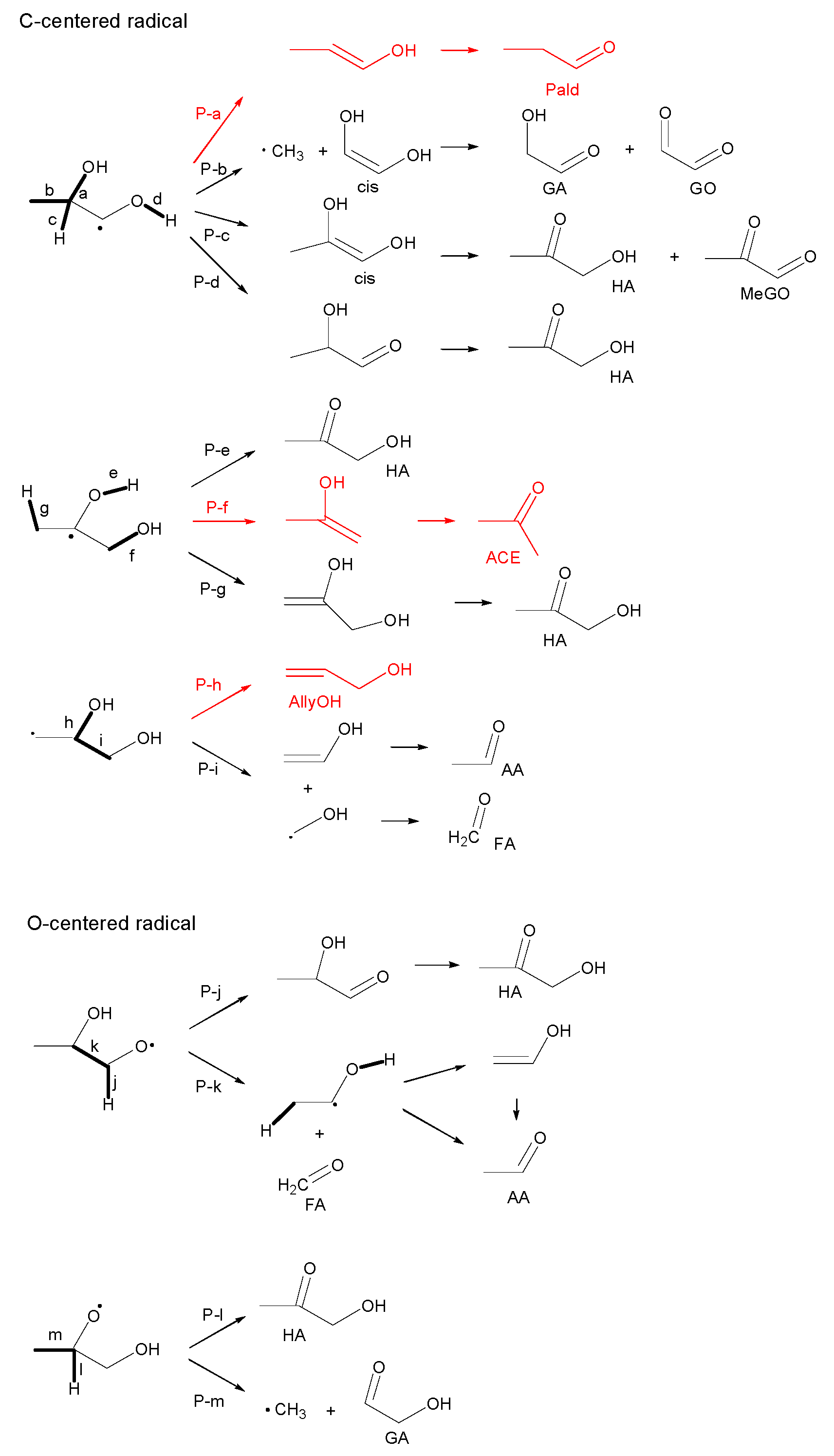
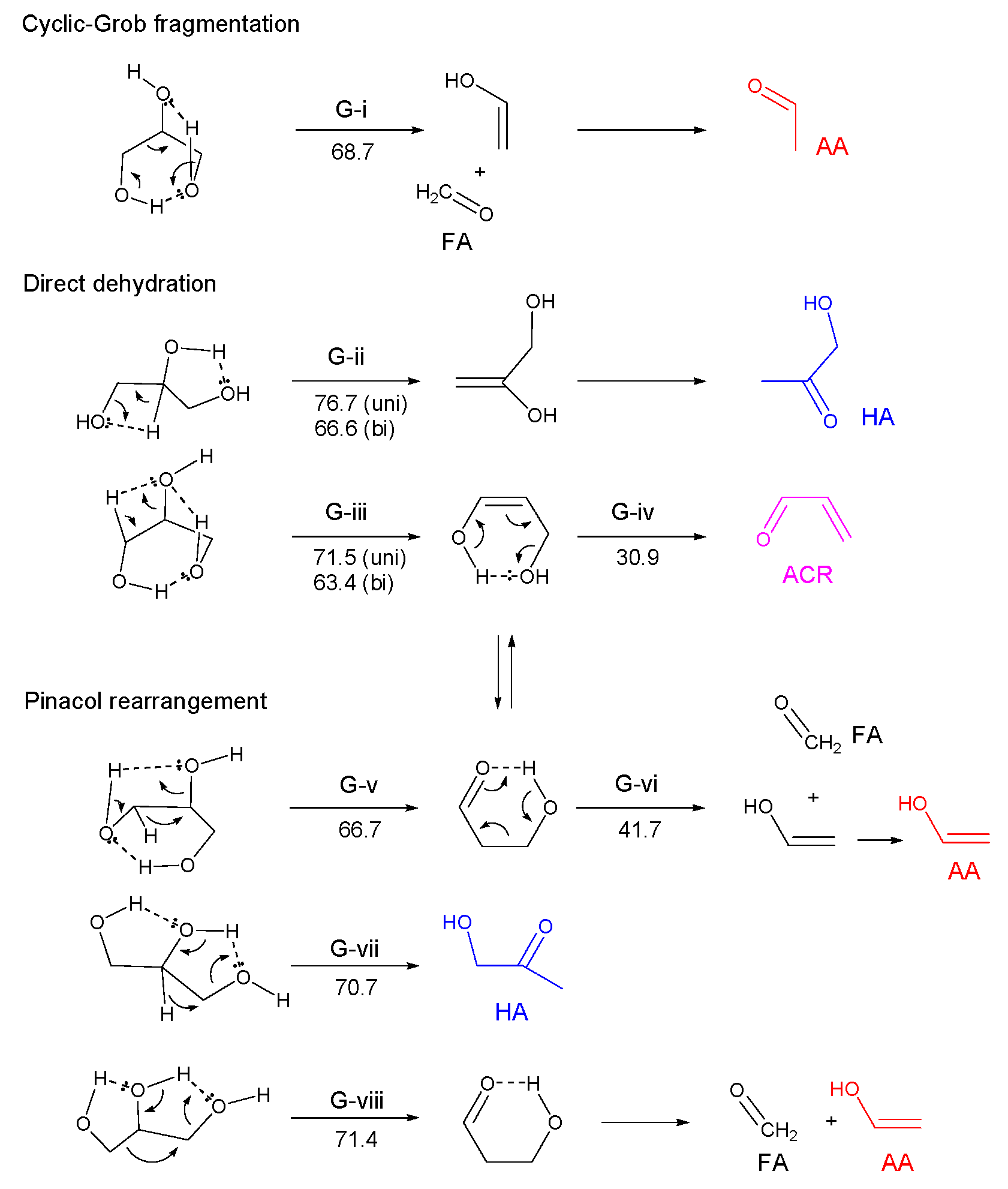
3.2.1. Ethylene Glycol (EG)
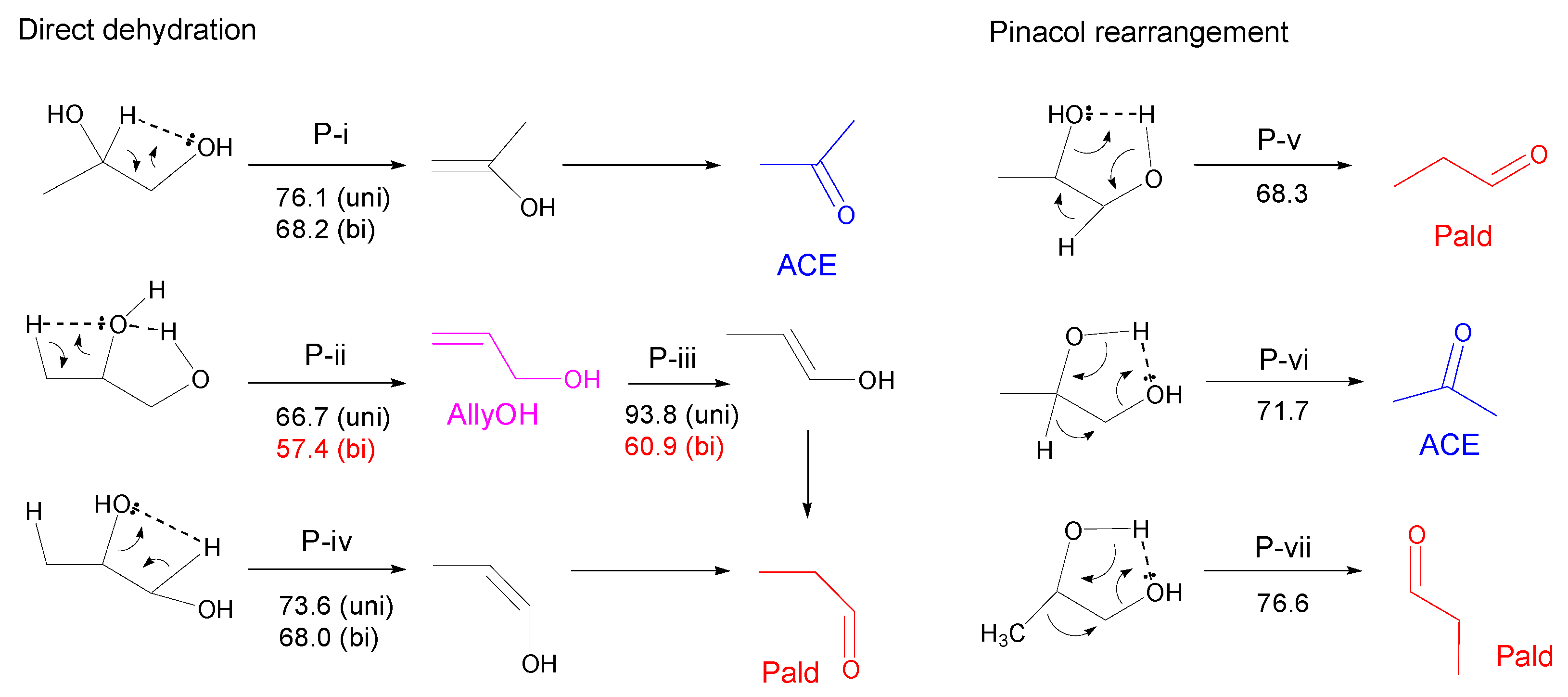
3.2.2. Propylene Glycol (PG)
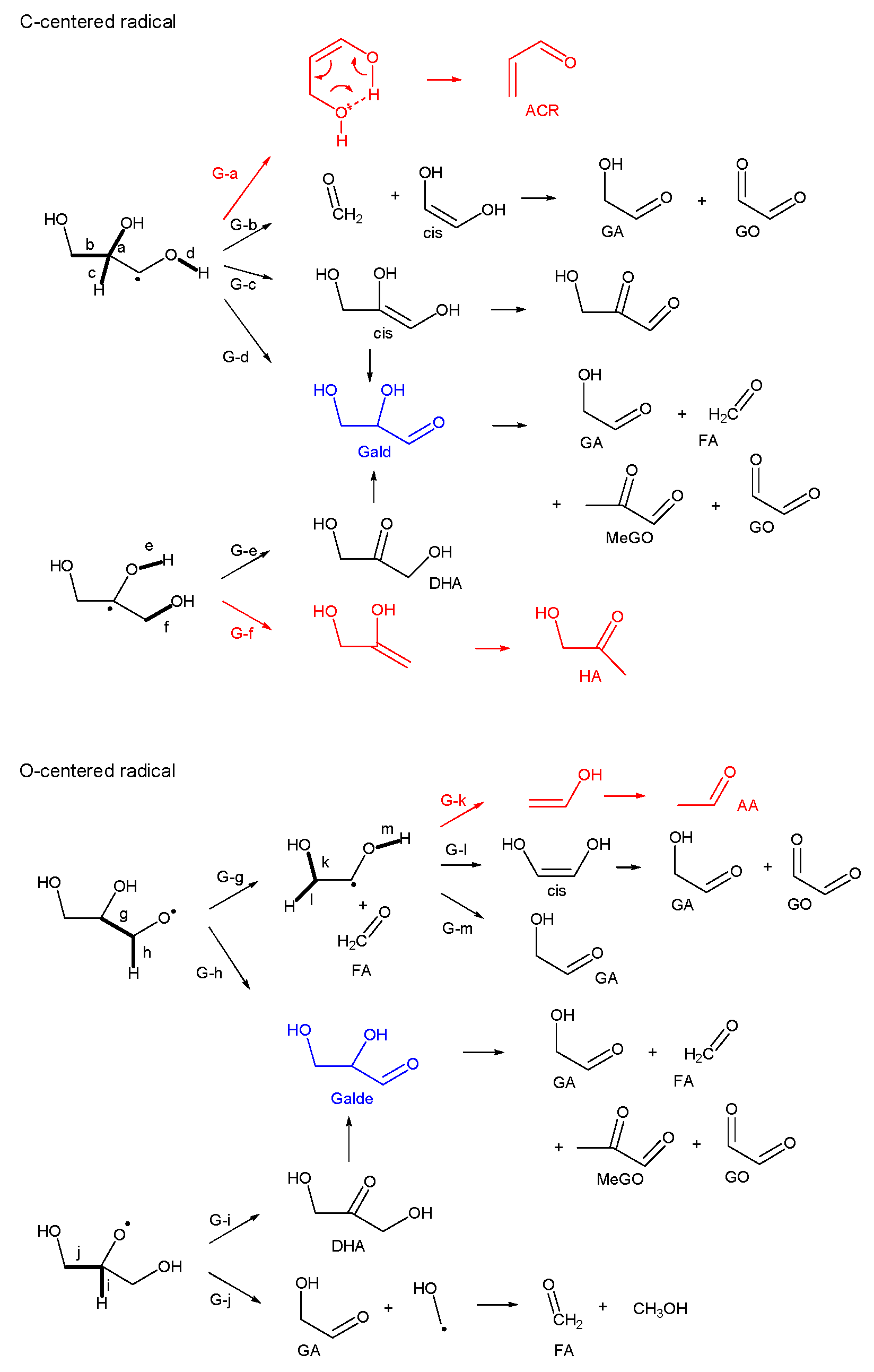
3.2.3. Glycerol (Gly)
3.3. Discussion for Syngas and Hydrocarbon Gas Production from Polyalcohol
4. Conclusions
