1. Introduction
Cohesive energy is an important quantity that is used to drive almost all the thermodynamical properties of materials [
1] and is defined as the energy needed to dissociate a solid to its neutral atomic components [
2]. Cohesive energy can be calculated by summing the total potential energies of all atoms in the solid materials. Many theoretical models were developed to investigate the size-dependent cohesive energy such as bond energy model [
3,
4], surface-area-difference model [
1], embedded-atom-method potential [
5], thermodynamic model [
6,
7], and a nonlinear, lattice type-sensitive model [
8,
9]. Other models were also proposed to calculate the cohesive energy based on the potential energy functions between two atoms inside metallic nanoparticles such as the Lennard–Jones (or L–J (12-6)) potential function [
10,
11], the Mie-type
potential function [
12], and the Morse potential function [
13]. Predicting cohesive energies and stabilities in solid particles can be enhanced by re-parameterization (the bond order parameters) of the analytic potential functions such as: A force field for zeolitic imidazolate framework-8 (ZIF-8) with structural flexibility [
14], parameterized analytical bond order potential for ternary the Cd–Zn–Te systems [
15].
Potential energy functions contain a certain number of parameters. For example, the Mie-type potential function contains two parameters
(where
) [
16]. The Morse potential function contains one parameter,
[
17]. These parameters determine the strength and the range of the interaction terms in the potential functions. The L–J potential function consists of two terms: The first term represents Pauli’s repulsion, whereas the second term represents the attractive dipole [
2].
Qi et al. [
10] found a disagreement between the calculated cohesive energies arising from the L–J (12-6) potential function and the experimental values of molybdenum (Mo) and tungsten (W) nanoparticles in a face-centered cubic structure. However, the calculated cohesive energies using the L–J (12-6) potential function agreed with the experimental values of Mo and W nanoparticles in a regular octahedron structure [
11]. Moreover, there was an agreement between the experimental values for the cohesive energy of Mo and W nanoparticles and the calculated cohesive energy using the Mie-type
potential function (with
and
) [
12] and the Morse potential function (with
) [
13]. The stability of nanoparticles using the Mie-type potential function
and the Morse potential function (
) was found to be a result of the softening in the repulsive interaction [
12,
13] in the potential functions. However, the interaction terms in the Mie-type potential function
and the Morse potential function (
) do not represent the Pauli repulsion and attractive dipole terms.
A modified Morse/long-range potential function was proposed to analyze the spectroscopic data of diatomic molecules N
2 [
18] and Co
2-H
2 complexes [
19]. The aim of the present work was to propose a new generalized Morse potential function that can predict the experimental values of cohesive energy of nanoparticles. The proposed potential function should contain a term that represents the Pauli repulsion interaction [
2].
The paper is organized as follows. In
Section 2 we describe the generalized Morse potential theory. In
Section 3, we describe a model to calculate the cohesive energy for nanoparticles based on the generalized Morse potential. Finally, in
Section 4, we discuss the numerical results with present conclusions.
2. Theory
The potential energy
that describes the bond between two atoms
and
in the nanoparticle separated by a distance
has a complicated form which can be written in series form as:
where
is the distance between the nearest two atoms in equilibrium case. The potential energy that describes the bond between the two atoms should satisfy the following requirements: (1) The potential should have one minimum point
at
(2) it should asymptotically go to zero as
, and (3) it should become infinite at
.
A well-known potential that satisfies the requirements is the Morse potential [
17]:
where
is a unitless parameter depending on type and structure of metallic nanoparticles [
20]. Lim [
21,
22] found that the parameter
can be expressed in terms of the Lennard–Jones parameters.
The Morse potential function contains two terms: The first term represents the attractive short-range interaction, whereas the second term represents the repulsive long-range interaction. However, the Morse potential function can be written using a summation form as follows:
The Morse potential function is the simplest form that was proposed to describe the interaction potential between two atoms in metallic nanoparticles [
13]. The form of the interaction potential function between two atoms in metallic nanoparticles is more complex than the Morse potential, the Morse potential was formulated by considering only the first three terms of the series in Equation (1) [
17]. However, if more terms are considered, then additional exponential terms of higher order emerge.
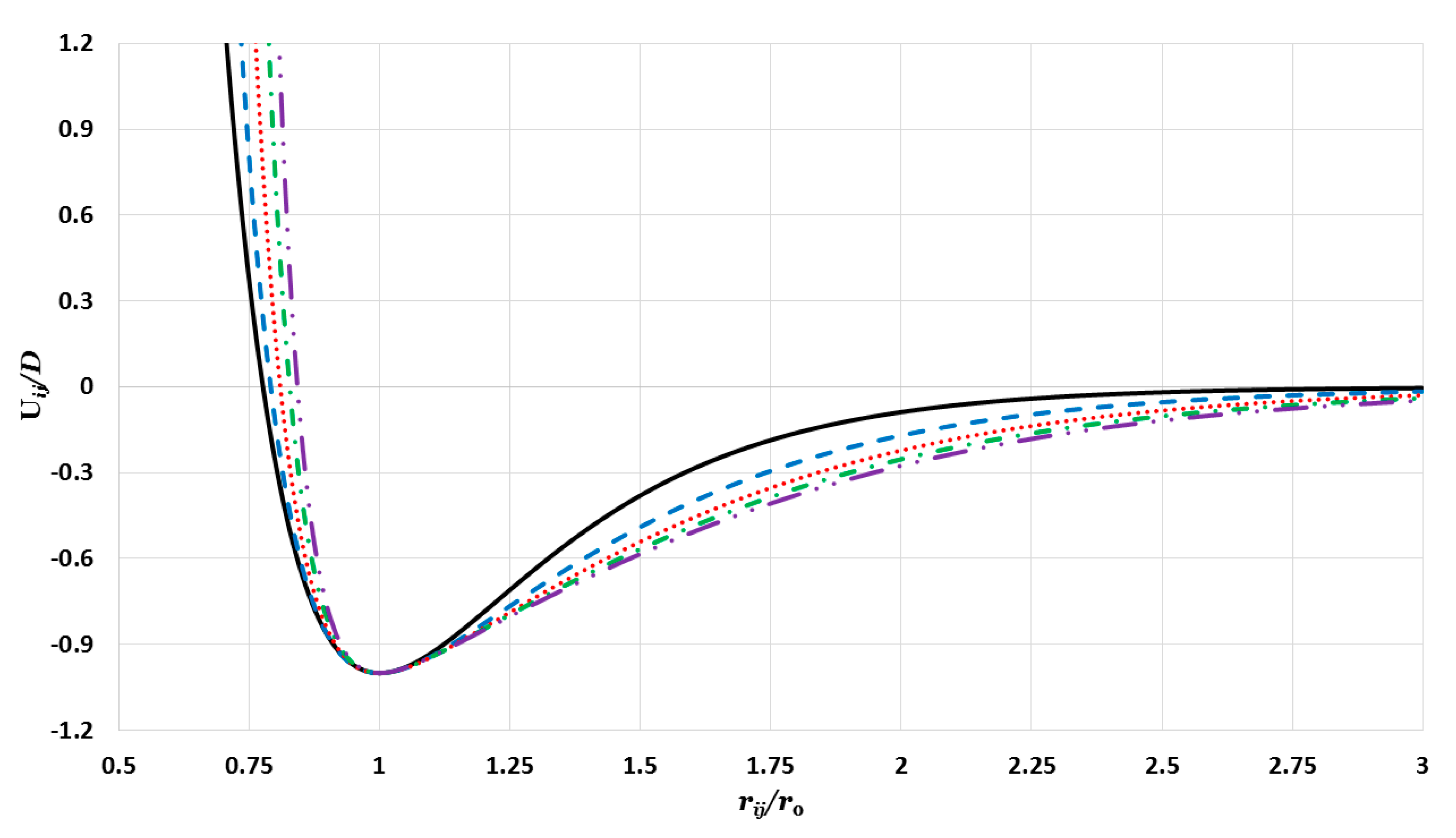
In this current work, a new generalized Morse potential function was proposed that includes more than two interaction terms. The additional terms are controlled by a new unitless parameter
(
) as follows:
For example, if , then the generalized Morse potential function will contain six terms. If , then the generalized Morse potential function becomes the ordinary Morse potential function.
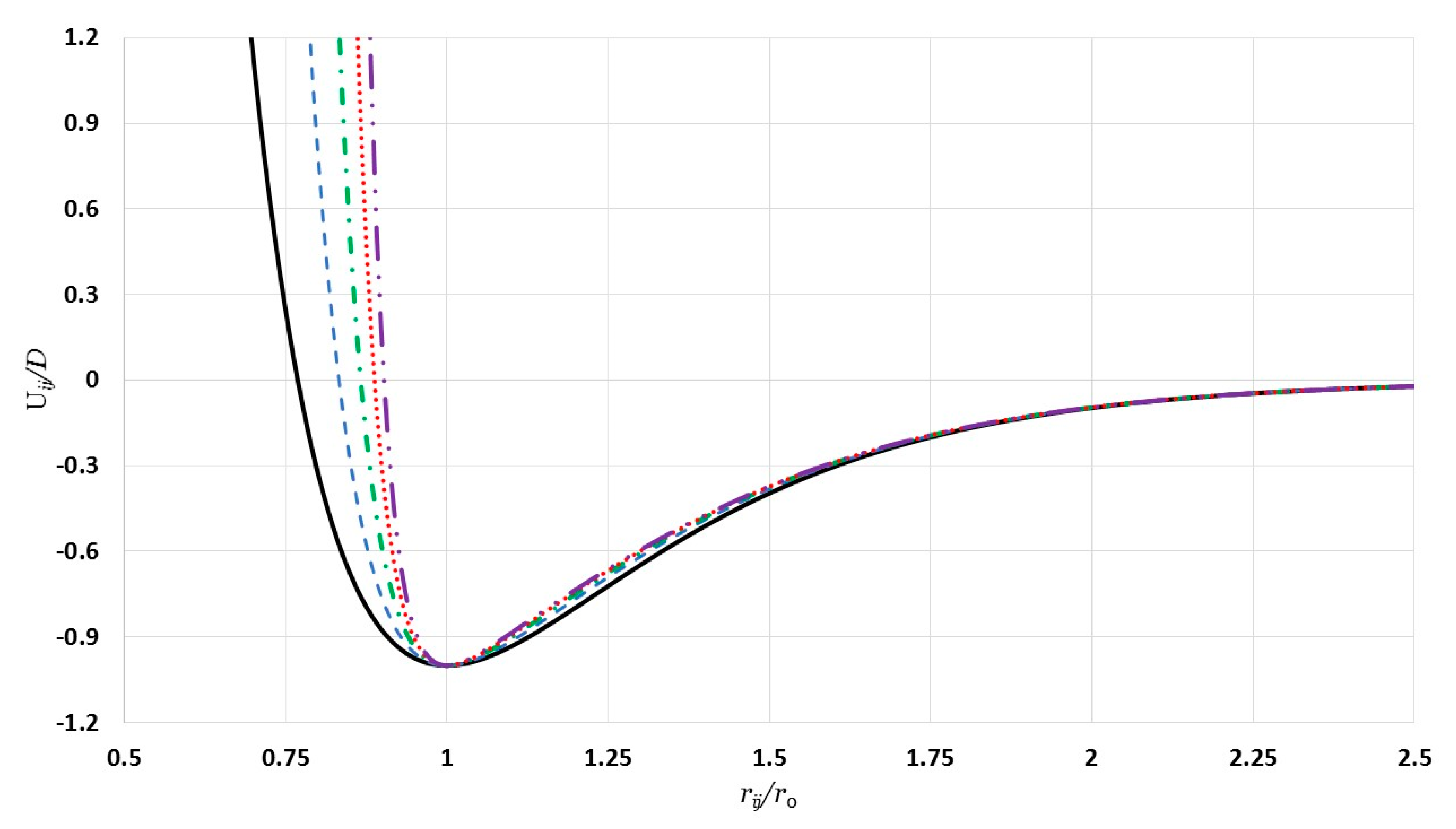
The curves in
Figure 1 represent the generalized Morse potential as a function of reduced separated distance between two atoms for a fixed value of
and different values of
. All potential curves in
Figure 1 satisfy all requirements of the potential energy between the two nearest atoms in a nanoparticle. As seen in
Figure 1, when
the repulsive wall becomes stiffer with little change in the range of the attractive interaction. Moreover, the repulsive walls of the potential curves converge as
and they become large, as seen, again, in
Figure 1.
3. The Model
The cohesive energy of the nanoparticles is obtained by the summation of the total energy of
atoms in a nanoparticle:
where
(
is the nearest distance between two atoms) and
is the reduced nearest distance between two atoms. Moreover,
’s (
) in Equation (6) represents the interaction terms of the potential. The range of the interaction terms varies from the shortest range (
) to the longest range (
).
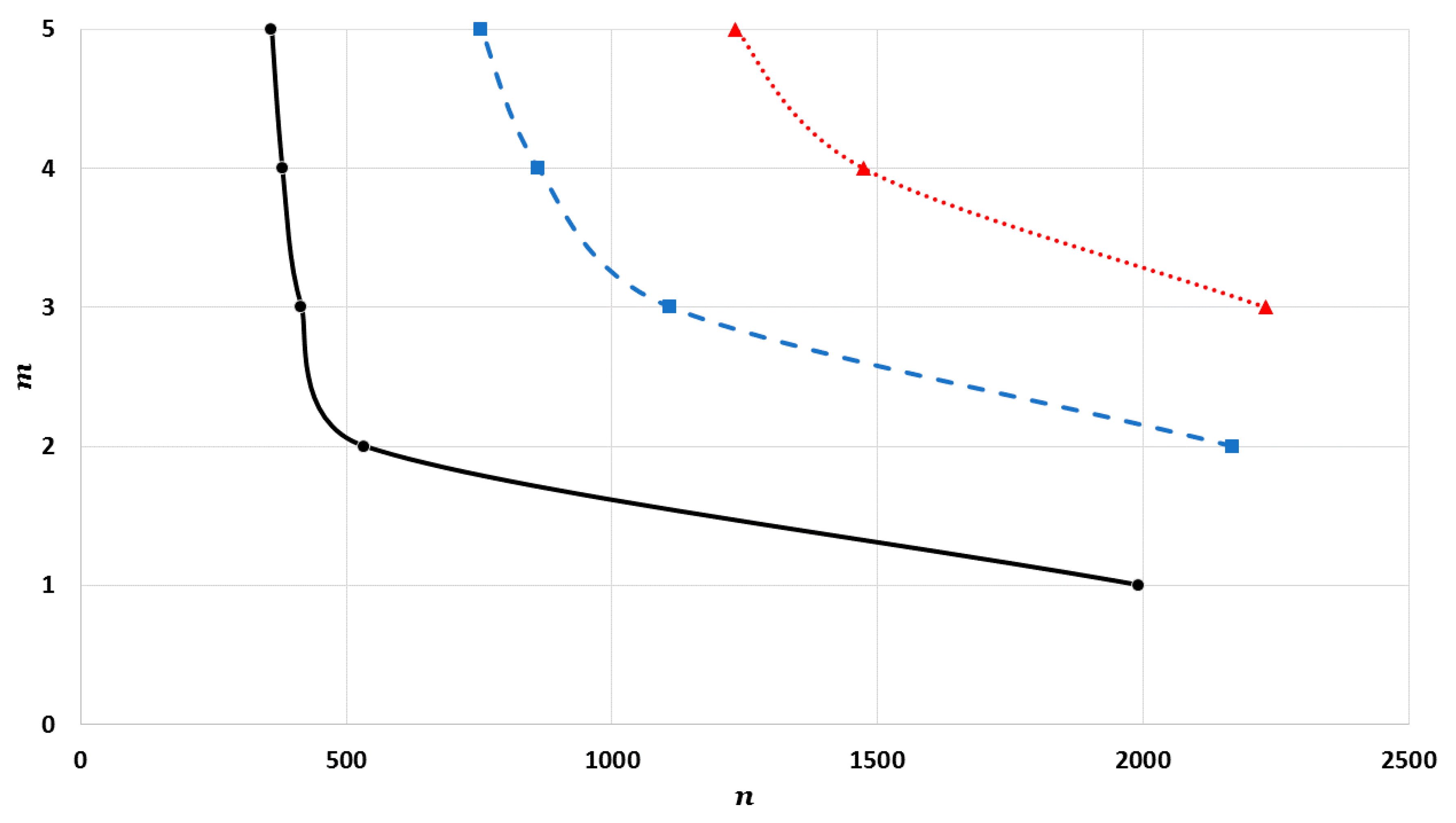
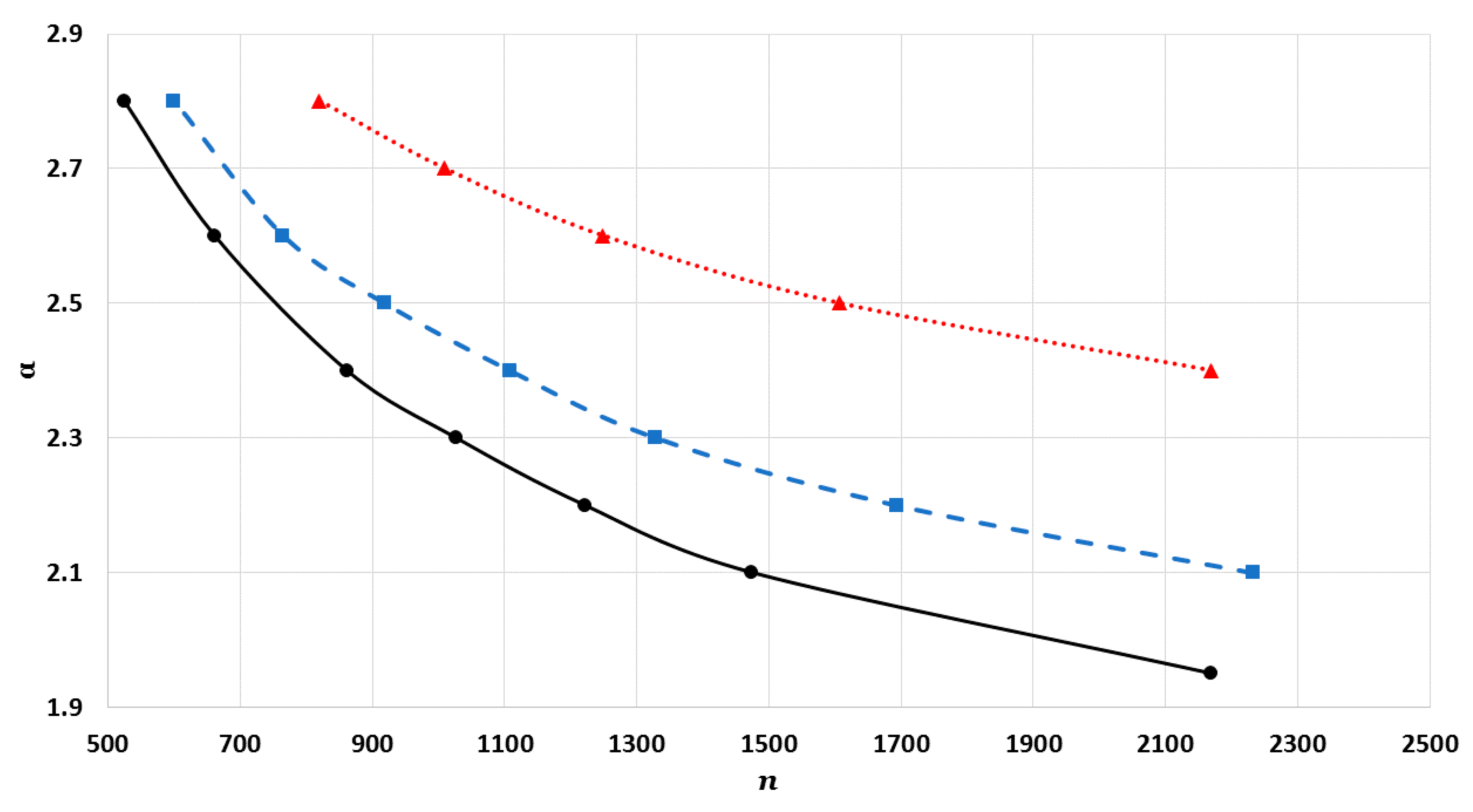
The cohesive energy of the nanoparticle is calculated in an equilibrium configuration for atoms. The equilibrium configuration is obtained by minimizing the total energy of atoms in the nanoparticle with respect to , where is the equilibrium reduced nearest distance between two atoms, which is obtained numerically.
The cohesive energy per atom in the equilibrium configuration is:
The relative cohesive energy of the nanoparticle is the ratio between the cohesive energy per atom and the cohesive energy of the corresponding bulk material
:
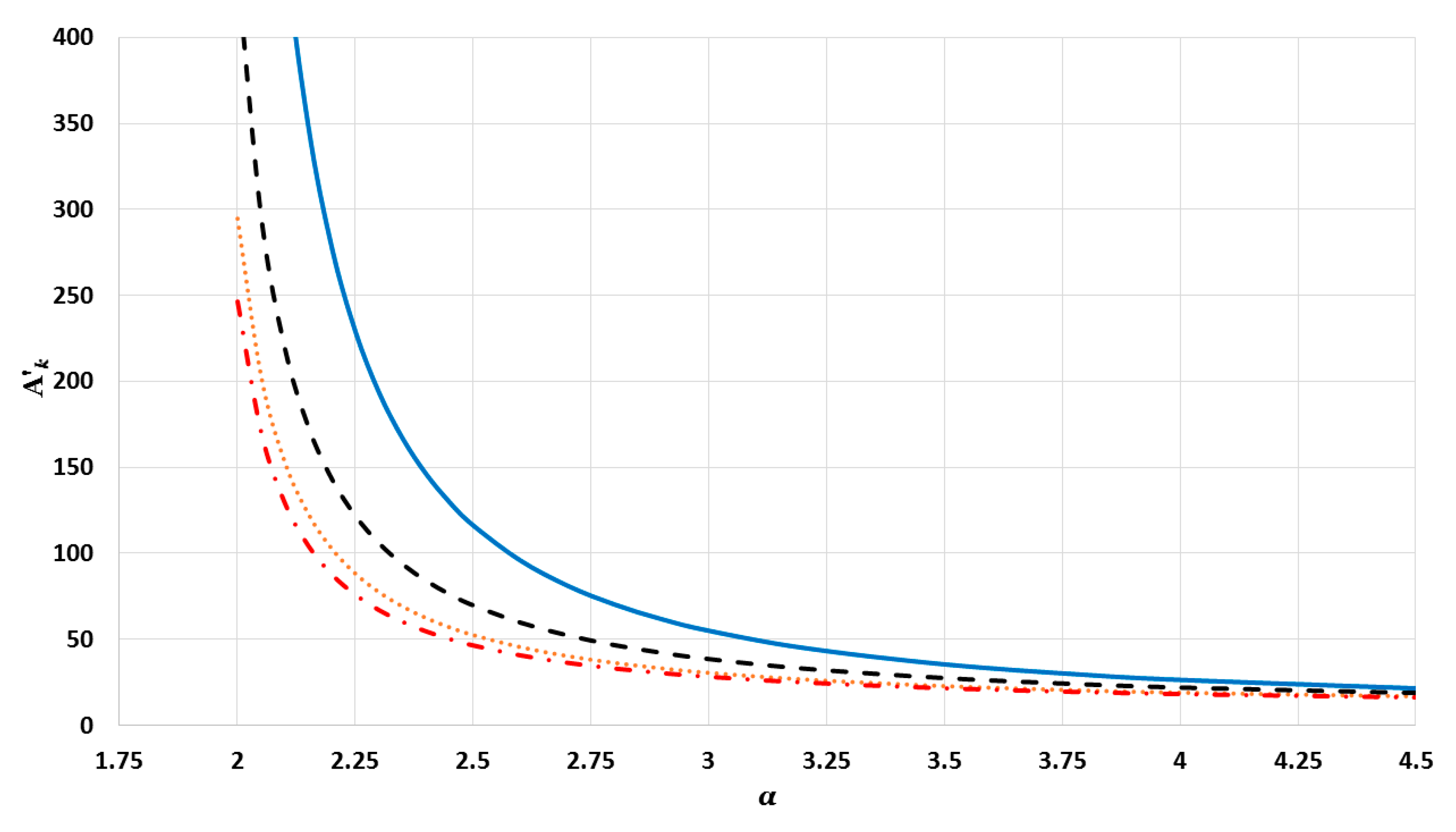
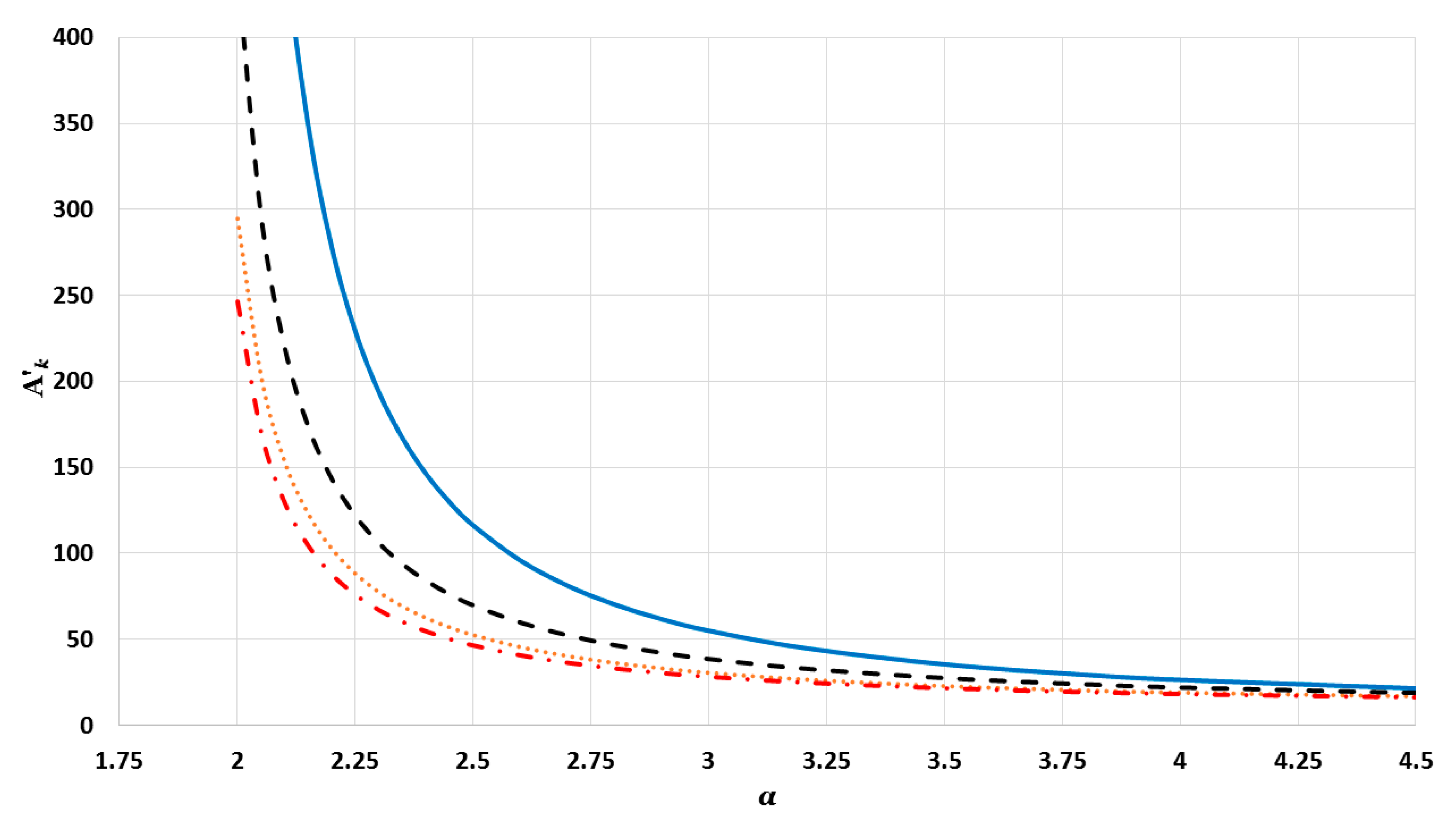
where
and
’s are the corresponding interaction terms of bulk metals (as
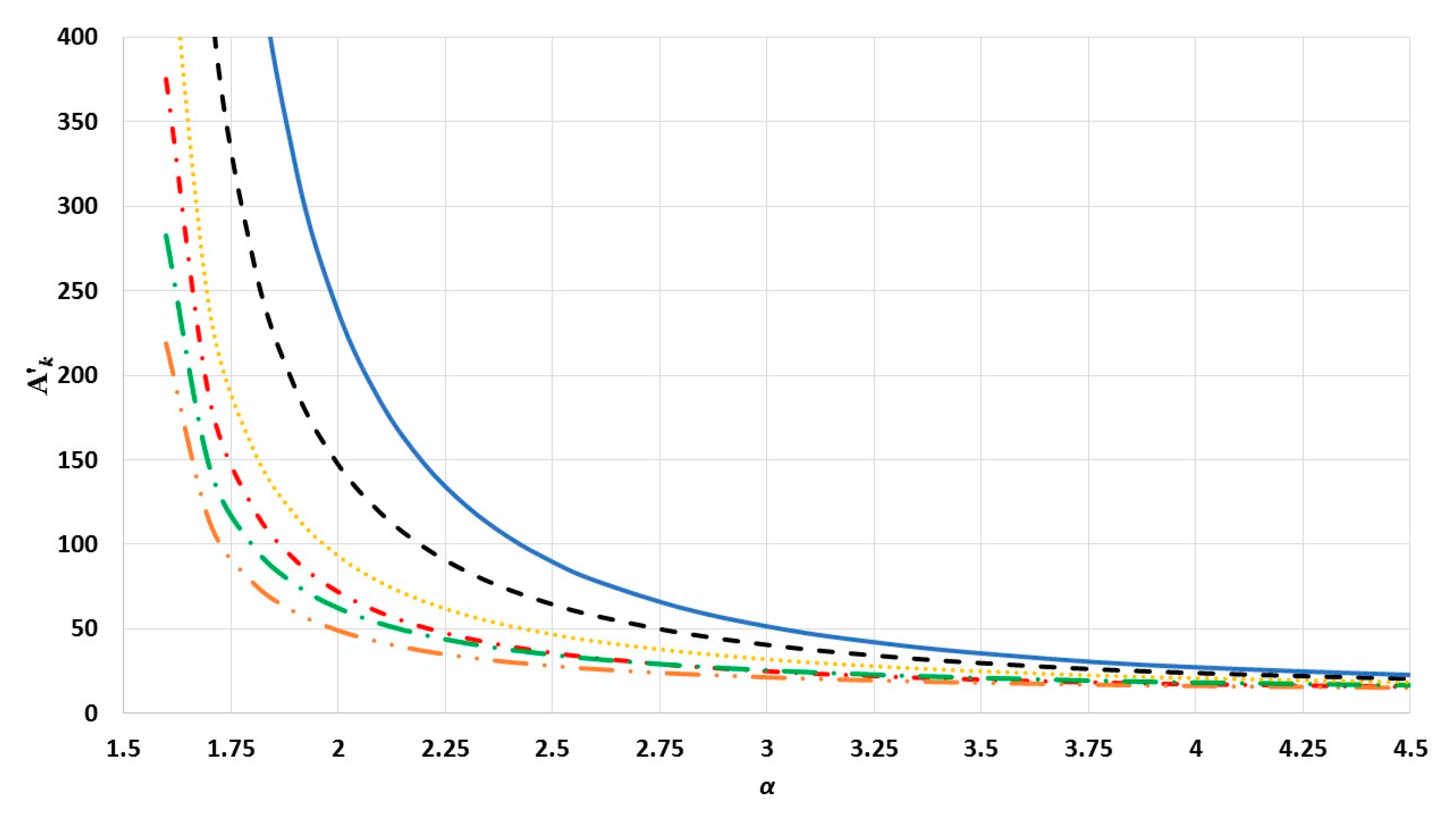
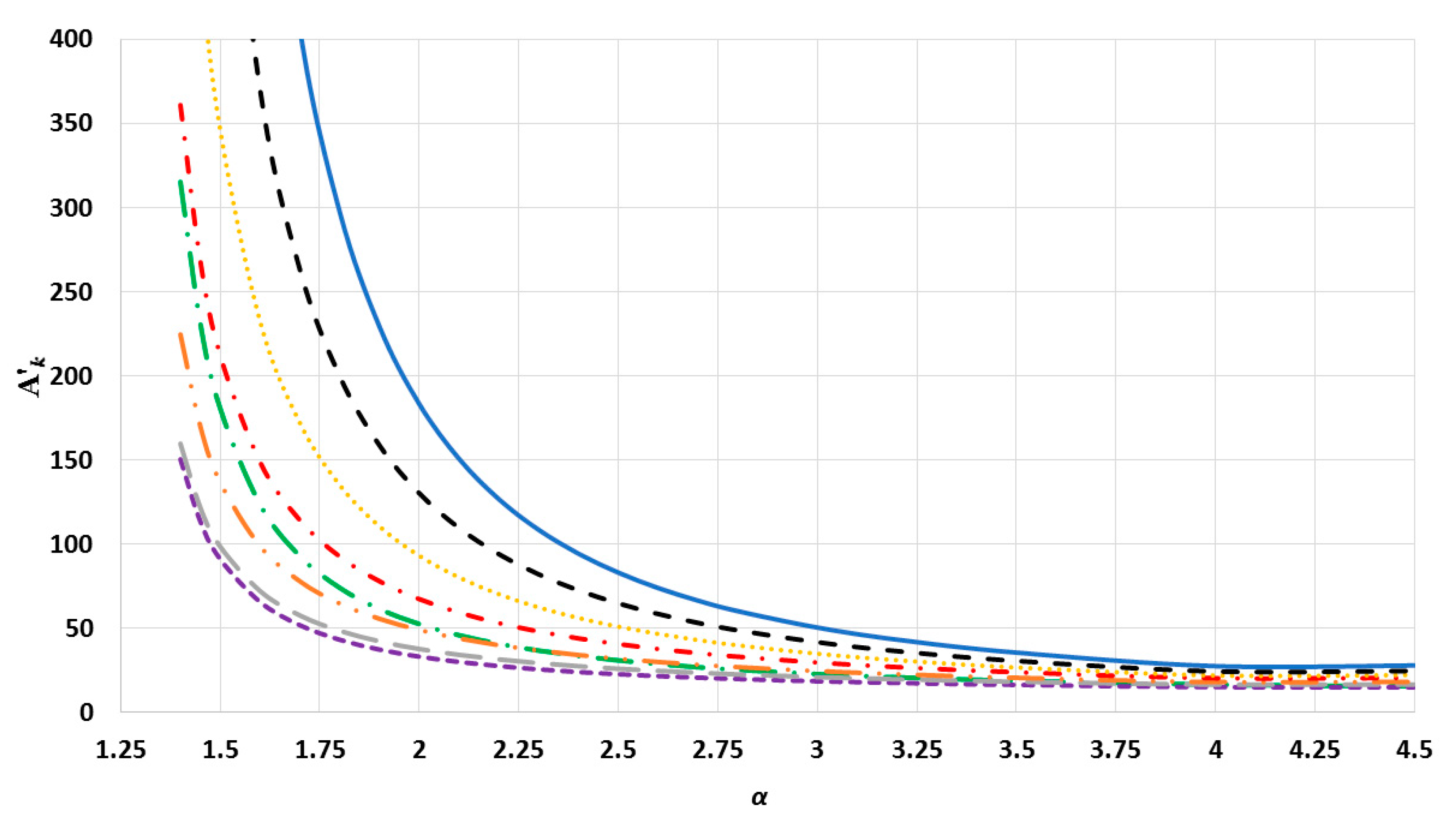
). The values of interaction terms
’s for given
vary with the
parameter, as seen in
Figure 2,
Figure 3 and
Figure 4. The values of interaction terms
’s grow rapidly to infinity as the value of
parameter decreases. Nevertheless, as this is not physically acceptable, a valid range for the
parameter is defined, such that the values of
’s are finite. It was thus found that for different cubic metallic structures, the valid range for the
parameter is approximately the same when
[
13]. Therefore, the values of the interaction terms
in
Figure 2,
Figure 3 and
Figure 4 are only for a face-centered cubic metallic structure.
4. Results and Discussion
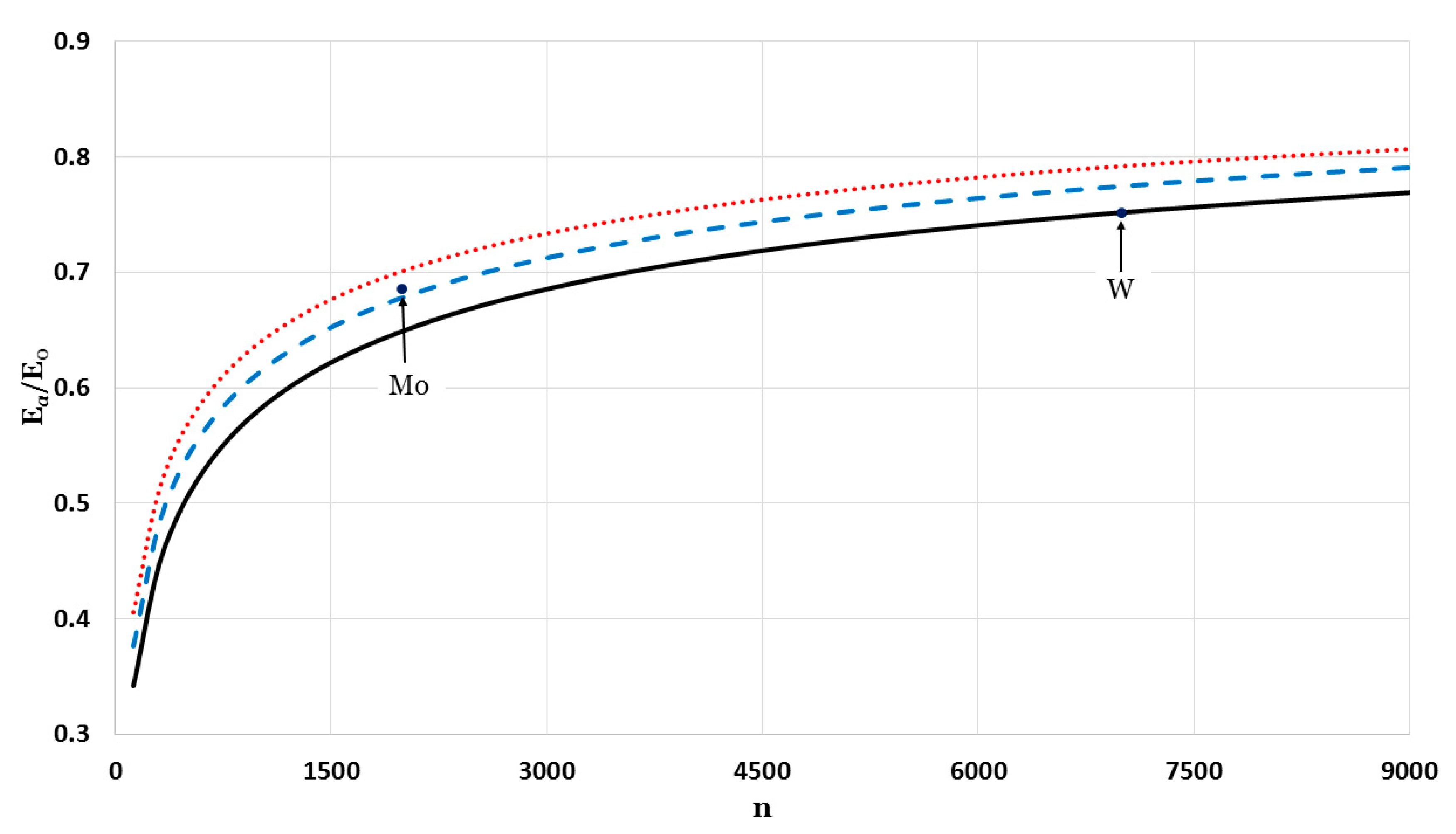
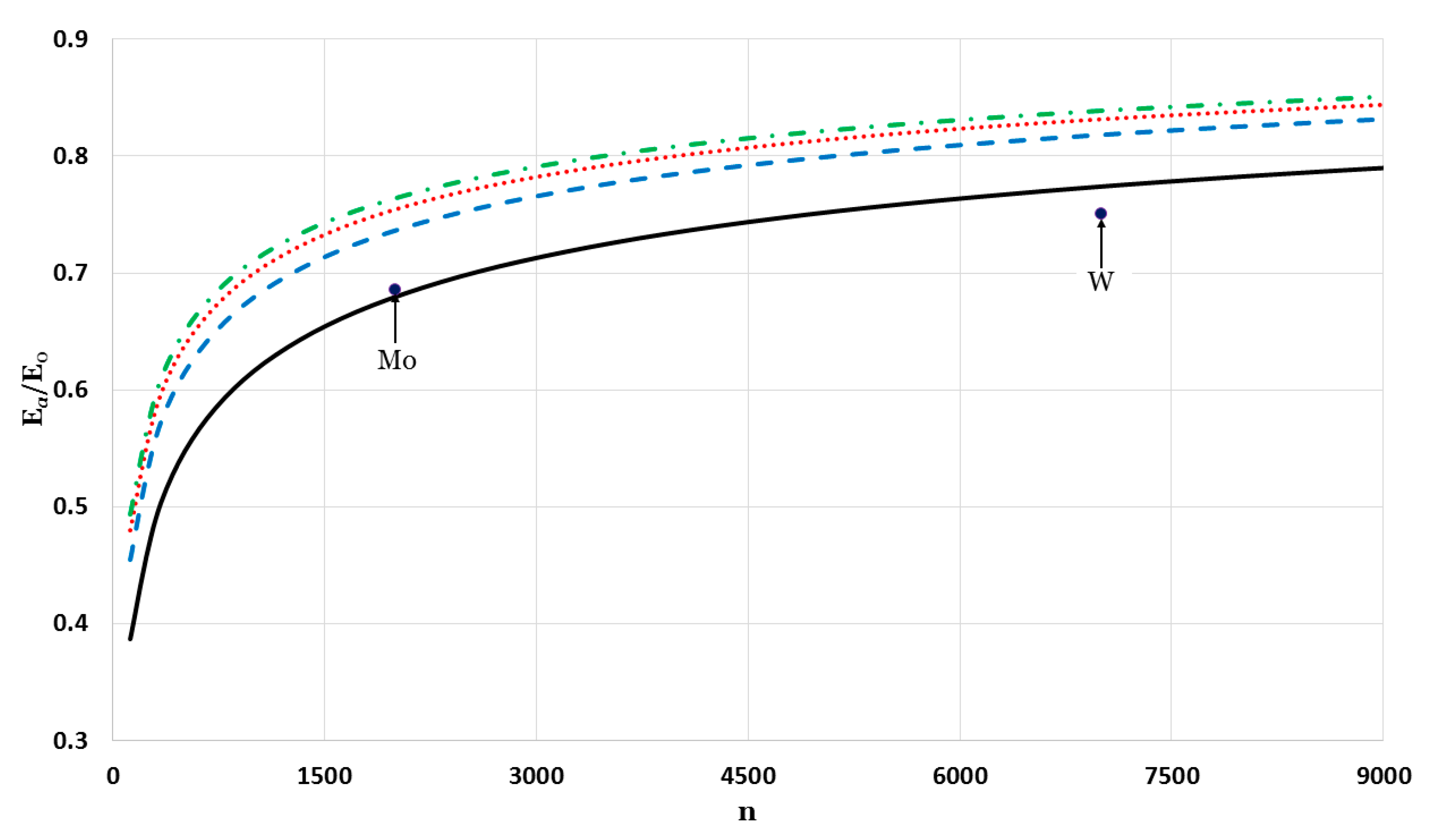
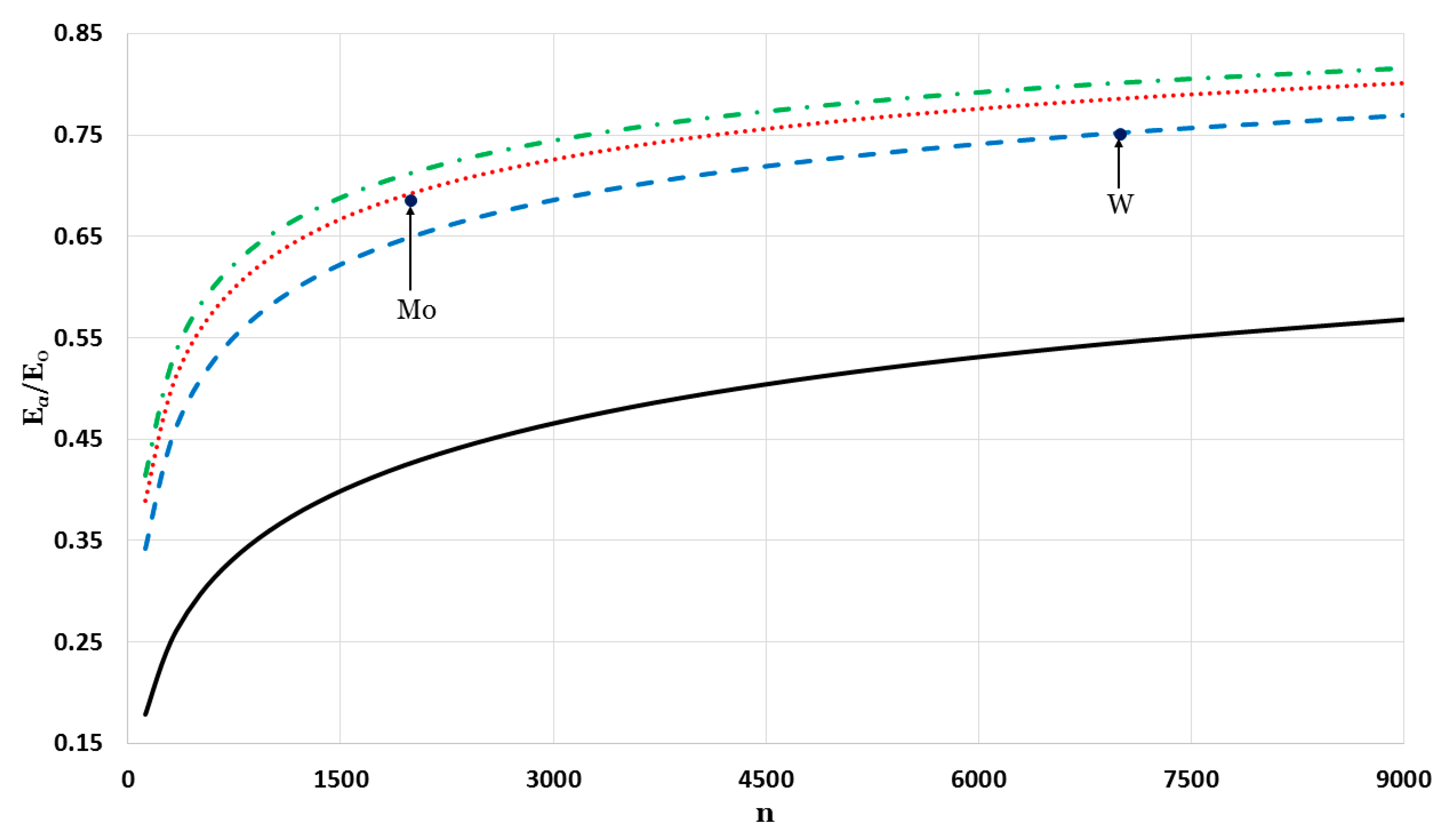
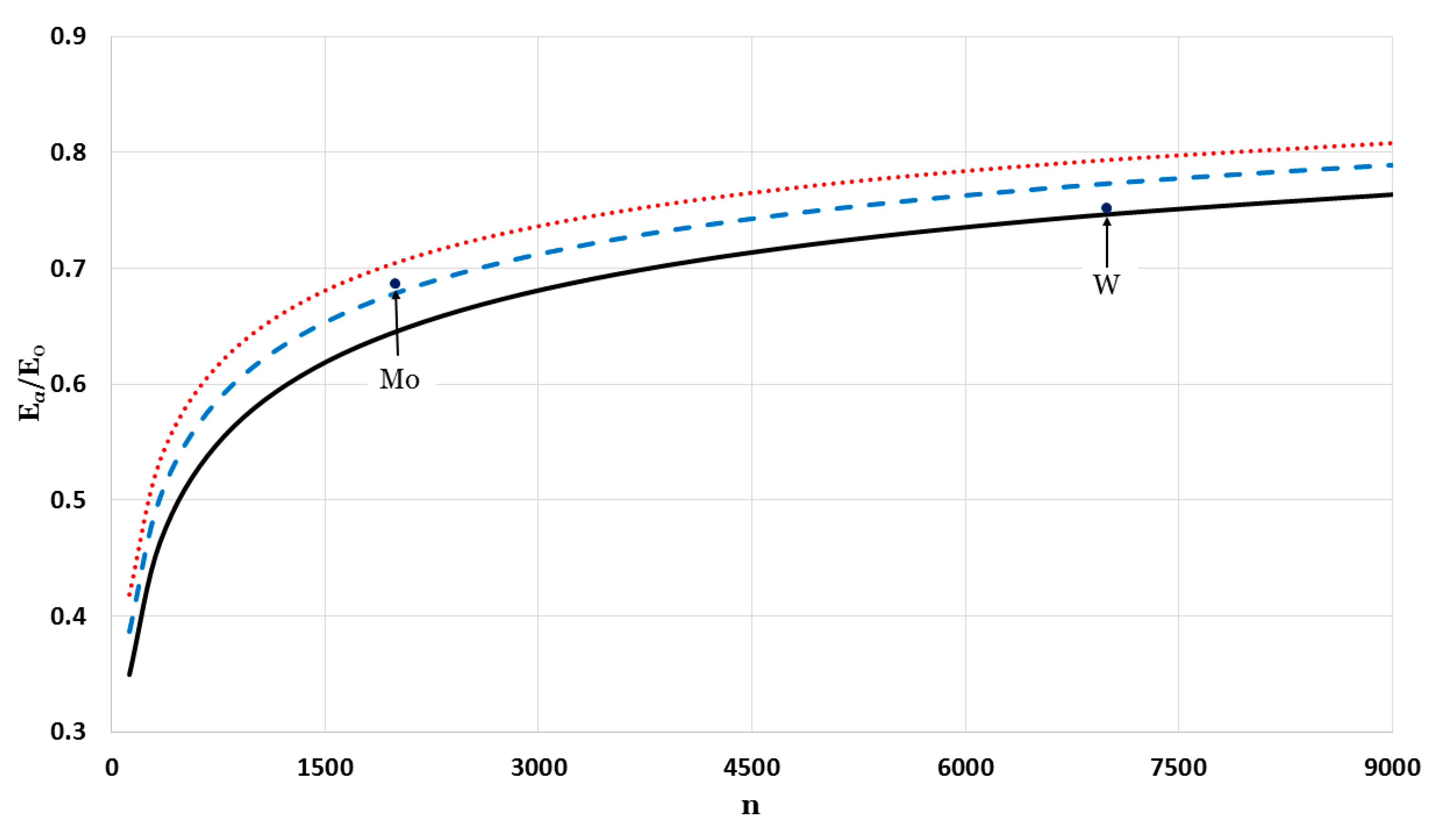
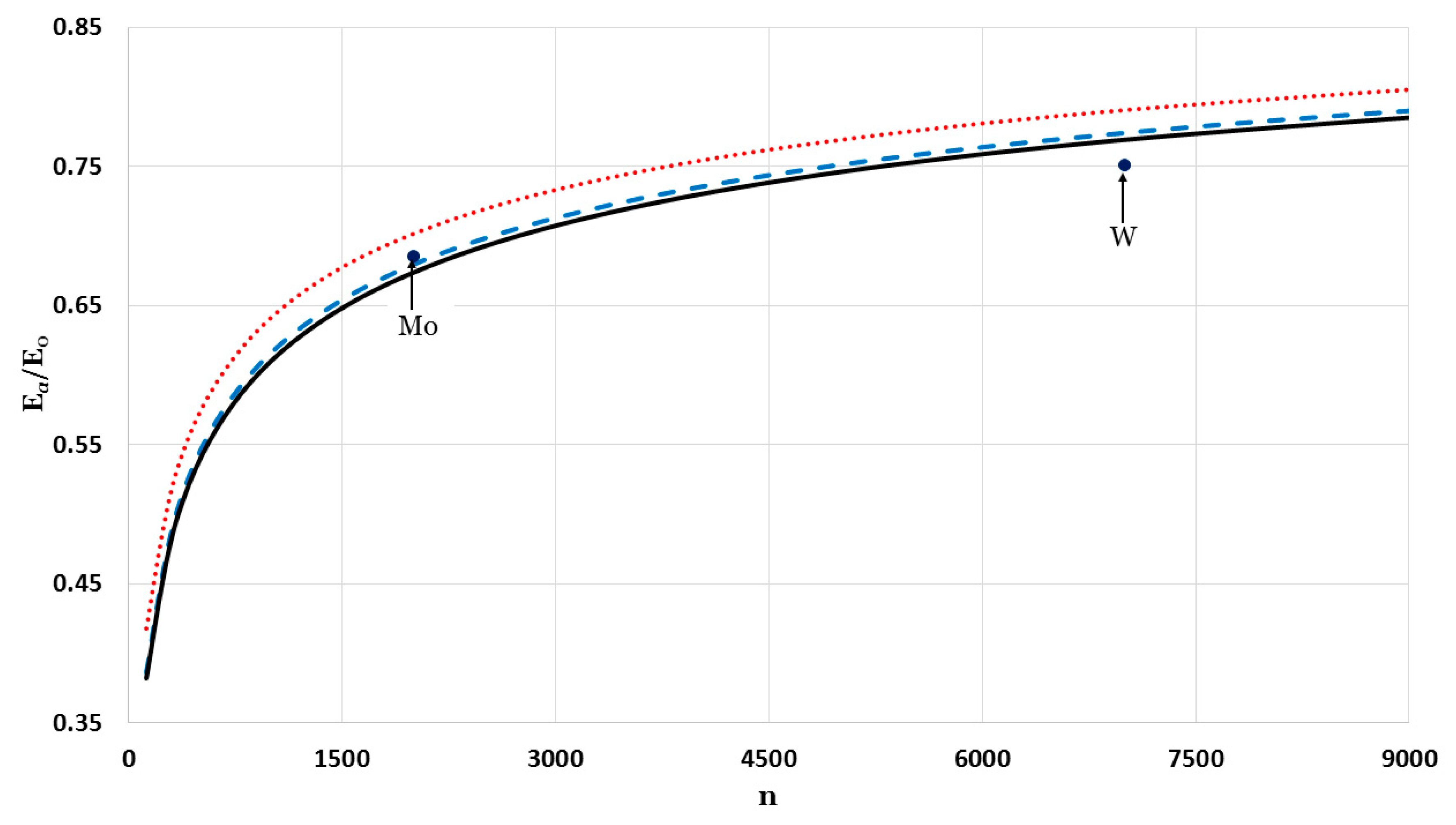
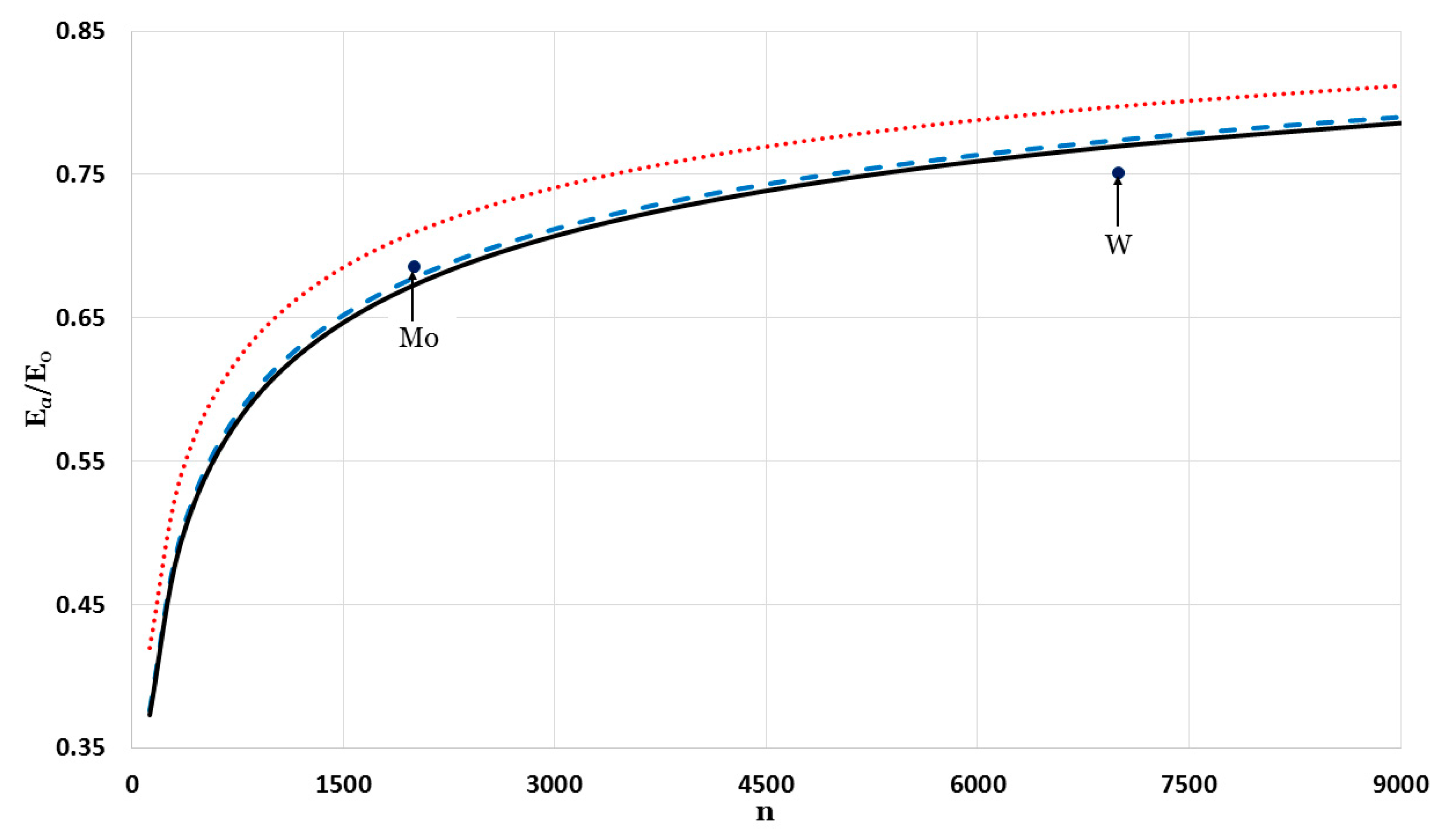
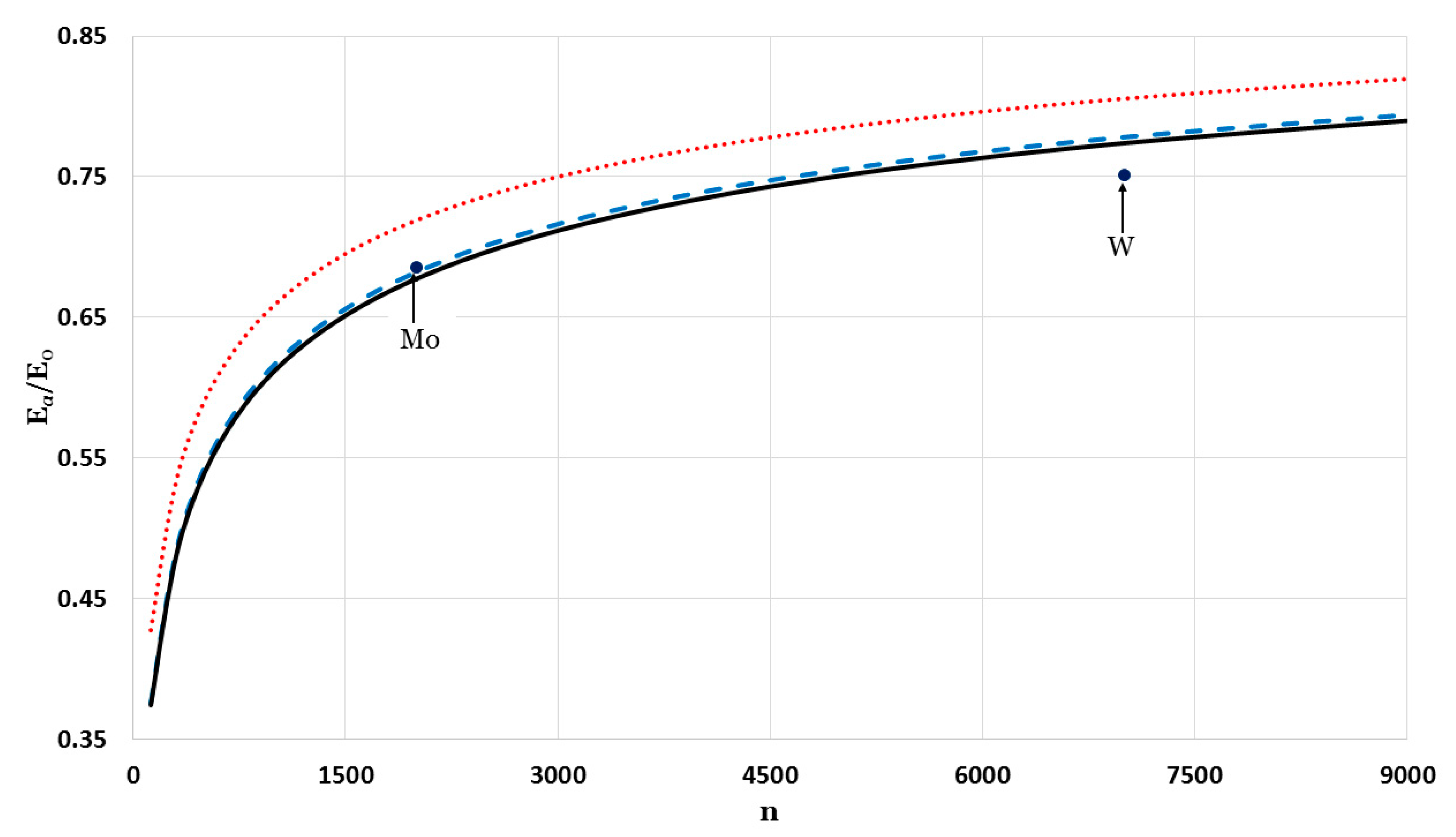
The relative cohesive energy of metallic nanoparticles in a face-centered cubic structure as a function of the number of atoms
was calculated by using the generalized Morse potential function for different values of
and a fixed value of
:
in
Figure 5 and
in
Figure 6. Additionally, the calculated relative cohesive energy of the nanoparticles with a face-centered cubic structure is comparable to the experimental values of the relative cohesive energy of Mo (−410 kJ/mol for
atoms [
23] and −598 kJ/mol for bulk [
24]) and W nanoparticles (−619 kJ/mol for
atoms [
23] and −824 kJ/mol for bulk [
24]). The calculations show the size dependence of the relative cohesive energy of the metallic nanoparticles. The values of relative cohesive energy were obtained from
Figure 5 and
Figure 6 for
and
atoms and summarized in
Table 1. As seen in
Table 1, the calculated values of the relative cohesive energy increased with the parameter
, due to the repulsive interaction becoming stiffer with
(as seen in
Figure 1). The calculated relative cohesive energies agreed with the experimental values for Mo nanoparticle when (
,
) and (
,
). On the other hand, when (
,
) the calculated relative cohesive energy agreed with the experimental value for W nanoparticles.
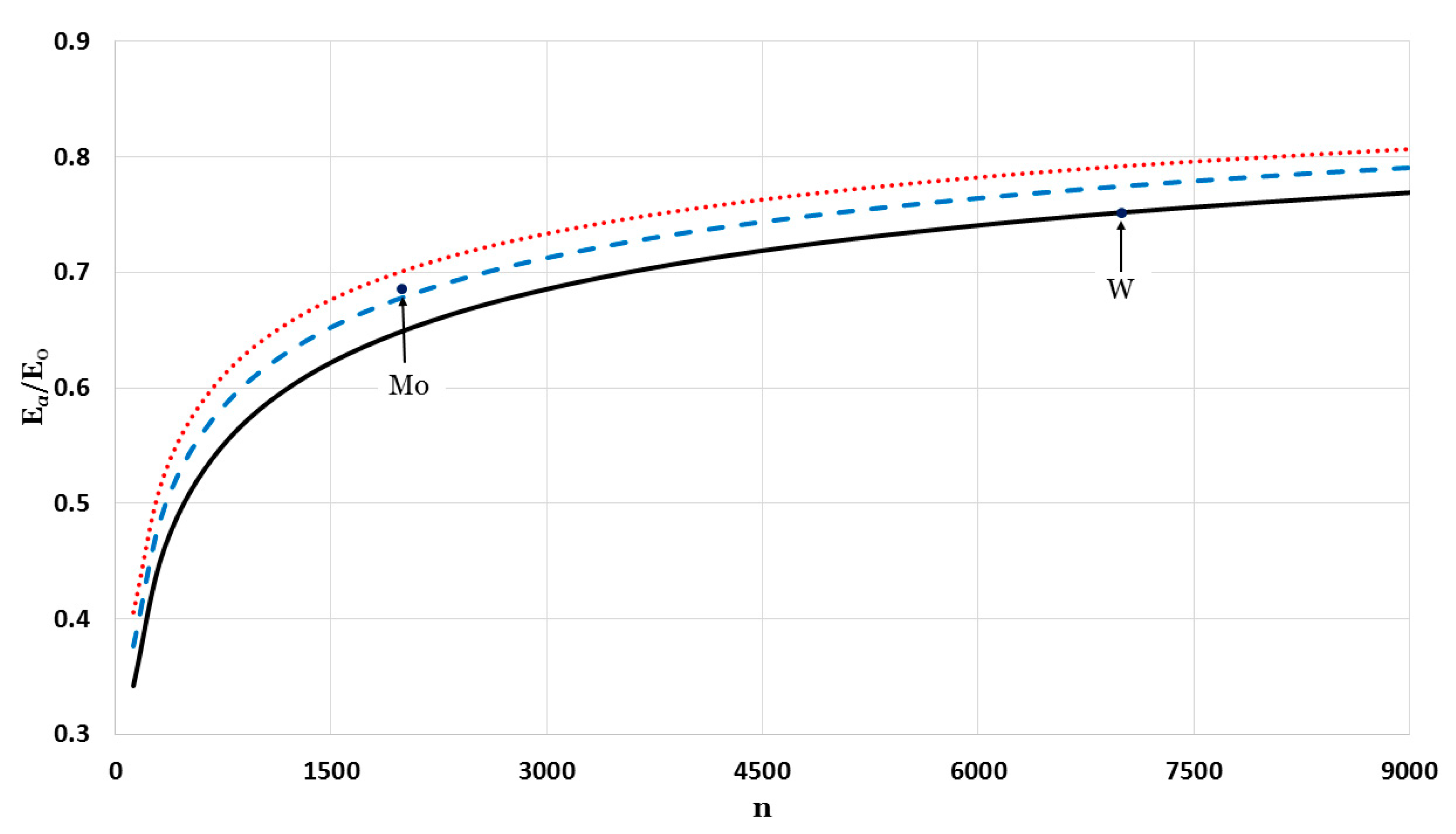
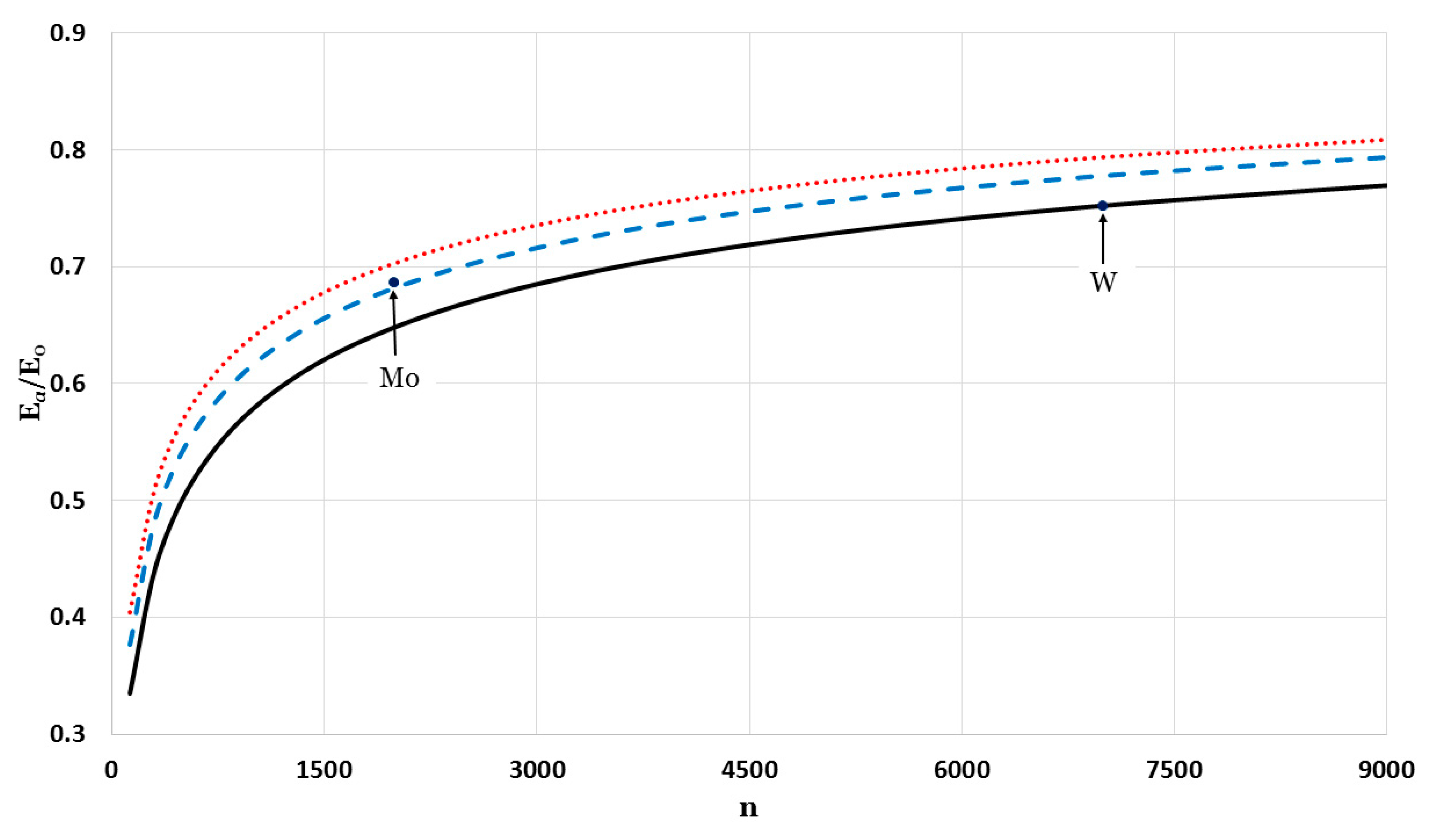
The potential function that is used to calculate the cohesive energy depends on two parameters,
and
. Different values of the parameter
gave different forms of the potential functions (as seen in
Figure 1). Consequently, the value of the parameter
will depend on type, structure, and value of
. The relative cohesive energies were calculated for a fixed value of
and different values of
:
Figure 7 (for
),
Figure 8 (for
), and
Figure 9 (for
). As seen in
Figure 7,
Figure 8 and
Figure 9, different values of
and
for the potential function, as summarized in
Figure 10, can be used to calculate the relative cohesive energies that are in agreement with the experimental values of Mo and W nanoparticles. The calculations show that the nanoparticles can be stable with a small value of
and a large value of
(due to the softening of the repulsive force in the potential function [
13]) or with a large value of
and small value of
(due to the strong attractive force in the potential function, which is discussed later in the article).
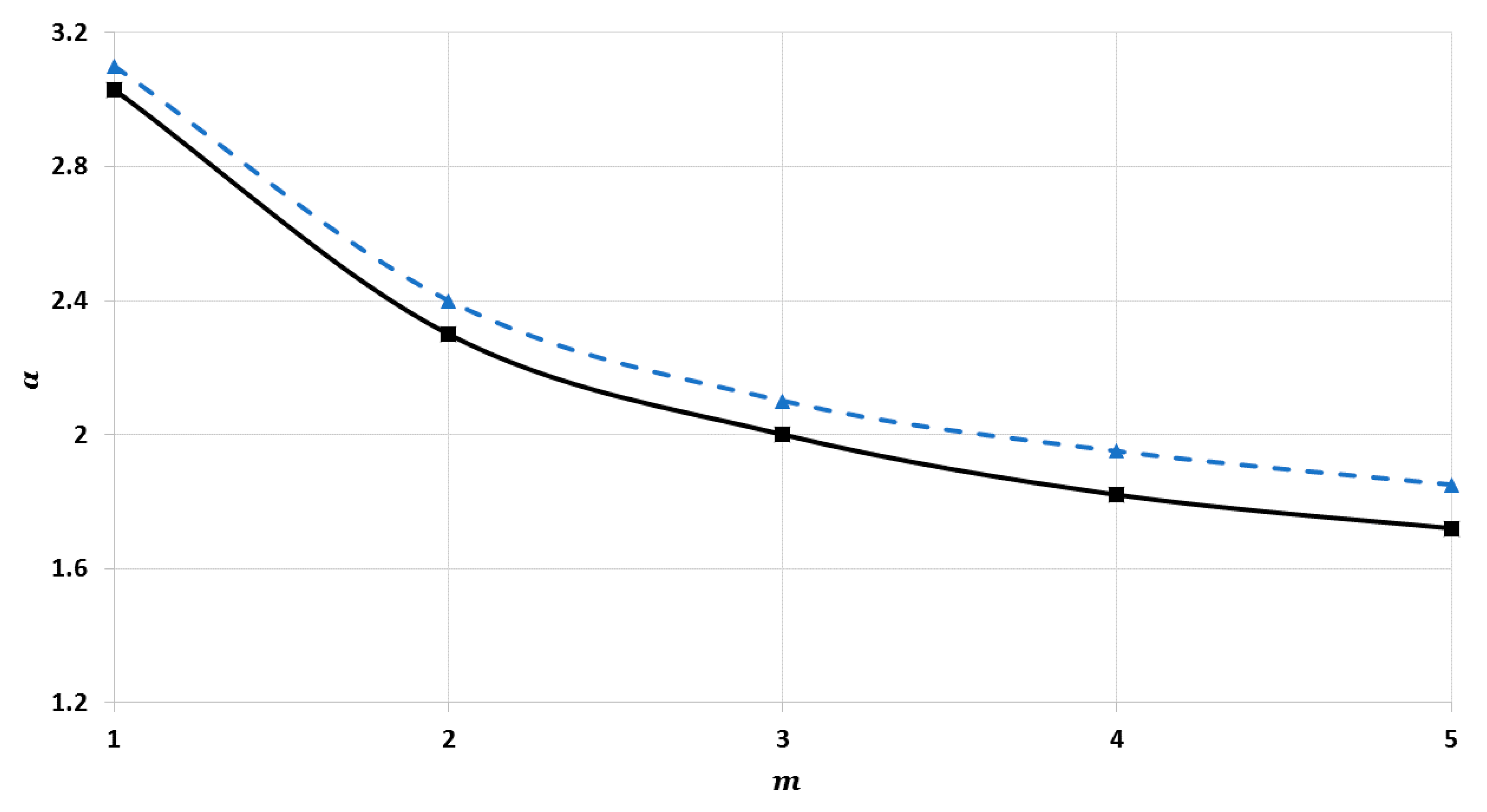
From another perspective, it was also found that the nanoparticles can be stable with a different number of atoms
with the same cohesive energy if the values of
and
of the potential function are changed, as seen in
Figure 11 (shows the relation between
and
for different values of
) and
Figure 12 (shows the relation between
and
for different values of
). Both figures show that the nanoparticles can be stabilized with a larger number of atoms having the same cohesive energy if the value of
is increased and the value of
is decreased. This result confirms that the long-range attractive force in the potential function plays an important role in the stability of the nanoparticles.
The stability of nanoparticles is a result of the balance between repulsive interactions and attractive interactions. It was found that the stability of nanoparticles using the Morse potential function (
) is due to the soft repulsive force and weak attractive force [
13]. This type of potential does not allow more bonds to form for each atom in the nanoparticle with distant surrounding atoms. Thus, only the few nearest surrounding atoms contribute to the stability of the nanoparticles. The curves in
Figure 13 represent the potential functions with different values of
and
that agree with the experimental value of the relative cohesive energy for Mo nanoparticles (as seen in
Figure 10). As it is clear in
Figure 10, when the parameter
in the generalized Morse potential function increases, the value of the
parameter becomes lower. Consequently, the repulsive interaction becomes stronger and the range of the attractive interaction becomes wider. However, the change in the attractive interaction range is weak when
due to the convergence in the
values. The enlargement of the attractive interaction range in the generalized potential function allows the atoms in the nanoparticles to have more bonds with the distant surrounding atoms. Consequently, the extra bonds play an important role in the stability of the nanoparticle.
The effect of nanoparticle structure in the cohesive energy was studied for different values of
and
such that:
and
in
Figure 14,
and
in
Figure 15, and
and
in
Figure 16. The calculated cohesive energies for a simple cubic structure are more obvious than body-centered cubic and face-centered cubic structures as predicted in other work [
12].
Melting point
is a size-dependent property of nanoparticles [
4,
6,
9]. Li et al. [
25] found that the cohesive energy is positively proportional to the melting point of the materials (
). Therefore, the melting point of nanoparticles as a function of the number of atoms
can be calculated using Equation (8), as follows:
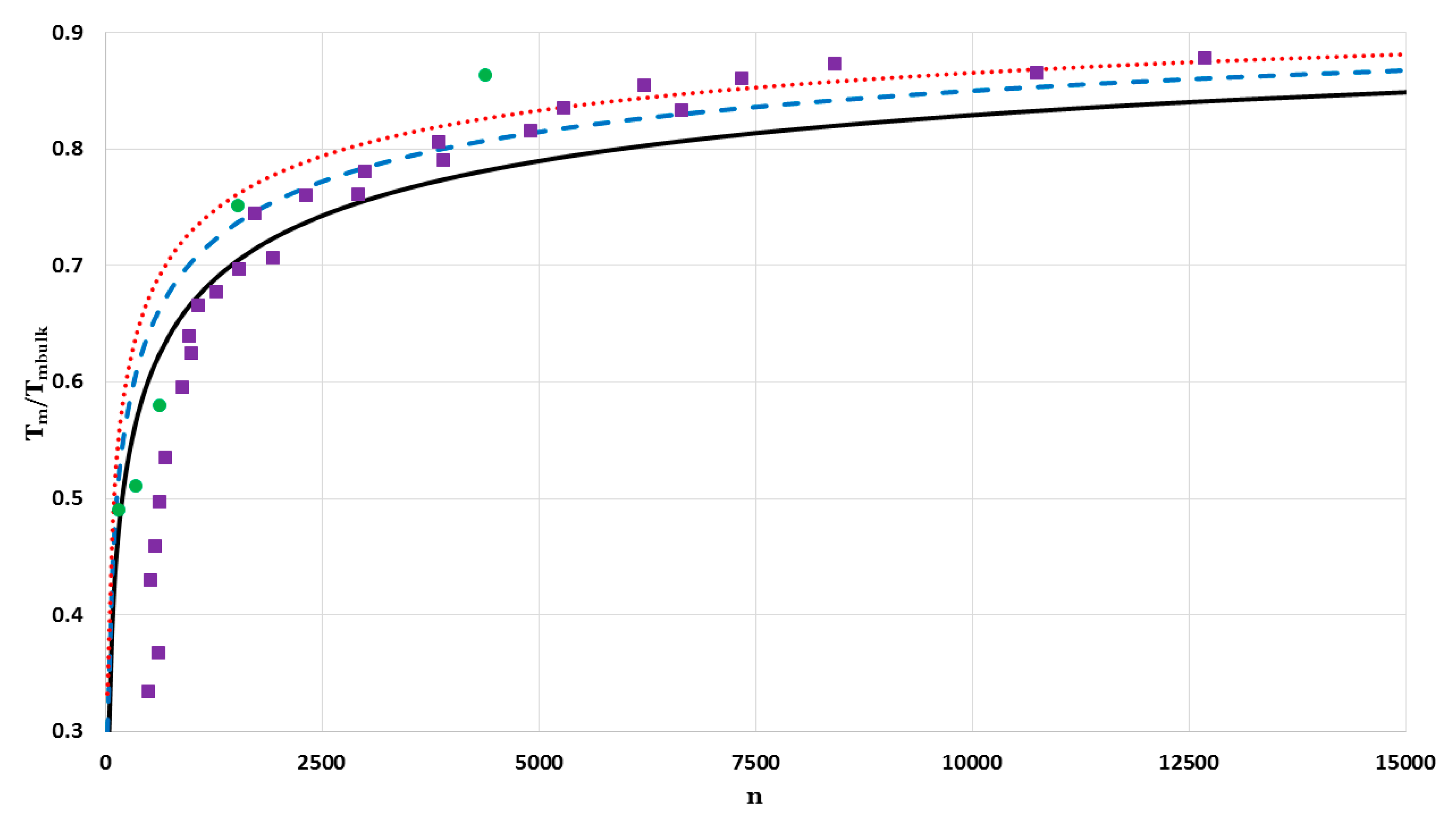
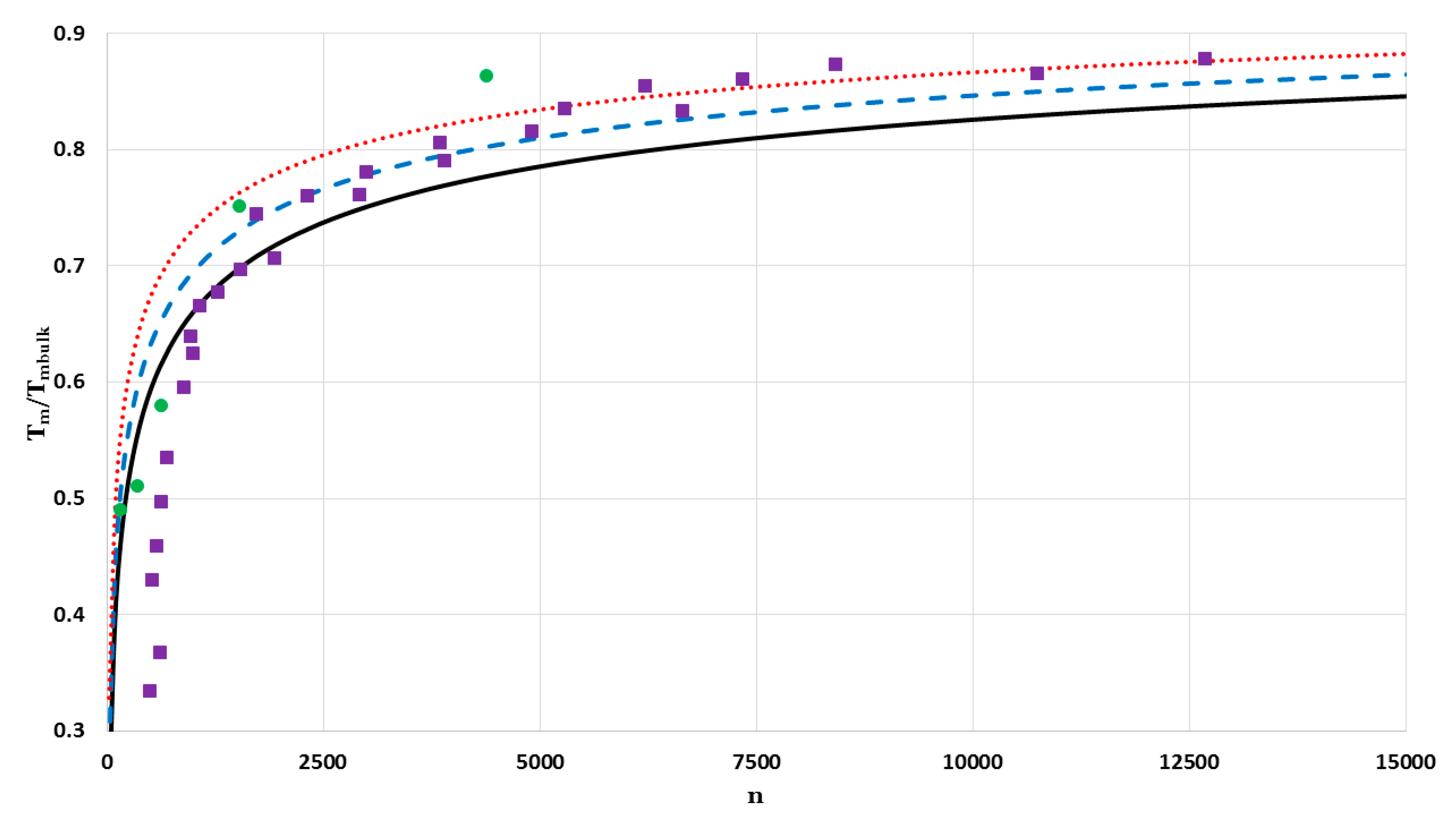
The values of
of nanoparticles in a face-centered cubic structure as a function of the number of atoms
are calculated for a fixed value of
and different values of
:
Figure 17 (for
) and
Figure 18 (for
). The calculated values of
are compared by two different sets of experimental data of melting points of Au (the mass density is
[
26] and the bulk melting point is
[
27]) nanoparticles in a face-centered cubic structure. The first set of the experimental data for melting points of Au measured by using a scanning electron-diffraction technique for nanoparticles on the amorphous carbon substrate [
27]. The second set of the experimental data for melting points of silica-encapsulated Au nanoparticle measured by using a differential thermal analysis (DTA) coupled to thermal gravimetric analysis (TGA) techniques [
28]. The melting points of Au nanoparticles in both sets of the experimental data are determined by their diameters
. The number of atoms
of Au nanoparticles in a face-centered cubic structure with given diameters can be determined through the relation:
[
9,
29] (where
is the atomic diameter and
is the lattice constant). The calculated values of
agree with the first set of the experimental data when
atoms (
) with little deviation when
atoms (
). The first set of the experimental data shows less values of melting points than those that are predicted by the model. The deviation arises due to the substrate effect on the melting point of very small Au nanoparticles [
30]. In the contrast, the silica shell had a small effect on the melting point of small Au nanoparticles, where the nanoparticles are considered as individual particles [
9,
28]. Therefore, the predicted values of
agree with the experimental data of the second set when
atoms (
).
5. Conclusions
In conclusion, the generalized Morse potential function for different values of
was used to account for the size dependence of the cohesive energy for different cubic metals: Simple cubic, body-centered cubic, and face-centered cubic structures. The generalized Morse potential function with
was used to predict the experimental values of the cohesive energy of W nanoparticles when
and Mo nanoparticles when
. If the value of
is approximated to be 2, then the generalized Morse potential function with
will have the following form:
The above potential function contains the following terms,
and
, that are equivalent to the Pauli repulsion and attractive dipole potentials, respectively, as in the Lennard–Jones potential proposed by Lim [
21,
22]. Consequently, the generalized Morse potential function with
can be the suitable potential to calculate the cohesive energy of nanoparticles. The stability of nanoparticles using a generalized Morse potential function with higher values of
(such as
) and low values of
(such as
) is due to the enlargement in the attractive interaction range rather than the softening of the repulsive interaction. The generalized Morse potential function with different values of
and
were used to calculate the melting point of nanoparticles in a face-centered cubic structure. The model using the generalized Morse potential function showed ability to predict the experimental data of melting points of Au nanoparticles with neglecting any surrounding effects, such as the effect of the substrate on the melting point of small Au nanoparticles.
A Lennard–Jones potential function with an additional two long-range attractive terms was proposed to calculate the energy bands of diatomic molecules [
31,
32]. Therefore, adding extra terms can also be applied to the Lennard–Jones potential function (as the generalized Morse potential function with
) to predict the experimental values of cohesive energy of W and Mo nanoparticles and the melting point of Au nanoparticles in face-centered cubic structures.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}