Enthalpies of Hydrate Formation and Dissociation from Residual Thermodynamics


Abstract
1. Introduction
2. Theoretical Analysis
Residual Thermodynamic Modelling of Hydrate Phase Transition
3. Results and Discussion
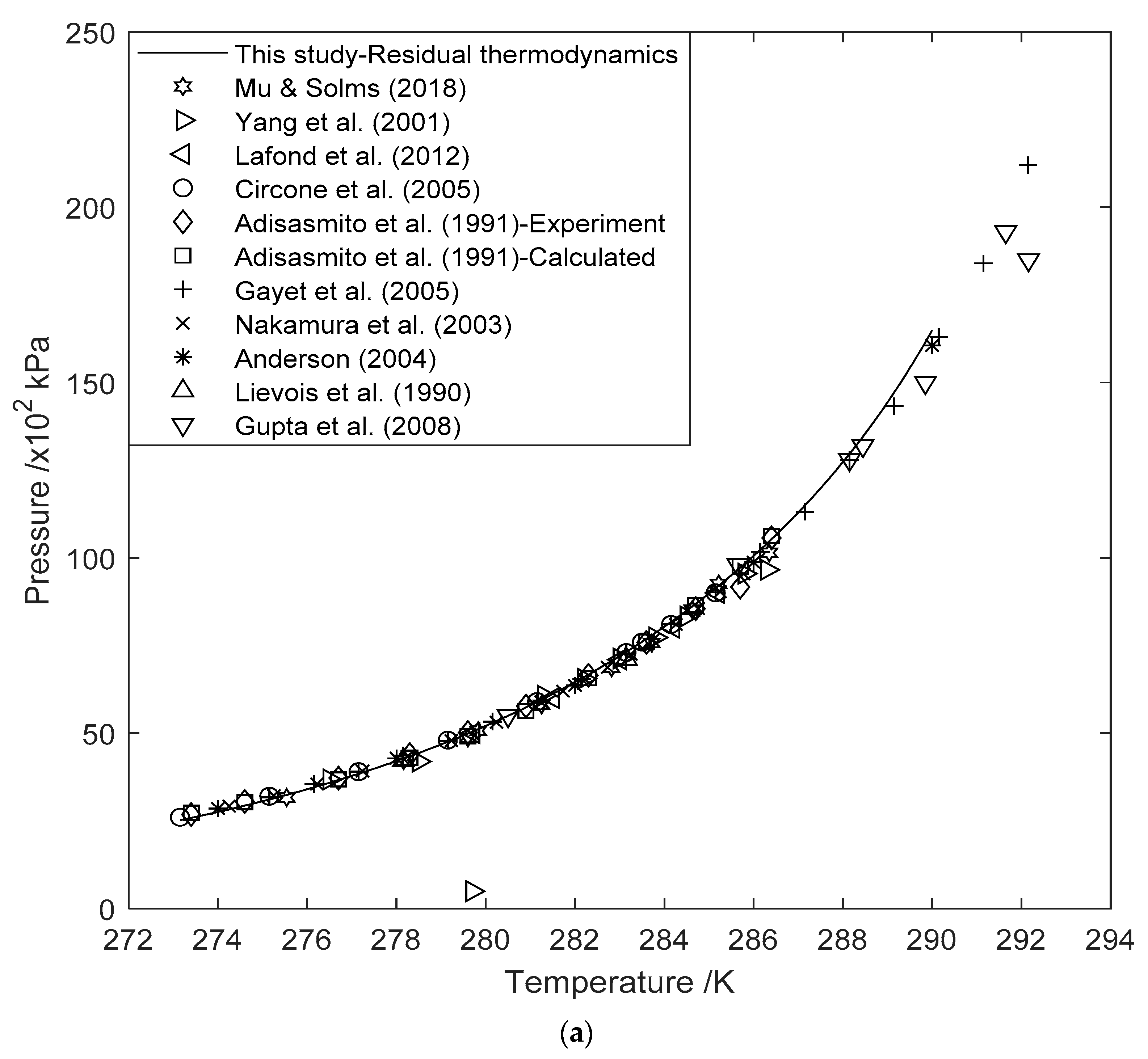
3.1. Hydrate Equilibrium Curves Using Residual Thermodynamics
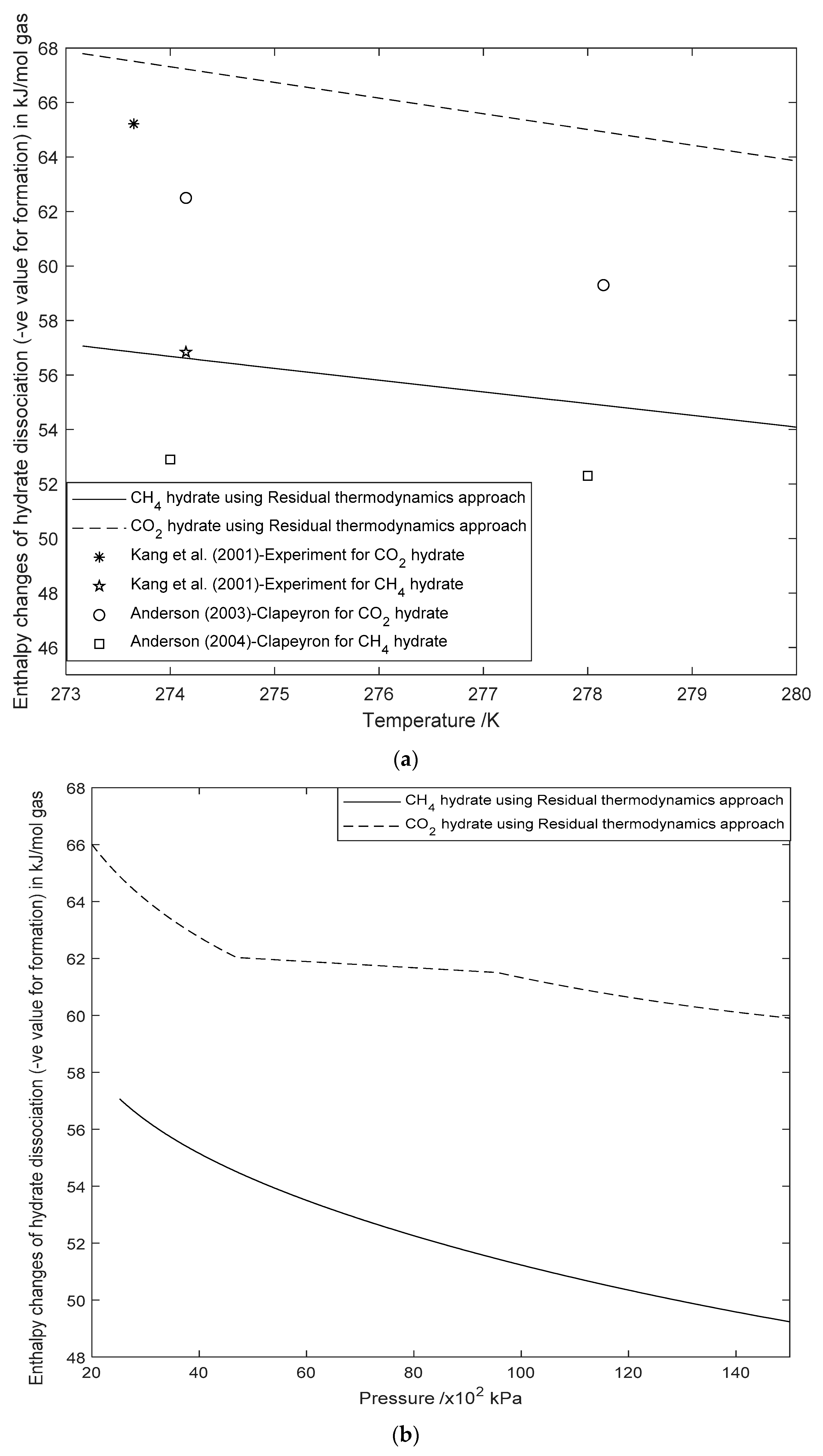
3.2. Enthalpy Changes of Hydrate Formation or Dissociation: Residual Thermodynamics versus Other Approaches
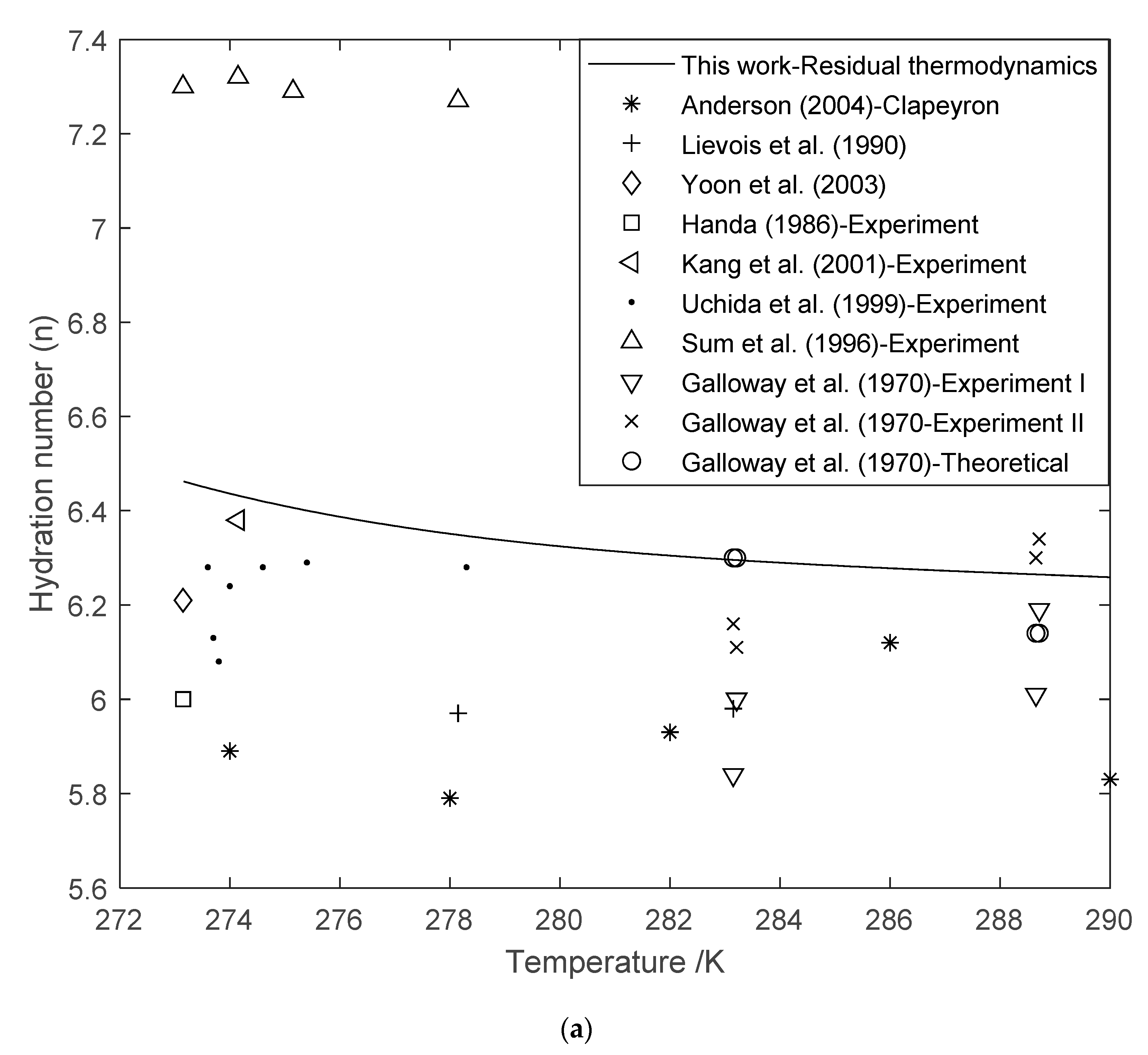
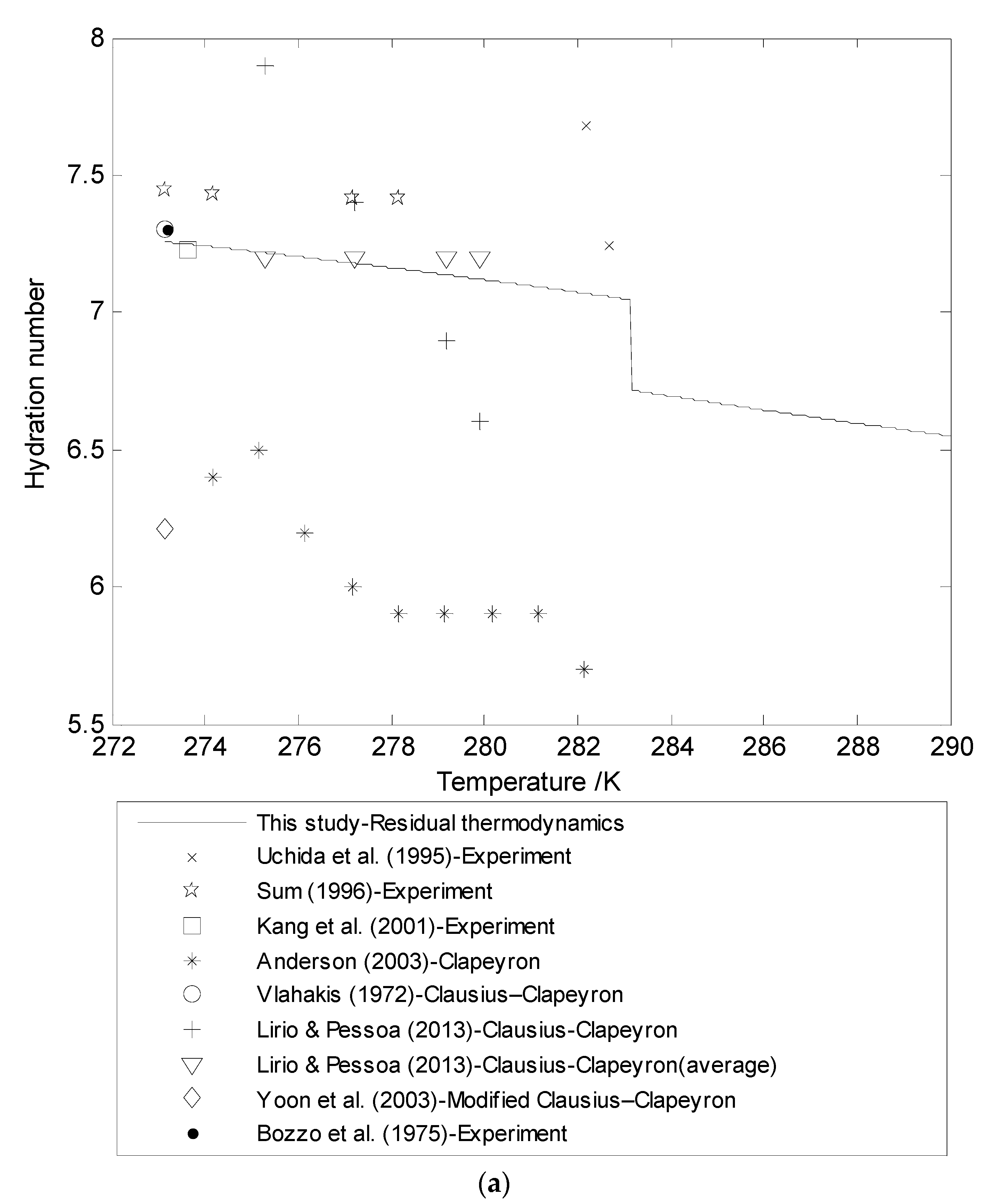
3.3. Hydration Number (n) Using Residual Thermodynamics versus Experimental Data
3.4. The Significance of Enthalpy Changes of Hydrate Formation or Dissociation in CH4-CO2 Swap
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Collett, T.S. Gas hydrates as a future energy resource. Geotimes 2004, 49, 24–27. [Google Scholar]
- Makogon, Y.F.; Holditch, S.A.; Makogon, T.Y. Natural gas-hydrates—A potential energy source for the 21st Century. J. Pet. Sci. Eng. 2007, 56, 14–31. [Google Scholar] [CrossRef]
- Fu, X.; Wang, J.; Tan, F.; Feng, X.; Wang, D.; He, J. Gas hydrate formation and accumulation potential in the Qiangtang Basin, northern Tibet, China. Energy Convers. Manage. 2013, 73, 186–194. [Google Scholar] [CrossRef]
- Anderson, B.J.; Kurihara, M.; White, M.D.; Moridis, G.J.; Wilson, S.J.; Pooladi-Darvish, M.; Gaddipati, M.; Masuda, Y.; Collett, T.S.; Hunter, R.B.; et al. Regional long-term production modeling from a single well test, Mount Elbert gas hydrate stratigraphic test well, Alaska North slope. Mar. Petroleum. Geol. 2011, 28, 493–501. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Y.; Wen, H.; Lu, Z.; Jia, Z.; Li, Y.; Li, Q.; Liu, C.; Wang, P.; Guo, X. Gas hydrates in the Qilian mountain permafrost, Qinghai, Northwest China. Acta. Geol. Sin. 2010, 84, 1–10. [Google Scholar] [CrossRef]
- Boswell, R. Is gas hydrate energy within reach? Science 2009, 325, 957–958. [Google Scholar] [CrossRef]
- Dunn, C.; Eshbaugh, M. Japan is the second largest net importer of fossil fuels in the world. US Energy Information Association. 7 November 2013. Available online: https://www.eia.gov/todayinenergy/detail.php?id=13711 (accessed on 7 December 2019).
- Liu, C.; Ye, Y.; Meng, Q.; He, X.; Lu, H.; Zhang, J.; Liu, J.; Yang, S. The characteristics of gas hydrates recovered from Shenhu Area in the South China Sea. Mar. Geol. 2012, 307, 22–27. [Google Scholar] [CrossRef]
- Lu, Z.; Zhu, Y.; Zhang, Y.; Wen, H.; Li, Y.; Liu, C. Gas hydrate occurrences in the Qilian Mountain permafrost, Qinghai province, China. Cold Reg. Sci. Technol. 2011, 66, 93–104. [Google Scholar] [CrossRef]
- Collett, T.S. Energy resource potential of natural gas hydrates. AAPG bulletin 2002, 86, 1971–1992. [Google Scholar]
- Klauda, J.B.; Sandler, S.I. Global distribution of methane hydrate in ocean sediment. Energ. Fuel 2005, 19, 459–470. [Google Scholar] [CrossRef]
- Milkov, A.V. Global estimates of hydrate-bound gas in marine sediments: how much is really out there? Earth-Sci. Rev. 2004, 66, 183–197. [Google Scholar] [CrossRef]
- Lirio, C.; Pessoa, F. Enthalpy of dissociation of simple and mixed carbon dioxide clathrate hydrate. Chem. Eng. Trans. 2013, 32, 557–582. [Google Scholar]
- Anderson, G.K. Enthalpy of dissociation and hydration number of methane hydrate from the Clapeyron equation. J. Chem. Thermodyn. 2004, 36, 1119–1127. [Google Scholar] [CrossRef]
- Kvamme, B.; Aromada, S.A.; Saeidi, N. Heterogeneous and homogeneous hydrate nucleation in CO2/water systems. J. Cryst. Growth 2019, 522, 160–174. [Google Scholar] [CrossRef]
- Kvamme, B.; Graue, A.; Aspenes, E.; Kuznetsova, T.; Gránásy, L.; Tóth, G.; Pusztai, T.; Tegze, G. Kinetics of solid hydrate formation by carbon dioxide: Phase field theory of hydrate nucleation and magnetic resonance imaging. Phys. Chem. Chem. Phys. 2004, 6, 2327–2334. [Google Scholar] [CrossRef]
- Kvamme, B.; Qasim, M.; Baig, K.; Kivelä, P.H.; Bauman, J. Hydrate phase transition kinetics from Phase Field Theory with implicit hydrodynamics and heat transport. Int. J. Greenh. Gas Control 2014, 29, 263. [Google Scholar] [CrossRef]
- Kivelä, P.H.; Baig, K.; Qasim, M.; Kvamme, B.R. Phase field theory modeling of methane fluxes from exposed natural gas hydrate reservoirs. AIP Conf. Proc. 2012, 1504, 351–363. [Google Scholar]
- Svandal, A.; Kvamme, B.; Grànàsy, L.; Pusztai, T.; Buanes, T.; Hove, J. The phase-field theory applied to CO2 and CH4 hydrate. J. Cryst. Growth 2006, 287, 486–490. [Google Scholar] [CrossRef]
- Tegze, G.; Pusztai, T.; Tóth, G.; Gránásy, L.; Svandal, A.; Buanes, T.; Kuznetsova, T.; Kvamme, B. Multiscale approach to CO2 hydrate formation in aqueous solution: Phase field theory and molecular dynamics. Nucleation and growth. J. Chem. Phys. 2006, 124, 234710. [Google Scholar] [CrossRef]
- Kvamme, B. Kinetics of Hydrate Formation from Nucleation Theory. Int. J. Off. Polar Eng. 2002, 12, 256. [Google Scholar]
- Kvamme, B.; Kuznetsova, T.; Bauman, J.M.; Sjöblom, S.; Avinash Kulkarni, A. Hydrate Formation during Transport of Natural Gas Containing Water and Impurities. J. Chem. Eng. Data 2016, 61, 936–949. [Google Scholar] [CrossRef]
- Aromada, S.A.; Kvamme, B. New approach for evaluating the risk of hydrate formation during transport of hydrocarbon hydrate formers of sI and sII. AIChE J. 2019, 65, 1097–1110. [Google Scholar] [CrossRef]
- Kvamme, B.; Aromada, S.A. Risk of Hydrate Formation during the Processing and Transport of Troll Gas from the North Sea. J. Chem. Eng. Data 2017, 62, 2163–2177. [Google Scholar] [CrossRef]
- Anderson, G.K. Enthalpy of dissociation and hydration number of carbon dioxide hydrate from the Clapeyron equation. J. Chem. Thermodyn. 2003, 35, 1171–1183. [Google Scholar] [CrossRef]
- Kvamme, B.; Aromada, S.A.; Gjerstad, P.B. Consistent Enthalpies of the Hydrate Formation and Dissociation Using Residual Thermodynamics. J. Chem. Eng. Data 2019, 64, 3493–3504. [Google Scholar] [CrossRef]
- van der Waals, J.H.; Platteeuw, J.C. Clathrate solutions. Advan. Chem. Phys. 1959, 2, 1–57. [Google Scholar]
- Sloan, E.D., Jr.; Koh, C.A. Clathrate Hydrates of Natural Gases; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Kvamme, B.; Lund, A.; Hertzberg, T. The influence of gas-gas interactions on the Langmuir constants for some natural gas hydrates. Fluid Phase Equilib. 1993, 90, 15–44. [Google Scholar] [CrossRef]
- Kvamme, B.; Førrisdahl, O.K. Polar guest-molecules in natural gas hydrates. Effects of polarity and guest-guest-interactions on the Langmuir-constants. Fluid Phase Equilib 1993, 83, 427–435. [Google Scholar] [CrossRef]
- Kvamme, B.; Tanaka, H. Thermodynamic stability of hydrates for ethane, ethylene, and carbon dioxide. J. Phys. Chem. 1995, 99, 7114–7119. [Google Scholar] [CrossRef]
- Kvamme, B. Feasibility of simultaneous CO2 storage and CH4 production from natural gas hydrate using mixtures of CO2 and N2. Can. J. Chem. 2015, 93, 897–905. [Google Scholar] [CrossRef]
- Kvamme, B. Thermodynamic Limitations of the CO2/N2 Mixture Injected into CH4 Hydrate in the Ignik Sikumi Field Trial. J. Chem. Eng. Data 2016, 61, 1280–1295. [Google Scholar] [CrossRef]
- Aromada, S.A. New Concept for Evaluating the Risk of Hydrate Formation during Processing and Transport of Hydrocarbons. Master’s Thesis, The University of Bergen, Bergen, Norway, 2017. [Google Scholar]
- Svandal, A. Modeling Hydrate Phase Transitions using Mean-Field Approaches. Ph.D. Thesis, University of Bergen, Bergen, Norway, 2006. [Google Scholar]
- Gibbs, J.W. The Collected Works of J. Willard Gibbs, Thermodynamics; Yale university Press: New Haven, CT, USA, 1928; Volume 1, pp. 55–353. [Google Scholar]
- Soave, G. Equilibrium constants from a modified Redlich-Kwong equation of state. Chem. Eng. Sci. 1972, 27, 1197–1203. [Google Scholar] [CrossRef]
- Mu, L.; Solms, N.V. Hydrate thermal dissociation behavior and dissociation enthalpies in methane-carbon dioxide swapping process. J. Chem. Thermodyn. 2018, 117, 33–42. [Google Scholar] [CrossRef]
- Yang, S.O.; Cho, S.H.; Lee, H.; Lee, C.S. Measurement and prediction of phase equilibria for water+ methane in hydrate forming conditions. Fluid Phase Equilib. 2001, 185, 53–63. [Google Scholar] [CrossRef]
- Lafond, P.G.; Olcott, K.A.; Sloan, E.D.; Koh, C.A.; Sum, A.K. Measurements of methane hydrate equilibrium in systems inhibited with NaCl and methanol. J. Chem. Thermodyn. 2012, 48, 1–6. [Google Scholar] [CrossRef]
- Circone, S.; Kirby, S.H.; Stern, L.A. Direct measurement of methane hydrate composition along the hydrate equilibrium boundary. J. Phys. Chem. B 2005, 109, 9468–9475. [Google Scholar] [CrossRef]
- Adisasmito, S.; Frank, R.J., III; Sloan, E.D., Jr. Hydrates of carbon dioxide and methane mixtures. J. Chem. Eng. Data 1991, 36, 68–71. [Google Scholar] [CrossRef]
- Gayet, P.; Dicharry, C.; Marion, G.; Graciaa, A.; Lachaise, J.; Nesterov, A. Experimental determination of methane hydrate dissociation curve up to 55 MPa by using a small amount of surfactant as hydrate promoter. Chem. Eng. Sci. 2005, 60, 5751–5758. [Google Scholar] [CrossRef]
- Nakamura, T.; Makino, T.; Sugahara, T.; Ohgaki, K. Stability boundaries of gas hydrates helped by methane—Structure-H hydrates of methylcyclohexane and cis-1, 2-dimethylcyclohexane. Chem. Eng. Sci. 2003, 58, 269–273. [Google Scholar] [CrossRef]
- Lievois, J.S.; Perkins, R.; Martin, R.J.; Kobayashi, R. Development of an automated, high pressure heat flux calorimeter and its application to measure the heat of dissociation and hydrate numbers of methane hydrate. Fluid Phase Equilib. 1990, 59, 73–97. [Google Scholar] [CrossRef]
- Gupta, A.; Lachance, J.; Sloan, E.D., Jr.; Koh, C.A. Measurements of methane hydrate heat of dissociation using high pressure differential scanning calorimetry. Chem. Eng. Sci. 2008, 63, 5848–5853. [Google Scholar] [CrossRef]
- Fan, S.-S.; Guo, T.-M. Hydrate formation of CO2-rich binary and quaternary gas mixtures in aqueous sodium chloride solutions. J. Chem. Eng. Data 1999, 44, 829–832. [Google Scholar] [CrossRef]
- Wendland, M.; Hasse, H.; Maurer, G. Experimental pressure− temperature data on three-and four-phase equilibria of fluid, hydrate, and ice phases in the system carbon dioxide− water. J. Chem. Eng. Data 1999, 44, 901–906. [Google Scholar] [CrossRef]
- Delahaye, A.; Fournaison, L.; Marinhas, S.; Chatti, I.; Petitet, J.P.; Dalmazzone, D.; Fürst, W. Effect of THF on equilibrium pressure and dissociation enthalpy of CO2 hydrates applied to secondary refrigeration. Ind. Eng. Chem. Res. 2006, 45, 391–397. [Google Scholar] [CrossRef]
- Fournaison, L.; Delahaye, A.; Chatti, I.; Petitet, J.P. CO2 hydrates in refrigeration processes. Ind. Eng. Chem. Res. 2004, 43, 6521–6526. [Google Scholar] [CrossRef]
- Sabil, K.M.; Witkamp, G.-J.; Peters, C.J. Estimations of enthalpies of dissociation of simple and mixed carbon dioxide hydrates from phase equilibrium data. Fluid Phase Equilib. 2010, 290, 109–114. [Google Scholar] [CrossRef]
- Ohgaki, K.; Makihara, Y.; Takano, K. Formation of CO2 hydrate in pure and sea waters. J. Chem. Eng. Jpn. 1993, 26, 558–564. [Google Scholar] [CrossRef]
- Kvamme, B.; Aromada, S.A. Alternative Routes to Hydrate Formation during Processing and Transport of Natural Gas with a Significant Amount of CO2: Sleipner Gas as a Case Study. J. Chem. Eng. Data 2018, 63, 832–844. [Google Scholar] [CrossRef]
- Aromada, S.A.; Kvamme, B. Production of Methane from Hydrate and CO2 Zero-Emission Concept. In Proceedings of the 10th EUROSIM2019 Congress, Logroño (La Rioja), Spain, 1–5 July 2019. [Google Scholar]
- Kang, S.-P.; Lee, H.; Ryu, B.-J. Enthalpies of dissociation of clathrate hydrates of carbon dioxide, nitrogen, (carbon dioxide+ nitrogen), and (carbon dioxide+ nitrogen+ tetrahydrofuran). J. Chem. Thermodyn. 2001, 33, 513–521. [Google Scholar] [CrossRef]
- Handa, Y. Compositions, enthalpies of dissociation, and heat capacities in the range 85 to 270 K for clathrate hydrates of methane, ethane, and propane, and enthalpy of dissociation of isobutane hydrate, as determined by a heat-flow calorimeter. J. Chem. Thermodyn. 1986, 18, 915–921. [Google Scholar] [CrossRef]
- Deaton, W.; Frost, E., Jr. Gas Hydrates and Their Relation to the Operation of Natural Gas Pipe-Lines; US Bur. Mines Monograph 8; U.S. Department of Energy Office of Scientific and Technical Information: Oak Ridge, TN, USA, 1946.
- Yoon, J.H.; Yamamoto, Y.; Komai, T.; Haneda, H.; Kawamura, T. Rigorous approach to the prediction of the heat of dissociation of gas hydrates. Ind. Eng. Chem. Res. 2003, 42, 1111–1114. [Google Scholar] [CrossRef]
- Sloan, E.; Fleyfel, F. Hydrate dissociation enthalpy and guest size. Fluid Phase Equilib. 1992, 76, 123–140. [Google Scholar] [CrossRef]
- De Roo, J.L.; Peters, C.J.; Lichtenthaler, R.N.; Diepen, G.A.M. Occurrence of methane hydrate in saturated and unsaturated solutions of sodium chloride and water in dependence of temperature and pressure. AIChE J. 1983, 29, 651–657. [Google Scholar] [CrossRef]
- Roberts, O.L.; Brownscombe, E.R.; Howe, L.S.; Ramser, H. Phase diagrams of methane and ethane hydrates. Petr. Eng. 1941, 12, 56. [Google Scholar]
- McLeod, H.O.; Campbell, J.M. Natural gas hydrates at pressures to 10,000 psia. J. Pet. Technol. 1961, 222, 590. [Google Scholar] [CrossRef]
- Skovborg, P.; Rasmussen, P. Comments on: Hydrate dissociation enthalpy and guest size. Fluid Phase Equilib. 1994, 96, 223–231. [Google Scholar] [CrossRef]
- Vlahakis, J. The Growth Rate of Ice Crystals: The Properties of Carbon Dioxide Hydrate; A Review of Properties of 51 Gas Hydrates; Washington U.S. Dept. of the Interior: Syracuse University, NY, USA, 1972; Volume 830, p. 14.
- Galloway, T.J.; Ruska, W.; Chappelear, P.S.; Kobayashi, R. Experimental measurement of hydrate numbers for methane and ethane and comparison with theoretical values. Ind. Eng. Chem. Fundam. 1970, 9, 237–243. [Google Scholar] [CrossRef]
- Uchida, T.; Hirano, T.; Ebinuma, T.; Narita, H.; Gohara, K.; