1. Background of the Work
Because of its carbon content, biomass is a potential source for renewable liquid fuels and organic chemicals with a lower carbon footprint compared to fossil products. Usually, biomass consisting of cellulose, hemicellulose, and lignin is rich in oxygen [
1]. Due to storage, distribution, and application reasons, the oxygen content for liquid fuels such as gasoline or diesel fuel is restricted, e.g., by fuel specifications. Substances containing oxygen in the molecule structure such as γ-valerolactone (GVL) and valeric esters [
2,
3] are discussed as alternative liquid fuels with preferable application behavior, e.g., reduced soot generation. The oxygen content of these substances is much lower than of usual biomass (C/O ratio for GVL = 2.5; for cellulose ≈ 1). Common processes, such as flash pyrolysis or one-step hydrothermal liquefaction are not able to produce liquid fuels that fulfill the specifications of standard fuels, especially concerning oxygen content [
4,
5]. A common way of upgrading fuel intermediates with high oxygen content, is hydrogenation. Because of the oxygen content, a treatment of biogenic intermediates by hydrogenation consumes a lot of hydrogen. The oxygen content of the intermediates is within a wide range, for flash pyrolysis from 35–40%
w/
w and for hydrothermal liquefaction from 8–22%
w/
w [
6]. For example, 20%
w/
w oxygen represents a need of hydrogen for deoxygenation and saturation in the range of 0.25–0.56 m
3 kg
−1 intermediate. This is a key factor that makes high quality biofuel provision via hydrogenation with gaseous hydrogen expensive.
An important part of available biomass potentials are substances with high moisture content. Because the water has to be removed before, normally by an energy intensive drying process, processes needing dry feedstock are very energy-inefficient with these streams. Therefore, processes with water as reaction medium are preferable.
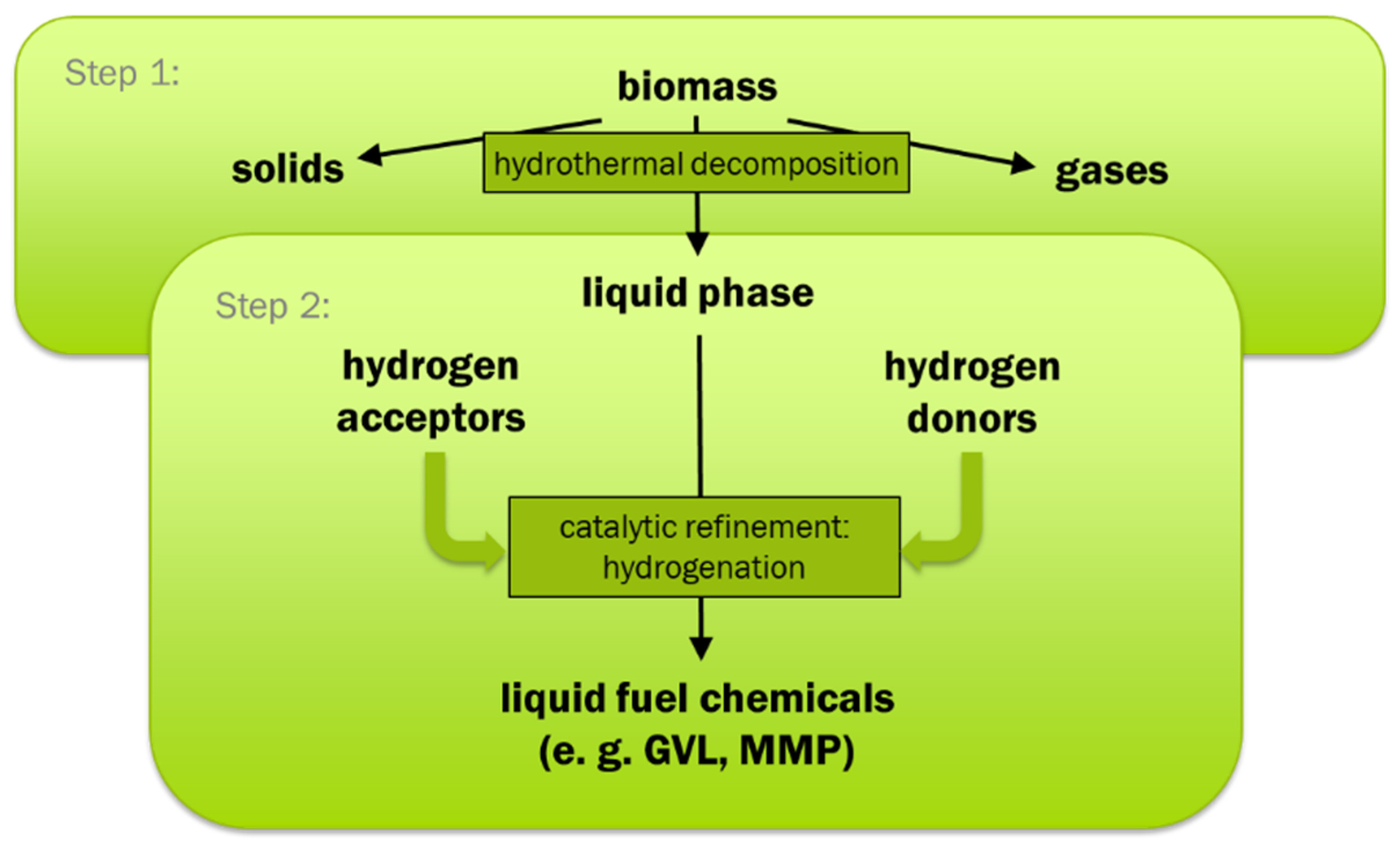
The scope of the research work was to investigate a new approach for the production of an upgraded liquid fuel with low oxygen content, with water as reaction medium, and without consumption of gaseous hydrogen. The approach for reaching this scope within this work is a combination of hydrothermal biomass decomposition and product upgrading in aqueous phase involving hydrogenation with hydrogen donor substances (in-situ hydrogenation).
For the hydrogenation, hydrodeoxygenation, and saturation of single substances without the application of gaseous hydrogen, hydrogenation with hydrogen donor substances is a common process [
7,
8]. In-situ hydrogenation can be a transfer hydrogenation with direct transfer of hydrogen from donor to acceptor or a hydrogenation with an immediate consumption of internal produced molecular hydrogen. In addition to the advantage of no need for external hydrogen, in-situ hydrogenation offers many other advantages such as no equipment for gaseous hydrogen provision and handling, smaller reactors, no reaction limitation because of mass transfer between gaseous and liquid phases and thereby milder reaction conditions (especially lower pressure) [
9,
10].
Watanabe [
11] describes the production of an oil with a low oxygen content out of biomass and formic acid (FA) in the presence of alkali carbonate using cobalt catalysts. Zhou [
12] explains that hydrogen transfer is a possible way to generate oxygen poor products. Hyde [
9] uses hydrogen, in-situ produced out of FA, for the hydrogenation of unsaturated carbohydrates, but under supercritical conditions. Gilkey [
13] gives an overview on the in-situ hydrogenation of several pure substances with FA at different catalysts.
For the transfer hydrogenation of levulinic acid (LA) with FA to GVL, some results are available. Deng [
14] mentioned that a GVL yield of 96% is possible while utilizing Ru catalysts. Following Braden [
15], an RuRe/C catalyst is more active in the presence of sulphuric acid than a Ru/C catalyst. Jing [
16] reaches 81% GVL yield with Ru/C catalyst, at 160 °C, and 180 min in presence of trietylamine. Metzker [
17] reaches 92% GVL yield after 15 h at 180 °C with a catalyst produced from carbonyliron.
Also, the in-situ hydrogenation of vanillin (V) to 2-methoxy-4-methylphenol (MMP) with FA was already explained. Singh [
18] found a yield of 99% at 130 °C and 6 h in water applying a Pd50Ag50/Fe
3O
4/NrGO catalyst.
Pileides [
19] explains that poor stability of solid catalysts under hydrothermal conditions is a major challenge of these approaches. A detailed investigation of catalyst stability was not described.
However, the addition of a hydrogen donor from an external source may not be an economic alternative to hydrogen addition. An inexpensive conversion process can be realized if it succeeds to use components from biomass as hydrogen donor for the hydrogenation of other parts in an integrated process.
A decomposition of biomass in hot compressed water, called hydrothermal decomposition, leads to a conversion of biomass macromolecules, like polysaccharides and lignin, mainly into smaller compounds. Typical products of the decomposition of carbohydrates are monomeric sugars, furan derivatives and carboxylic acids such as LA and FA [
20,
21,
22,
23,
24,
25,
26]. With lignin as raw material, mainly aromatic compounds such as phenols are formed [
27]. Some of the conversion products, especially FA, are hydrogen donor substances, applicable in in-situ hydrogenation as described above. Because of the presence of solid particles in the hydrothermal process, the possibilities for applying heterogeneous catalysts are very limited. Homogeneous catalysts, especially catalysts affecting the pH, which are known to promote hydrothermal decomposition, have been tested.
The products of step 1 include both, typical hydrogen donors such as FA and substances that can form liquid fuel components by reaction with hydrogen such as phenols, unsaturated hydrocarbons, and LA. Based on this the new approach is a two-step process with hydrothermal decomposition as step 1 forming hydrogen donors and acceptors. In step 2, the aim is to use the formed hydrogen donors for the hydrogenation of the products of the hydrothermal decomposition step that are easy to hydrogenate to form fuel components. That is why no external reactants such as added hydrogen should be needed. The concept in principle is shown in
Figure 1.
Hyde [
9] emphasizes the opportunities of the integration of the steps of hydrothermal biomass conversion. Within the literature, some multistage approaches for hydrothermal fuels production from biomass are described [
28,
29,
30,
31].
The approach described in this paper differs in main points from the state of the art explained briefly:
Intermediate production of hydrogen is not the main target, this is accepted but the direct hydrogen transfer is aimed because of the above-mentioned advantages;
The whole biomass is applied, without a lignin–carbohydrate separation, the upgrading of products of lignin degradation is investigated as well, which implies the use of the lignin part as chemical feedstock;
Instead of supercritical water as in the process described by Hyde [
13] and others, subcritical water is used, which implies milder reaction conditions combined with the possibility for a reduction of material strain;
There is no hydrogenation of sugars to polyols as initial step planned. This implies a decrease of the O/C ratio without hydrogen consumption during hydrothermal decomposition.
The present article mainly focuses on the introduction as well as the proof of this multi-stage concept for biomass conversion and aims to give a whole overview of the possible process chain. Details of some experimental investigation of partial steps are also published in previous publications.
For proof of concept, the following issues were investigated:
Hydrothermal decomposition of model substances and real biomass (step 1) with the aim to
- o
Maximize the yield of desired products, e.g., organic acids, phenols and unsaturated hydrocarbons which can be converted via hydrogenation with hydrogen donor substances in step 2; and
- o
Minimize solid by-products, e.g., insoluble humins;
Selection of catalyst supports which are stable under hydrothermal conditions, represented by low decrease of the Brunauer–Emmett–Teller (BET) surface in hydrothermal experiments;
Test of selected catalysts with model mixtures representing the products of step 1;
Design of a laboratory plant as model for the whole process, considering the two steps of the planned process;
Test of the whole process chain in laboratory scale.
2. Materials and Methods
In this section, the approach concerning realization of the experiments is described. Because of the different equipment, the subsections in this part of the paper refer exactly to the subsections in the results part.
2.1. Materials and Methods for Hydrothermal Degradation of Biomass
For the investigation of the hydrothermal degradation of real biomass, experiments were performed with grass suspensions. The typical grass sample was dried at 313 K until complete dryness, milled with a ball mill, and sieved to a particle size of <0.1 mm. This grass powder was used as a suspension of 1% w/w in water (stirred overnight for soaking). Experiments were done as not-catalyzed as well as homogeneously catalyzed reactions—for the catalyzed reactions, 5% w/w of K2CO3 (according to the whole suspension mass) was added to the suspension.
The continuous reactor (cf.
Figure 2, left) used for the biomass degradation experiments is a coiled tube (CTR: 4 m length, 7 mm inner diameter) with a preheater and decompression vent. Advantages of this kind of reactor in contrast to a batch autoclave are its fast heating by using a heating band, plug flow, and fast cooling behavior. Residence time in the reactor is controlled by velocity regulation of the pump. The grass suspension in the reactor was processed at temperatures of 553, 573, and 593 K with autogenous pressure at different residence times.
2.2. Preparation and Characterization of Solid Catalysts
As solid catalysts Al2O3, ZrO2 and activated carbon (AC) based supports with Pt, Ru, Pd as well as Fe, Co, Ni as metal components were chosen. Prior to the catalytic experiments, all catalysts were reduced at 623 K under a flow of H2 (2.0 cm3 min−1) in N2 (8.0 cm3 min−1) for 4 h.
Characterization of the catalysts followed the instructions given by Al-Naji et al., 2020 [
32].
The catalyst stability was investigated during a hydrogenation of LA with FA (xcat. = 0.24% w/w in reactant solution, T = 493 K, cLA = 22.06 g L−1, cFA = 27.16 g L−1, cV = 4.56 g L−1, n = 700 min−1 stirring speed, and autogenous pressure).
2.3. Materials and Methods for Hydrogenation of Model Substances
The investigations of the hydrogenation of model substances and real biomass degradation products were subdivided into different parts.
Part 1—The experiments of part 1 were carried out in a 500 mL stainless steel batch reactor (
Berghof Products + Instruments GmbH, Eningen, Germany; reactor type BR-500, max. pressure 20 MPa at max. temperature 573 K, cf.
Figure 2, right with adjustable reactor head and fixed reactor vessel (tightly closed with a PTFE seal ring) within a heating mantle box. On top of the reactor head, the stirrer motor was mounted and connected to the stirrer axis via magnet coupling. The stirrer itself is characterized as straight blade gas-entrainment stirrer with a fixing ring at the bottom of the inner reactor vessel. The heating mantle box was connected to an external thermostat (
Julabo GmbH, Seelbach, Germany; thermostat type SL-6) and provided controlled reactor heating via a suitable thermo oil. If needed, fast cooling was provided by an independent cryostat (
Julabo GmbH, Seelbach, Germany; cryostat type F 25 MA). The pressure within the reactor was controlled and adjusted via a pressure valve (
Spectrolab, Division of KPG Design Group GmbH, Täferrot, Germany; valve type LM 51 6) connected to the gas (N
2, H
2) supply tubing.
Experimental studies with a model mixture were done in the presence of a commercially available Ru/AC catalyst and gaseous hydrogen. Based on literature data and preliminary experimental tests, the following process conditions were chosen for further investigations: T = 433–473 K, p = 3.4 MPa with added gaseous hydrogen, Ru/AC catalyst (5% w/w Ru on AC powder), xcat. = 1% w/w in reactant solution, cFF = 5 g L−1; cLA = 20 g L−1; cV = 7.5 g L−1; cFA = 25 g L−1. Main target products were GVL, MMP, and 2-methyltetrahydrofuran (MTHF). Samples from the experiments conducted were taken in certain intervals.
Part 2—The catalytic experiments were carried out in a 300 mL stainless steel batch reactor (Parr Instruments Company, Moline, IL, USA, reactor type 4560) [
32]. The reactor was equipped with a head stirrer and temperature, pressure and stirring speed were monitored externally. To investigate step 2 in the absence of gaseous hydrogen, a two component aqueous mixture containing LA and FA (c
LA = 22.06 g L
−1, c
FA = 27.16 g L
−1) was converted under the following conditions: 0.3 g of the reduced catalyst and 125 mL of the mentioned aqueous solution of LA and FA were processed after purging the air out of the reactor system by nitrogen. The experiments were then carried out at 493 K with a stirring speed of 700 min
−1 under autogenous pressure. Liquid samples were taken in certain intervals (starting time = 0 = time, when the desired temperature was reached, typically within 30 min) and centrifuged to separate the solid from the liquid sample.
Part 3—In order to mimic the complexity of real lignocellulosic biomass-derived hydrolysates, a model mixture containing LA, V, and FA has been used in further experiments in the Parr mini-reactor (see above; xcat. = 0.24% w/w in reactant solution, T = 493 K, cLA = 22.06 g L−1, cFA = 27.16 g L−1, cV = 4.56 g L−1, n = 700 min−1 stirring speed, autogenous pressure).
2.4. Development of the Two Stage Hydrothermal Laboratory Plant
The developed reactor concept for the hydrogenation of biomass with internally produced hydrogen donor substances in continuous mode is divided into two stages according to the intended way of process and—as the development was a special part of the project—described in
Section 3.4 in detail.
2.5. Materials and Methods for the Investigations on the Laboratory Plant
Part 1—The investigations for the first stage were carried out in the developed laboratory plant, outlined in
Section 3.4. For the investigation of the hydrothermal degradation of real biomass according to the experiments from
Section 3.1, experiments were performed with grass suspensions, which were prepared in the same way as described in
Section 2.1. Experiments were done as not-catalyzed as well as homogeneously catalyzed reactions – for the catalyzed reactions 1%
w/
w of K
2CO
3 (according to the whole suspension mass) was added to the suspension.
Part 2—The experiments for the second stage were done with an aqueous model mixture containing LA, FA and V (cLA = 21.5 g L−1, cFA = 25.6 g L−1, cV = 5 g L−1). The solid catalyst 1Pt/60Boehmite-40SiO2 was activated with hydrogen at 473 K before starting the experiment, afterwards the reactor was flushed with nitrogen. Because of the long reaction time, the experiment was carried out in batch mode at 493 K for 6 h with a stirring speed of 700 min−1.
Part 3—The real product mixture from the first stage degradation of grass suspension (cf. part 1) was converted in the second stage of the developed laboratory plant with the above mentioned solid catalyst and 10 MPa hydrogen for 8 h and at 493 K in a batch mode with a stirring speed of 100 min−1.
2.6. Analysis Methods
The product samples of the hydrothermal degradation steps (
Section 2.1) were analyzed by HS-GC-FID (organic acids, 5-methylfurfural (MF)) [
33] and HPLC-DAD/RID (LA [
34] and 5-hydroxymethylfurfural (HMF) resp. furfural (FF) [
35]). Samples of the
Section 2.3, part 1, were analyzed by HPLC-RI (column: Eurokat Pb, particle size 10 μm, 30 × 8 mm pre-column, 300 × 8 mm separating column; T
oven = 338 K; eluent: degassed, deionized H
2O; eluent flow: 0.7 mL min
−1).
During the experiments, described in the
Section 2.2 and
Section 2.3, parts 2 and 3, the conversion of LA, FA, and V (X
LA, X
FA, and X
V) as well as the yields of GVL, pentanoic acid (PA), and 2-methoxy-4-methylphenol (MMP; Y
GVL, Y
PA, and Y
MMP) were calculated by signal area integration, as outlined in [
32].
For analysis of phenolic compounds and other emerging interesting components by using GC-MS (cf.
Section 3.1 and
Section 3.5) the samples were treated as follows: 700 µL of the product solution were extracted with 400 µL toluene by intensive mixing. After phase separation (after 20 min) the toluene phase of the sample was analyzed by GC-MS. A ZB-5HT Inferno column (30 m × 0.25 mm × 0.25 µm; Phenomenex) was used. The injection temperature was 523 K (split ratio 10:1, injection volume 1 µL). The constant gas flow was 1.2 mL min
−1. The column temperature program was set to 323 K→(5 K min
−1)→498 K→(30 K min
−1)→573 K (for 5 min). All masses from 50 to 450 u were registered. The device is calibrated by manufacturing a mixed calibration solution (with 4-tertbutylphenol as ISTD) of the components in the following ranges of concentration: phenol (2.701–270.1 mg L
−1), o-cresol (2.610–261.0 mg L
−1), sum of m-/p-cresol (4.574–457.4 mg L
−1), guaiacol (2.777–277.7 mg L
−1), syringol (2.309–230.9 mg L
−1), eugenol (2.597–259.7 mg L
−1), V (2.645–264.5 mg L
−1) and syringaldehyd (1.297–129.7 mg L
−1). Other peaks were only qualitatively identified (by comparison with the NIST-database and in the case of GVL by comparison with the pure substance).
3. Results of the Investigation to the Key Questions
3.1. Hydrothermal Degradation of Biomass
Based on extensive laboratory tests for the hydrothermal conversion of model substances such as glucose, cellulose, xylan, pure lignin, and V the hydrothermal degradation of a suspension of grass as real biomass in water was investigated (cf.
Section 2.1).
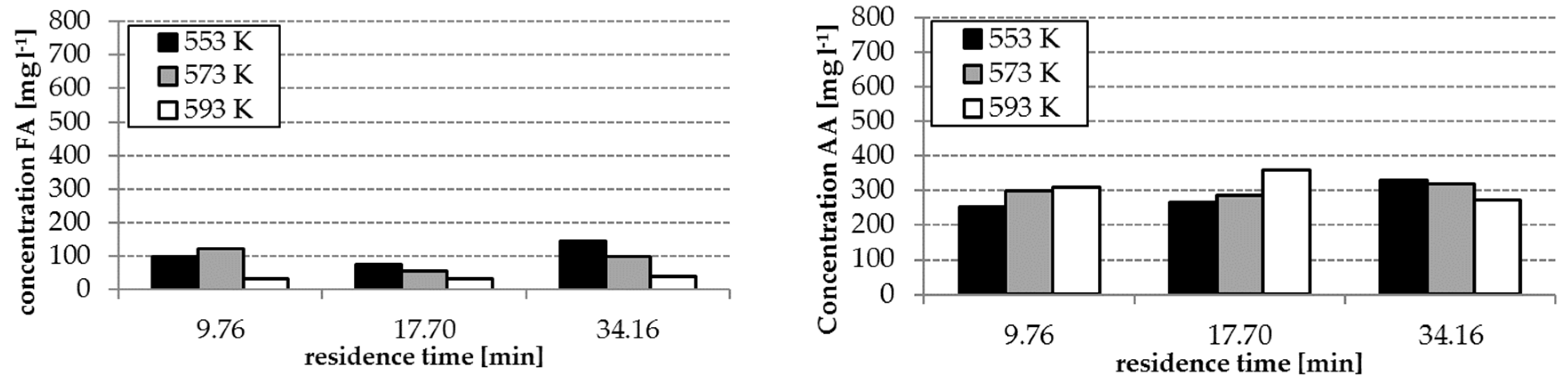
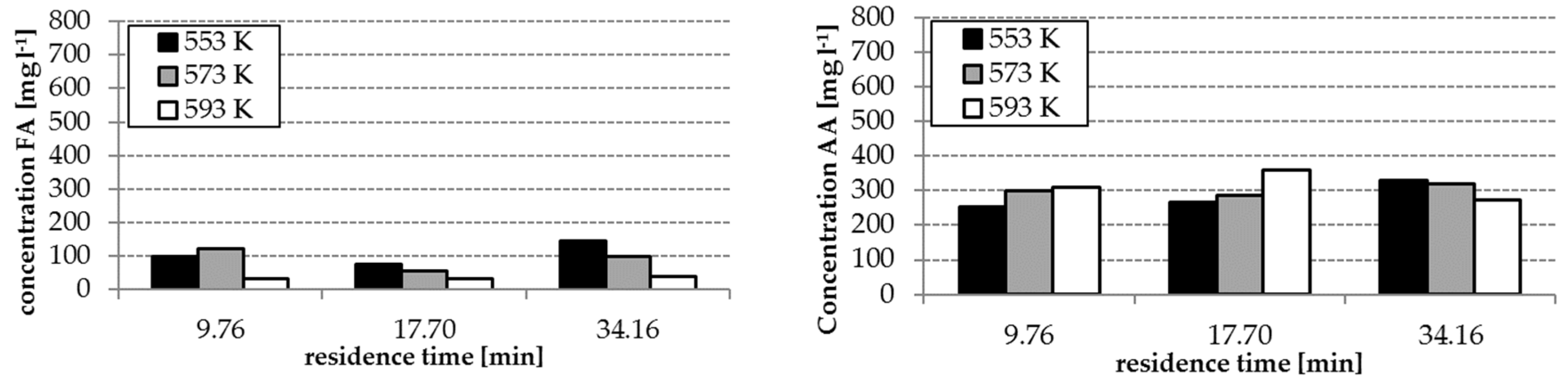
The analysis results of the experiment with and without homogenous catalyst at different reaction temperatures are shown in
Figure 3 and
Figure 4. The concentrations of FA and acetic acid (AA) without catalyst were only slightly influenced by temperature and residence time (34–140 mg L
−1 FA, 250–360 mg L
−1 AA). By using the catalyst K
2CO
3, the FA concentrations increased with increasing temperature and residence time (
Figure 3). However, FA concentrations in the range of 300–536 mg L
−1 were reached. The effect of the presence of the catalyst on the yields of AA is lower as observed during formation of FA—only concentrations in a range of 250–450 mg L
−1 were reached.
Alternatively, the formation of furan derivatives was considerably influenced by the catalyst. In the absence of the catalyst, furan derivatives were measured (HMF: 2.67–8.23 mg L
−1, MF: 0.48–1.35 mg L
−1, FF: 4.49–13.8 mg L
−1); by using the catalyst, no furan derivatives were detected. The dosage of K
2CO
3 causes a significant increase of the potassium (K) content of the suspension and an increase of the pH value (3 mg L
−1 K and a pH of 5 in a suspension without catalyst versus 288 mg L
−1 K and a pH of 10.6–11.1 in a suspension with 5%
w/
w K
2CO
3). According to [
36,
37], the alkaline pH is responsible for the increased contents of FA and AA because an initial alkaline pH value of the hydrothermal conversion of cellulose leads to the formation of mainly carboxylic acids and not any furan derivatives.
As phenolic compounds up to 5.6 mg L−1 phenol, 20.7 mg L−1 guaiacol, and 5.2 mg L−1 of V were measured under these conditions, absence of the catalyst had no influence on the formation of phenolic compounds (9.8 mg L−1 phenol, 7.3 mg L−1 guaiacol, and 7.3 mg L−1 V).
The compound LA was only formed in the absence of the catalyst with concentrations in a range of 7–15 mg L−1. By using a catalyst, the formation of LA did not take place.
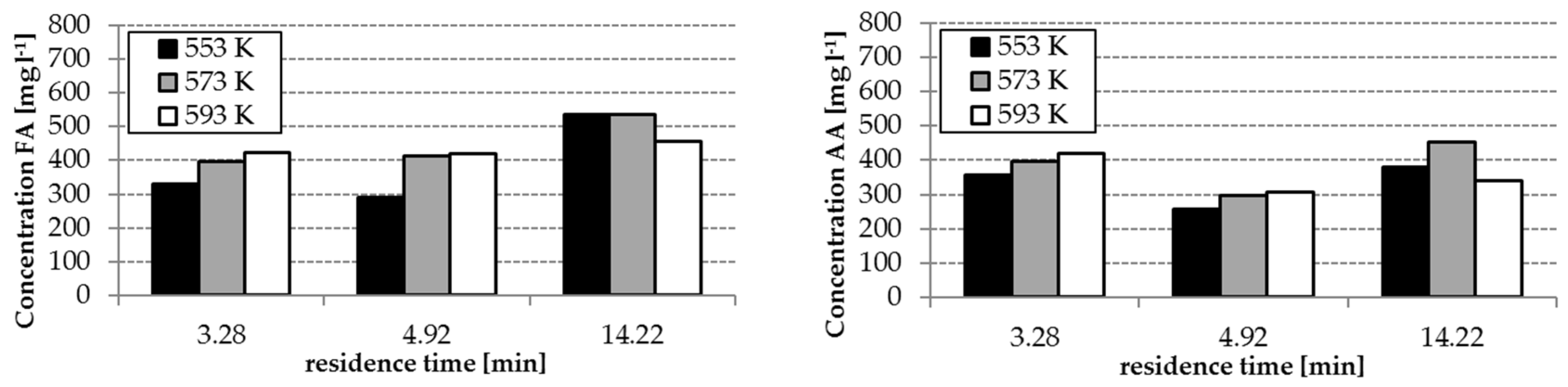
Reaction temperature, residence time and the absence/presence of K
2CO
3 have an influence on the composition of the hydrothermal degradation product solution. The highest concentrations of FA and AA as possible hydrogen donors for the step 2 of the process were received with temperatures of 553 and 573 K, a residence time of 14.22 min and the application of K
2CO
3. (cf.
Figure 4). With these conditions, the formation of HMF, which tends to polymerization, can be avoided. This is advantageous for the protection of the solid catalyst in step 2 of the whole process (cf.
Section 3.4)
3.2. Upgrading of Degradation Products by Hydrogenation in the Presence of Solid Catalysts
Within degradation during step 1, hydrogen acceptors (e.g., FF, HMF, LA, and V) and hydrogen donors (e.g., FA) are formed. In step 2, as explained in
Section 1, a hydrogenation with internally produced hydrogen is very much favored above a process by utilization of molecular hydrogen. Possible reaction pathways can be described as
For upgrading of the product solution derived from step 1 to GVL and MMP as main products, a catalyst is needed and must fulfill special requirements. Under hydrothermal conditions, a lot of catalysts are not stable because of the increased pressure, the high temperature, and, as a consequence, the properties of water. That is why adapted catalysts especially with high stability under hydrothermal conditions within step 2 had to be an outcome of the research. For first tests of the stability of these catalysts, it is still advantageous to work in a hydrogen atmosphere. For further investigations, tests with multicomponent model mixtures of FA as hydrogen donor and different hydrogen acceptors, which are characteristic for the product solution of step 1, have taken place.
For further processing the product from hydrothermal biomass degradation (step 1), bifunctional, mono, and bimetallic catalysts with enhanced hydrothermal stability were focused on three systems, i.e., Al2O3, ZrO2, and activated carbon (AC)-based supports with Pt, Ru, and Pd and Fe, Co, and Ni as metal components. The supports were selected based on previous preliminary tests regarding stability with LA and FA (from cellulose/hemicellulose) and V (from lignin) as common products of hydrothermal biomass degradation, which represent a hydrogen accepting and a hydrogen delivering substance.
Gamma-Al2O3 (γ-Al2O3) based catalysts show a low hydrothermal stability with a rapid phase transformation to boehmite under the conditions applied in the aqueous phase hydrogenation, associated with a decrease in specific BET surface area (ABET) from 280 m² g−1 to 60 m² g−1 after 24 h. Nevertheless, the supported γ-Al2O3 catalysts remain active over 24 h. Interestingly, boehmite-supported catalysts with different mass fractions of SiO2 provide an alternative with significantly higher stability. Also, stabilization of γ-Al2O3 by silylation results in a catalyst with a less pronounced degree of phase transformation and decrease in ABET from 265 m² g−1 to 120 m² g−1.
Catalysts based on commercial ZrO
2 as support show reasonable stability, but relatively low activity mainly due to comparably low specific surface area A
BET 65 m² g
−1 and acid site density (ASD) 9 µmol g
−1. Hence, different mesoporous ZrO
2 based catalysts with tailor made A
BET (140–220 m² g
−1) and ASD (76–136 µmol g
−1) were investigated. The mesoporous ZrO
2 with highest A
BET and ASD were the most active and stable (cf.
Section 2.4).
Although AC is often successfully used for hydrothermal processes [
12,
13,
16], our experiments show a different picture—AC was found to be an unstable catalyst support under the conditions applied accompanied by a significant decrease in A
BET (1200 m² g
−1 to 110 m² g
−1, after 24 h) as well as partial phase transformation. However, hydrothermal reactions often take place in much less than 24 h, so that the use of a carbon-based carrier is not generally excluded. Furthermore, AC can be stabilized by deposition precipitation of Nb showing only a slight decrease in A
BET (1100 m² g
−1 to 980 m² g
−1) after 24 h of catalytic experiment.
3.3. Hydrogenation of Model Substances
For the investigation of the hydrogenation as step 2, experiments with different aqueous model mixtures in order to mimic the product range of step 1 have been conducted.
Part 1—Experimental studies with a model mixture containing FF, LA, FA and V were carried out in presence of a commercially available Ru/AC catalyst and gaseous hydrogen (cf.
Section 2.3, part 1). The choice for Ru was made due to its generally lower costs than Pt and the reports on other comparable chemical reactions. Thus, gaseous hydrogen was initially added (instead of any inert gas) to the reaction mixture, to make sure that hydrogenation toward the desired products will result in measurable yields and allow conclusions on the influence of process parameters.
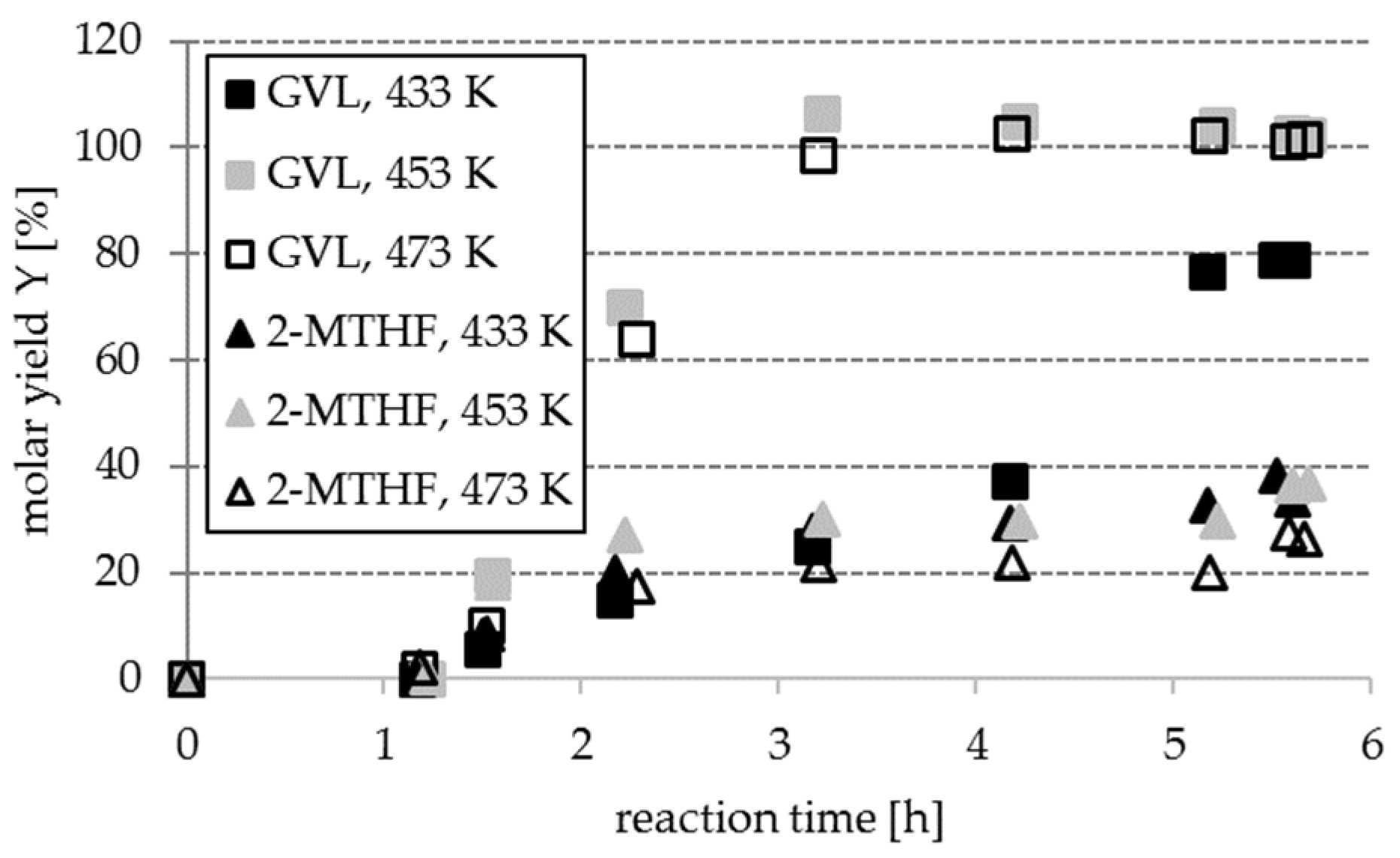
The investigation of the temperature influence elucidated that quantitative conversion of LA to GVL (98.7% and towards 100%) were obtained at 453 K and 473 K after 190 min (c
GVL,190min = 17.0 g L
−1 and 18.3 g L
−1), whereas MTHF molar yields were lower at 453 K and lowest at 473 K, compared to 433 K (38%, based on reactant FF), cf.
Figure 5.
Part 2—Further there were experiments in the absence of gaseous hydrogen were carried out (cf.
Section 2.3, part 2). In contrast to the aqueous phase hydrogenation with externally supplied hydrogen for which Ru is the most active metal [
38,
39], Pt was found to be the most active metal for the aqueous phase hydrogenation of the model mixture. In comparison to the monometallic supported Pt, bimetallic catalysts showed an up to 20% higher GVL yield after 24 h reaction time. The highest GVL yield of 90% is observed in presence of Pt-Co/γ-Al
2O
3. Non-noble metal catalysts, i.e., Fe and Co, alone do not show any significant hydrogenation activity. In presence of the stable 3Pt-9Co on silylated and calcined (1073 K) γ-Al
2O
3, an only slightly lower GVL yield compared to 3Pt-9Co/γ-Al
2O
3 (without silylation and calcination) is achieved, resulting in 60% (without silylation and calcination) and 47% (with silylation and calcination) after 6 h, respectively. In the presence of Pt catalysts supported on mesoporous ZrO
2 the GVL yield increased with increasing A
BET as well as ASD and amounts to 78% after 24 h for the catalyst with the highest A
BET and ASD (1.6Pt/ZrO
2), which was also found to be hydrothermally stable after two catalytic experiments (cf.
Section 2.3). At higher reaction temperatures up to 533 K, the hydrogenation in the presence of 1.6Pt/ZrO
2 also results in the formation of PA as a product of consecutive GVL hydrogenation (see also [
32]). However, PA is not found in presence of the Al
2O
3- or AC-based catalysts at comparable reaction conditions. An overview of the results is shown in
Table 1.
In experimental tests using a HMF, LA, V, and FA mixture, a considerable amount of solid by- products was formed leading to a fast catalyst deactivation. HMF was found to be the main source for solids formation, i.e., insoluble humins [
40]. Therefore, the reaction conditions of hydrothermal degradation in step 1 were adjusted and a nearly HMF-free product solution could be achieved (cf.
Section 2.1).
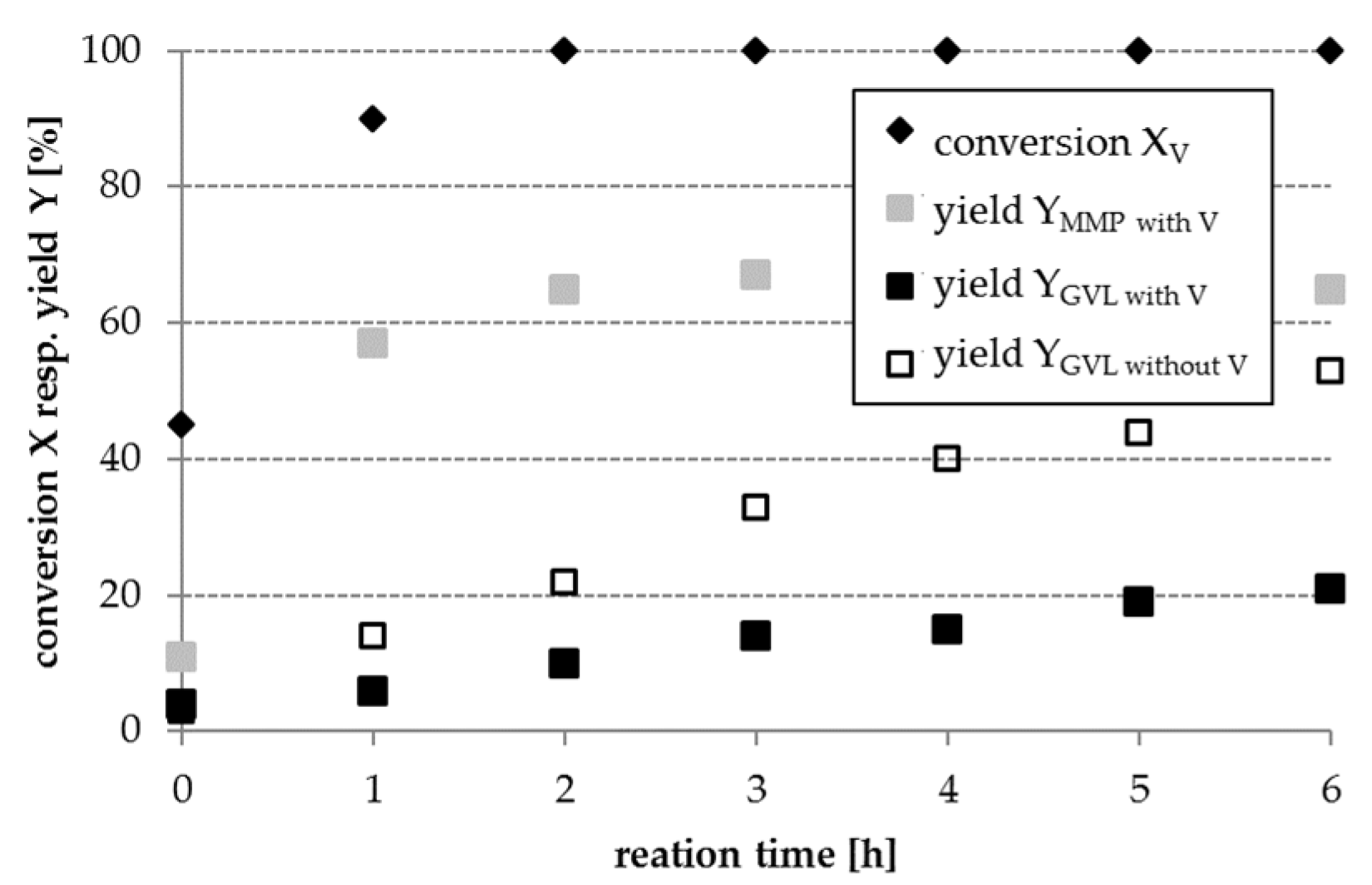
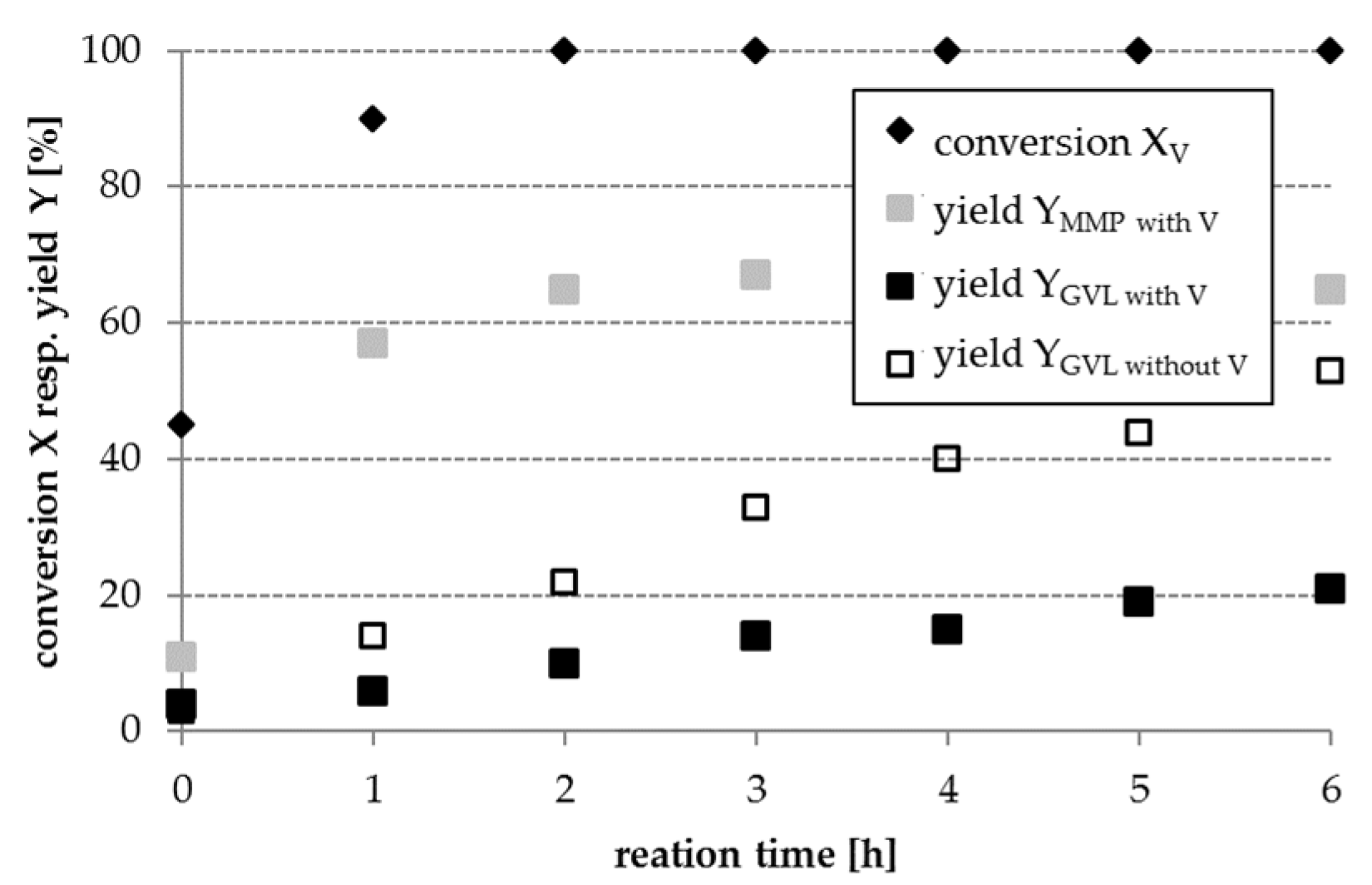
Part 3: In order to mimic the complexity of real lignocellulosic biomass-derived hydrolysates, a model mixture containing LA, V, and FA has been used in further experiments (cf.
Section 2.3, part 3). When converting this model mixture, the GVL yield obtained is significantly lower as achieved during the conversion of LA with FA in the absence of V (cf.
Figure 5). This result can be attributed to a competitive adsorption of LA and V on the catalyst surface. The reduction in GVL yield is different for various catalysts and shown in
Figure 6 as an example for a Pt/AC catalyst. The hydrogenation product of V, MMP, was obtained over all investigated catalysts and with a yield of up to 60% in the presence of Pt/AC after 6 h reaction time.
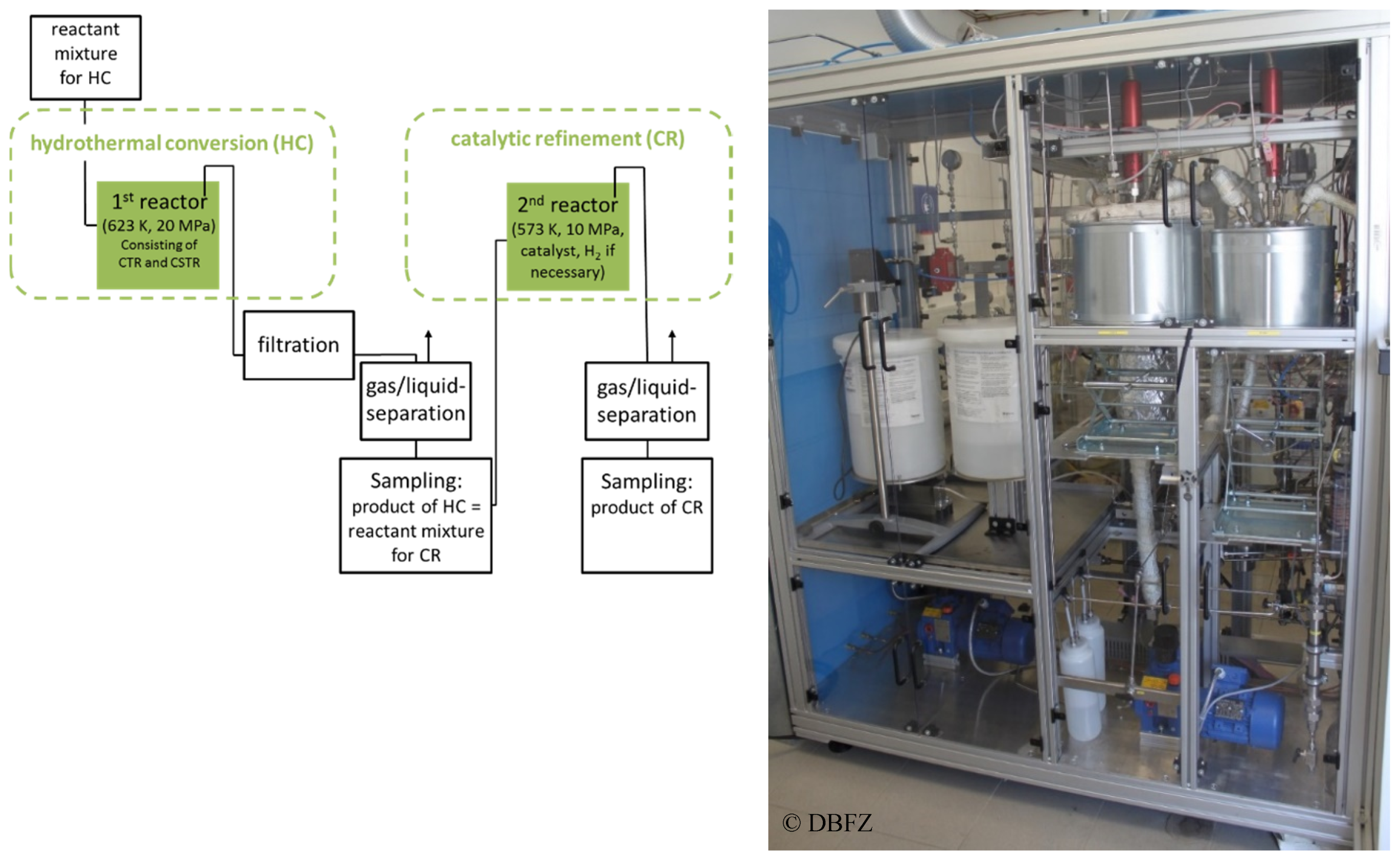
3.4. Reactor Design, Reactor Modeling, and Reactor Implementation
The developed reactor concept for the hydrogenation of biomass with internally produced hydrogen donor substances in continuous mode is divided into two stages according to the intended way of process (cf.
Figure 7). The first reactor stage for hydrothermal degradation of biomass without catalyst or homogeneously catalyzed consists of a combination of a continuous tubular reactor (CTR) and stirred tank reactor (CSTR). The second reactor stage for the heterogeneously catalyzed hydrogenation of the product from the second stage is designed as a stirred tank reactor (STR) as well.
The biomass is fed to the stainless steel continuous tubular reactor with a suitable pump (flow
max = 3 L h
−1) which can handle a suspension of solid biomass with defined particle size in a liquid phase. The CTR (stainless steel X6CrNiMoTi17-12-2; following the definition in DIN EN 10088-3) with a length of 5.3 m can be electrically heated up to 623 K by a heating band. Downstream of the CTR media is fed into a 200 mL continuous top-stirred tank reactor (CSTR, stainless steel X6CrNiMoTi17-12-2). The temperature of this reactor can be electrically adjusted up to 623 K. The working pressure can be controlled in the range up to 20 MPa. Between the first reactor system and the reactor for the catalytic refinement (stage 2) a filtration and separation unit for removing solid by-products is installed (cf.
Figure 7).
After filtration, the liquid phase is transferred to the 2nd reaction stage by a high-pressure pump (flowmax = 3 L h−1). It enters the reactor from the top. This reactor, made from stainless steel (stainless steel X6CrNiMoTi17-12-2), possessed an usable internal volume of 500 mL and is equipped with a gassing stirrer. The temperature can be set up to 573 K by an electrical heating. A maximum working pressure of 10 MPa can be set. Hydrogen can be automatically dosed to the reactor if necessary. To retain the solid catalyst inside the vessel, a special formed catalyst basket can be used.
3.5. Investigation of Whole Process Chain on the Laboratory Scale
The reactor concept was applied as a laboratory plant (cf.
Section 2.5) with the aim to investigate the whole process chain up to the gas separation from the raw second stage product solution. Because of implementation problems, the experiments in the first and the second stages of the plant were not conducted successively, but independently.
Part 1—After first experiments concerning the degradation of biomass with glucose and cellulose solutions as model substances the hydrothermal degradation of grass suspensions was investigated with and without K
2CO
3. The highest yields were achieved with a dosage of 1%
w/
w K
2CO
3 at a temperature of 573 K after a residence time of 4.84 min. The concentrations of organic acids amount to 400–423 mg L
−1 FA, 502–528 mg L
−1 AA, 254–278 mg L
−1 LA and 1216–1311 mg L
−1 lactic acid. No furan derivatives were detected. In contrast to the experiments with 5%
w/
w K
2CO
3 in the tubular reactor (cf.
Section 3.1), during the experiment with 1%
w/
w K
2CO
3 in the laboratory plant little amounts of LA were produced. The carbon related yields of the organic acids can be calculated to 2.4% FA, 2.3% AA, 0.6% LA, and 3.8% lactic acid (the carbon content of the grass was analyzed to 44.65%
w/
w C).
Part 2—1Pt/60Boehmite-40SiO2 was used as catalyst for the heterogeneously catalyzed aqueous phase hydrogenation without gaseous hydrogen in the second stage. After 6 h in batch mode at 493 K, the concentrations of the most interesting components in the product solution were as follows (the conversion rates concern to the initial concentrations):
LA was converted up to 7.2–9.2 g L−1, which corresponds to a conversion rate of 57–67%;
FA was converted up to 10.5–11.6 g L−1, which corresponds to a conversion rate of 55–59%;
V was converted up to 1.3–2.4 g L−1, which corresponds to a conversion rate of 53–74%.
By GC-MS GVL, benzaldehyde, benzyl alcohol, MMP and 2-methoxyphenol could be qualitatively identified. GVL was identified by NIST database and by direct comparison with the pure substance. Benzaldehyde and benzyl alcohol are substances, which were contained in the raw as well as in the product solution.
Part 3—In contrast to the model mixture (content: LA, FA, and V), the mixture converted here contained lower concentrations of the compounds LA and FA and also other compounds. Components like AA, lactic acid (up to 1.3 g L−1) and little concentrations of long-chained organic acids or alcohols were identified. In this experiment, it could be shown that the concentrations of the organic acids LA and FA clearly were diminished with conversion rates of 55–61% (LA) and 36–43% (FA), respectively. Only MMP as one of the desired products could be analyzed qualitatively. GVL was not detectable in the product solution.
4. Discussion of the Approach Based on the Results of the Undertaken Investigations
In
Section 1, issues to be investigated for proof of concept have been defined. The hydrothermal conversion of grass suspension as the chosen example for real biomass was processed in a tubular reactor and furthermore the identified parameters have been adapted for the conversion process in a new laboratory plant. Suitable process parameters to increase the yields of FA, LA and AA while avoiding the formation of HMF and solid by-products as product of the first step could be determined but have to get more optimized in subsequent experiments.
The stability of the catalyst has proven to be crucial for the concept. With 1Pt/60Boehmite-40SiO2 a reasonable catalyst for the aqueous phase hydrogenation as second step was found. Further investigations for catalyst selection and optimization are necessary such as experiments with other suitable catalysts with improved stability towards the conditions in the aqueous reaction mixture from step 1 as well as higher activity and selectivity for the desired target products. In this respect both more stable supports and more stable active components should be identified. In addition, effective catalysts without noble metal are necessary before commercialization.
The scope of the work during step 2 (catalytic upgrading) was the production of value-added products without externally supplied hydrogen. As a result, the possible conversion of the chosen aqueous model mixture (V, FA, and LA) on the selected catalyst could be shown. Especially for the hydrogenation of LA to GVL very high yields up to 90% could be reached (cf.
Section 3.3). In experiments with more than one substance to be hydrogenated, especially with LA and V, the yield of products for one substance is influenced by the presence of the other substance.
In the new experimental plant, the two-step-process could be implemented on the laboratory scale. Investigations for the process could be executed. The whole process on the basis of the defined representative mixture was thereby proven, and the formation of the target components GVL and MMP was qualitatively shown. Step 2, without added gaseous hydrogen, could be shown to be suitable for the conversion of model mixtures also within the new experimental plant. The new experimental plant could also be successfully used for step 2 with feedstock from real biomass, but so far, it has only been used with hydrogen for maintaining the process pressure.
In further research work, the processing of real biomass (especially separation of solid byproducts) within the whole chain has to be optimized. Based on this, data for engineering of the next up-scaling step has to be determined. Because further work for the investigation of the processing of real biomass within the whole chain has to be done and extrapolation of the results is limited by the laboratory plant dimension, the energy balance can only be done based on estimations. The theoretically possible efficiency of chemical conversion of carbohydrate to paraffin is 94%, losses because of solids formation can be up to 12% as reported in [
41] and losses during pre-heating and because of other by-products are estimated as 15% following own experiences. Based on these estimations, an overall efficiency of 70% results.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}