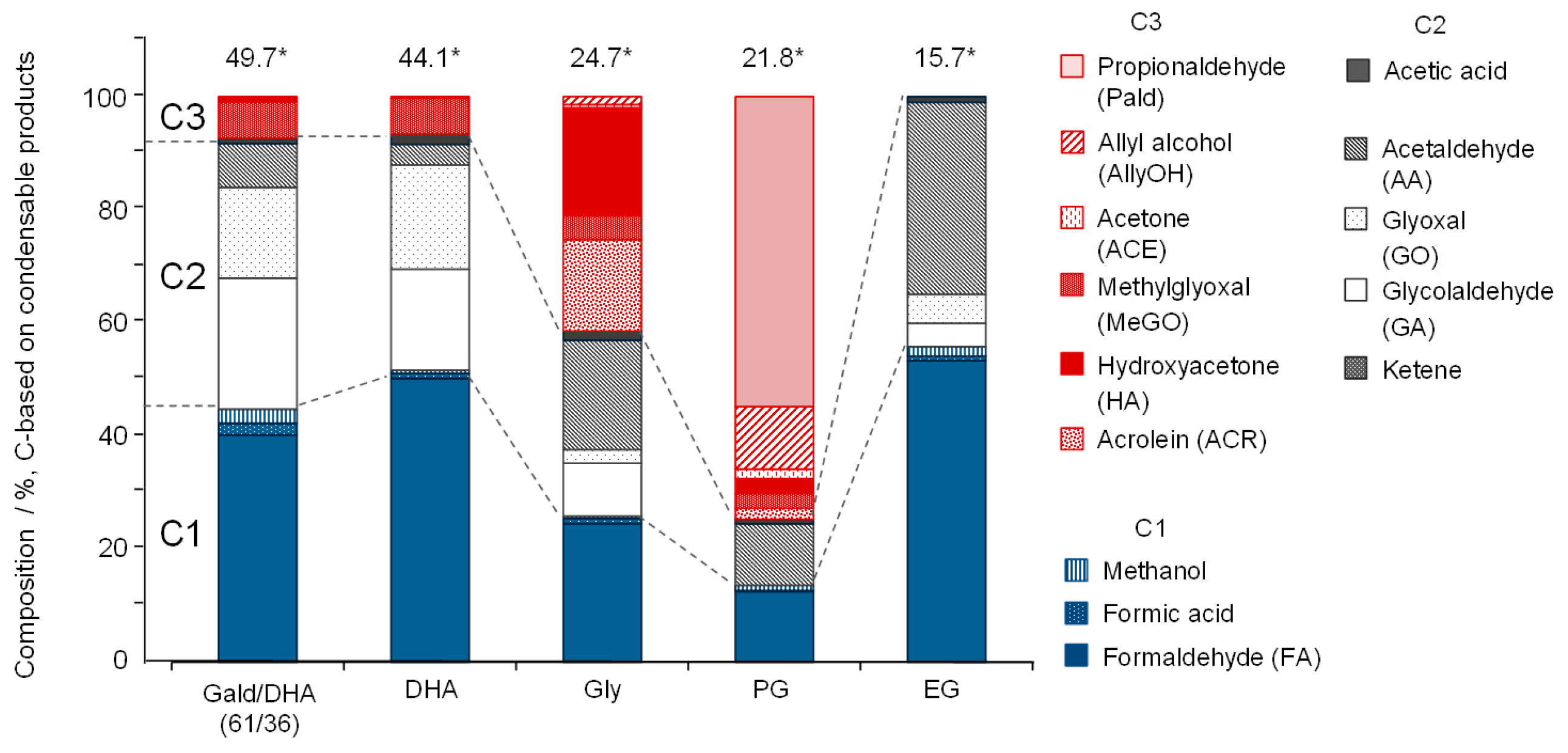
- All polyalcohols become reactive at 600 °C and produce condensable products in 15.7–24.7% yields (C-based), corresponding to 33.9–38.4% based on the amounts of reacted polyalcohols. Because the polyalcohols are completely converted into non-condensable gases at 800 °C, the condensable products observed at 600 °C act as gas-forming intermediates.
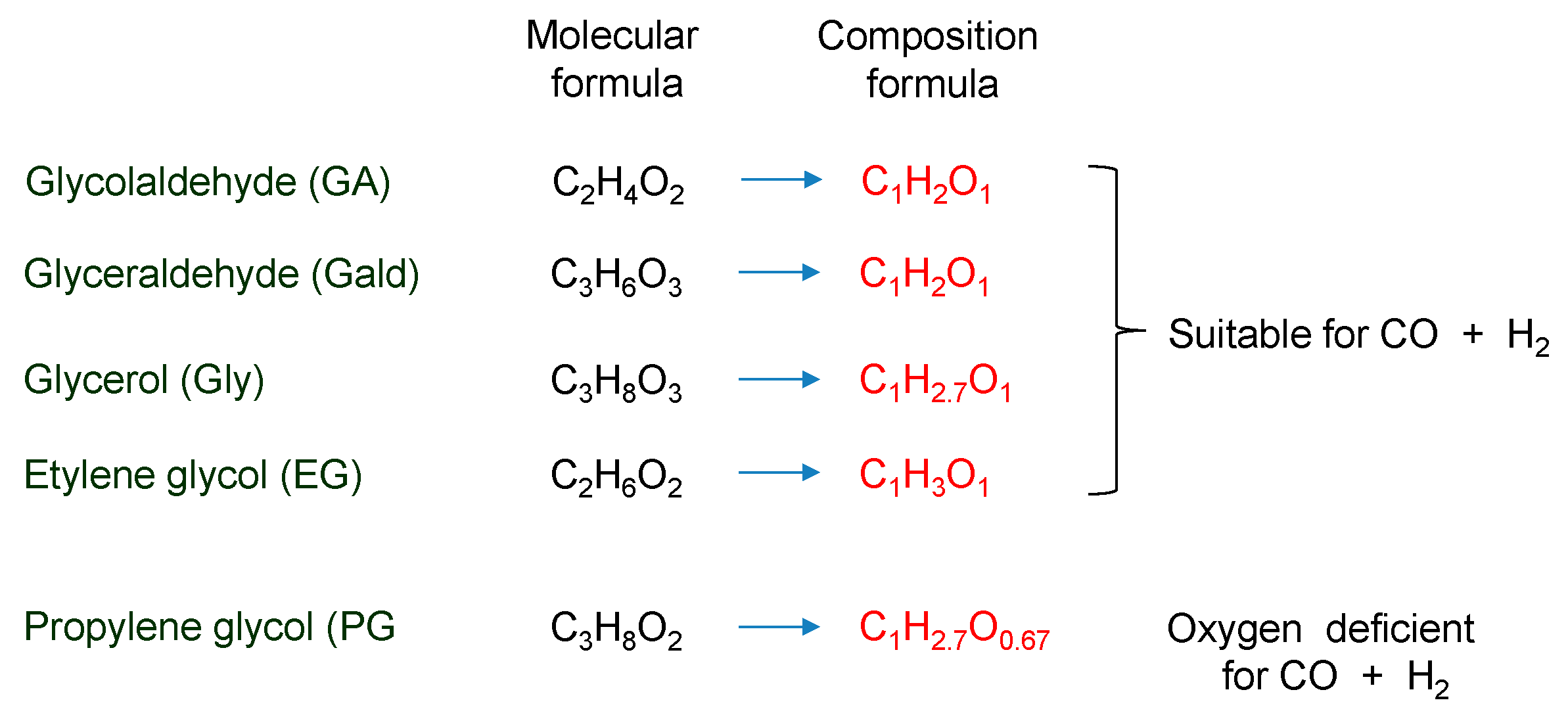
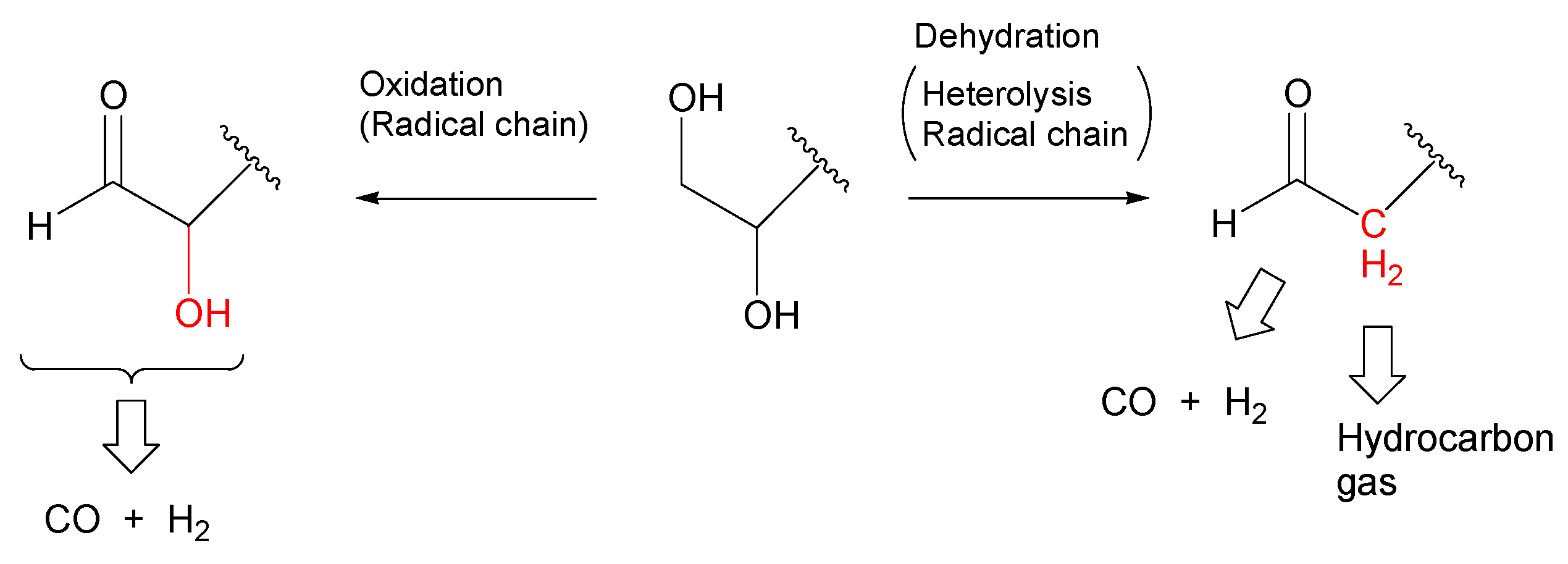
- Most of the condensable products are aldehydes, which can be formed from by dehydration and the following keto–enol tautomerization. Loss of some oxygen atoms as water is also suggested by the elemental compositions of gas and gas-forming intermediates.
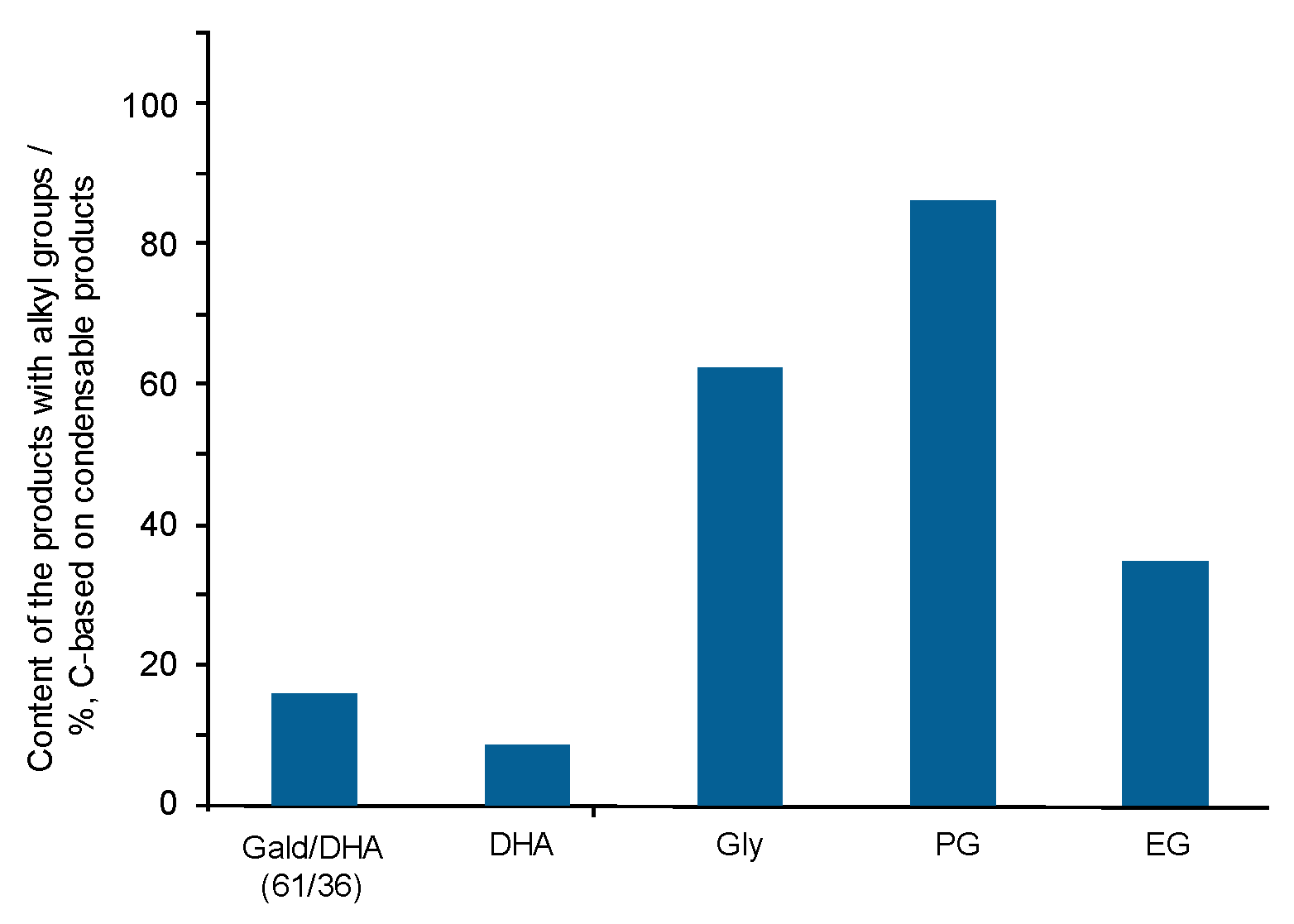
- Yields of gas-forming intermediates (600 °C, mostly aldehydes) bearing alkyl groups are proportionally correlated to the yields of hydrocarbon gases (800 °C), suggesting the alkyl groups being converted into hydrocarbon gases.
- Fragmentation pathways of aldehydes via acyl radicals can explain the formation of characteristic hydrocarbons from polyalcohols (methane from ethylene glycol, ethylene and acetylene from glycerol and propylene glycol).
- Glyceraldehyde directly fragments into CO and H2 with a smaller contribution of dehydration reaction to form methyl glyoxal leading to the methane production, which explains why glyceraldehyde produces syngas selectively.
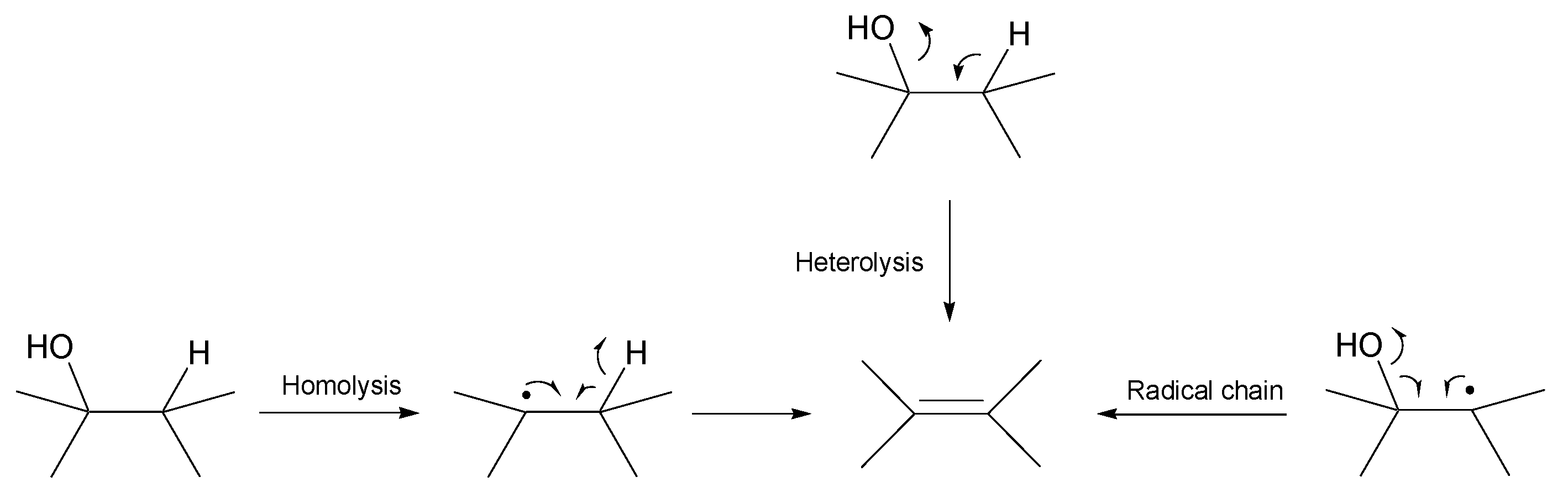
- Theoretical calculations indicate heterolysis mechanisms for polyalcohol dehydration along with the contribution of radical chain mechanisms.
- By assuming the bimolecular mechanisms for heterolytic dehydration reactions, the formations of gas-forming intermediates are explainable more reasonably.
- These findings provide insight into upgrading gasification processes and controlling the gas compositions between hydrocarbons and syngas for sustainable utilization of biomass in biofuel and biochemical applications.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Gly | glycerol |
| Gald | glyceraldehyde |
| HC | hydrocarbon |
| GA | glycolaldehyde |
| MeGO | methylglyoxal |
| HA | hydroxyacetone |
| BDE | bond dissociation energy |
| PG | propylene glycol |
| DHA | 1,3-dihydroxyacetone |
| FA | formaldehyde |
| Pald | propionaldehyde |
| ACR | acrolein |
| AllyOH | allyl alcohol |
| Uni | unimolecular |
| EG | ethylene glycol |
| OX | oxygenated |
| AA | acetaldehyde |
| GO | glyoxal |
| ACE | acetone |
| Ea | activation energy |
| bi | bimolecular |
References
- Yoshida, T.; Oshima, Y. Partial oxidative and catalytic biomass gasification in supercritical water: A promising flow reactor system. Ind. Eng. Chem. Res. 2004, 43, 4097–4104. [Google Scholar] [CrossRef]
- Maschio, G.; Lucchesi, A.; Stoppato, G. Production of syngas from biomass. Bioresour. Technol. 1994, 48, 119–126. [Google Scholar] [CrossRef]
- Stein, Y.S.; Antal, M.J., Jr.; Jones, J.M. A study of the gas-phase pyrolysis of glycerol. J. Anal. Appl. Pyrolysis 1983, 4, 283–296. [Google Scholar] [CrossRef]
- Fukutome, A.; Kawamoto, H.; Saka, S. Gas- and coke/soot-forming reactivities of cellulose-derived tar components under nitrogen and oxygen/nitrogen. J. Anal. Appl. Pyrolysis 2014, 108, 98–108. [Google Scholar] [CrossRef]
- Fukutome, A.; Kawamoto, H.; Saka, S. Gas-phase reactions of glyceraldehyde and 1,3-dihydroxyacetone as models for levoglucosan conversion during biomass gasification. ChemSusChem 2016, 9, 703–712. [Google Scholar] [CrossRef]
- Fukutome, A.; Kawamoto, H.; Saka, S. Molecular mechanisms for the gas-phase conversion of intermediates during cellulose gasification under nitrogen and oxygen/nitrogen. Chem. Ind. Chem. Eng. Q. 2016, 22, 343–353. [Google Scholar] [CrossRef]
- Fukutome, A.; Kawamoto, H.; Saka, S. Processes forming gas, tar, and coke in cellulose gasification from gas-phase reactions of levoglucosan as intermediate. ChemSusChem 2015, 8, 2240–2249. [Google Scholar] [CrossRef]
- Hosoya, T.; Kawamoto, H.; Saka, S. Different pyrolytic pathways of levoglucosan in vapour- and liquid/solid-phases. J. Anal. Appl. Pyrolysis 2008, 83, 64–70. [Google Scholar] [CrossRef]
- Matsuoka, S.; Kawamoto, H.; Saka, S. What is active cellulose in pyrolysis? An approach based on reactivity of cellulose reducing end. J. Anal. Appl. Pyrolysis 2012, 93, 24–32. [Google Scholar] [CrossRef]
- Kawamoto, H.; Ueno, Y.; Saka, S. Thermal reactivities of non-reducing sugars in polyether-Role of intermolecular hydrogen bonding in pyrolysis. J. Anal. Appl. Pyrolysis 2013, 103, 287–292. [Google Scholar] [CrossRef]
- Kawamoto, H.; Hosoya, T.; Ueno, Y.; Shoji, T.; Saka, S. Thermal stabilization and decomposition of simple glycosides in the presence of aromatic substances in closed ampoules: The role of OH•••π hydrogen bonding. J. Anal. Appl. Pyrolysis 2014, 109, 41–46. [Google Scholar] [CrossRef]
- Fukutome, A.; Kawamoto, H.; Saka, S. Kinetics and molecular mechanisms for the gas-phase degradation of levoglucosan as a cellulose gasification intermediate. J. Anal. Appl. Pyrolysis 2017, 124, 666–676. [Google Scholar] [CrossRef]
- Kumar, A.; Jones, D.D.; Hanna, M.A. Thermochemical biomass gasification: A review of the current status of the technology. Energies 2009, 2, 556–581. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kozu, H.; Yamagata, S.; Murao, N.; Ohta, S.; Shiya, S.; Ohba, T. Effect of superficial velocity on tar from downdraft gasification of biomass. Energy Fuels 2005, 19, 1186–1191. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chemistry of acyl radicals. Chem. Rev. 1999, 99, 1991–2070. [Google Scholar] [CrossRef]
- Castañeda, R.; Iuga, C.; Álvarez-Idaboy, J.R.; Vivier-Bunge, A. Rate constants and branching ratios in the oxidation of aliphatic aldehydes by OH radicals under atmospheric conditions. J. Mex. Chem. Soc. 2012, 56, 316–324. [Google Scholar] [CrossRef]
- Olivella, S.; Solé, A. Mechanisms for the reactions of hydroxyl radicals with acrolein: A theoretical study. J. Chem. Theory Comput. 2008, 4, 941–950. [Google Scholar] [CrossRef]
- Raúl Alvarez-Idaboy, J.; Mora-Diez, N.; Boyd, R.J.; Vivier-Bunge, A. On the importance of prereactive complexes in molecule-radical reactions: Hydrogen abstraction from aldehydes by OH. J. Am. Chem. Soc. 2001, 123, 2018–2024. [Google Scholar] [CrossRef]
- Nimlos, M.R.; Blanksby, S.J.; Qian, X.; Himmel, M.E.; Johnson, D.K. Mechanisms of glycerol dehydration. J. Phys. Chem. A 2006, 110, 6145–6156. [Google Scholar] [CrossRef]
- Dagaut, P.; Sarathy, S.M.; Thomson, M.J. A chemical kinetic study of n-butanol oxidation at elevated pressure in a jet stirred reactor. Proc. Combust. Inst. 2009, 32, 229–237. [Google Scholar] [CrossRef]
- Jodkowski, J.T.; Rayez, M.T.; Rayez, J.C.; Bérces, T.; Dóbé, S. Theoretical study of the kinetics of the hydrogen abstraction from methanol. 3. Reaction of methanol with hydrogen atom, methyl, and hydroxyl radicals. J. Phys. Chem. A 1999, 103, 3750–3765. [Google Scholar] [CrossRef]
- Fletcher, A.N. Self-association of methanol vapor. Evidence for dimers and tetramers. J. Phys. Chem. 1971, 75, 1808–1814. [Google Scholar] [CrossRef]
- Cheam, V.; Farnham, S.B.; Christian, S.D. Vapor phase association of methanol. Vapor density evidence for trimer formation. J. Phys. Chem. 1970, 74, 4157–4159. [Google Scholar]
- Renner, T.A.; Kucera, G.H.; Blander, M. A study of hydrogen bonding in methanol vapor by measurement of thermal conductivity. J. Chem. Phys. 1977, 66, 177–184. [Google Scholar] [CrossRef]
- Mitev, V.M.; Stefanov, B.; Ivanov, L.M.; Georgiev, G.M. Infrared absorption spectra of methanol vapor: Dimer and tetramer contributions. J. Mol. Struct. 1985, 129, 11–15. [Google Scholar] [CrossRef]
- Dixon, J.R.; George, W.O.; Hossain, M.F.; Lewis, R.; Price, J.M. Hydrogen-bonded forms of methanol IR spectra and ab initio calculations. J. Chem. Soc. Faraday Trans. 1997, 93, 3611–3618. [Google Scholar] [CrossRef]
- Huisken, F.; Stemmler, M. Infrared molecular beam depletion spectroscopy of size-selected methanol clusters. Eur. Phys. J. D 1992, 24, 277–287. [Google Scholar] [CrossRef]
- Shi, Y.J.; Consta, S.; Das, A.K.; Mallik, B.; Lacey, D.; Lipson, R.H. A 118 nm vacuum ultraviolet laser/time-of-flight mass spectroscopic study of methanol and ethanol clusters in the vapor phase. J. Chem. Phys. 2002, 116, 6990–6999. [Google Scholar] [CrossRef]
- Parsons, A.F. An Introduction to Free Radical Chemistry; Blaclwell Sciemce Ltd.: London, UK, 2000; p. 65. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight (Da) | Density (g/cm3) | Melting Point (°C) | Boiling Point (°C) | |
|---|---|---|---|---|
| Glycerol | 92.09 | 1.261 | 17.8 | 290 |
| Propylene glycol | 76.09 | 1.036 | −59 | 188 |
| Ethylene glycol | 62.07 | 1.113 | −12.9 | 197 |
| Glyceraldehyde | 90.08 | 1.455 | 145 | 228 * |
| 1,3-Dihydroxyacetone | 90.08 | 1.283 | 90 | 214 * |
| kcal mol−1 |  | ||||
| ΔG‡ | ΔH‡ | TΔS‡ | |||
| EG (uni) | 25 °C | 71.3 | 71.6 | 0.3 | |
| 400 °C | 70.7 | 72.1 | 1.4 | ||
| 600 °C | 70.3 | 72.2 | 1.9 | ||
| EG-EG (bi) | 25 °C | 61.0 | 59.3 | −0.7 | |
| 400 °C | 63.1 | 59.3 | −2.8 | ||
| 600 °C | 64.3 | 59.4 | −3.9 | ||
| EG + H2O (bi) | 25 °C | 66.5 | 65.3 | −1.2 | |
| 400 °C | 68.2 | 65.1 | −3.1 | ||
| 600 °C | 69.1 | 65.2 | −3.9 | ||
| kcal mol−1 |  | ||||
| ΔG‡ | ΔH‡ | TΔS‡ | |||
| DHA (uni) | 25 °C | 66.4 | 65.7 | −0.7 | |
| 400 °C | 67.2 | 65.6 | −1.6 | ||
| 600 °C | 67.8 | 65.5 | −2.3 | ||
| DHA-DHA (bi) | 25 °C | 45.4 | 43.3 | −2.1 | |
| 400 °C | 48.4 | 42.6 | −5.8 | ||
| 600 °C | 50.2 | 42.4 | −7.8 | ||
| DHA + H2O (bi) | 25 °C | 48.9 | 46.6 | −2.3 | |
| 400 °C | 52.3 | 45.6 | −6.7 | ||
| 600 °C | 54.3 | 45.4 | −8.9 | ||
| kcal mol−1 |  | ||||
| ΔG‡ | ΔH‡ | TΔS‡ | |||
| AllyOH (uni) | 25 °C | 94.2 | 93.5 | −0.7 | |
| 600 °C | 95.7 | 93.2 | −2.5 | ||
| AllyOH (bi) | 25 °C | 61.6 | 60.6 | −1.0 | |
| 600 °C | 63.2 | 61.2 | −2.0 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukutome, A.; Kawamoto, H. Dehydration Leads to Hydrocarbon Gas Formation in Thermal Degradation of Gas-Phase Polyalcohols. Energies 2020, 13, 3726. https://doi.org/10.3390/en13143726
Fukutome A, Kawamoto H. Dehydration Leads to Hydrocarbon Gas Formation in Thermal Degradation of Gas-Phase Polyalcohols. Energies. 2020; 13(14):3726. https://doi.org/10.3390/en13143726
Chicago/Turabian StyleFukutome, Asuka, and Haruo Kawamoto. 2020. "Dehydration Leads to Hydrocarbon Gas Formation in Thermal Degradation of Gas-Phase Polyalcohols" Energies 13, no. 14: 3726. https://doi.org/10.3390/en13143726
APA StyleFukutome, A., & Kawamoto, H. (2020). Dehydration Leads to Hydrocarbon Gas Formation in Thermal Degradation of Gas-Phase Polyalcohols. Energies, 13(14), 3726. https://doi.org/10.3390/en13143726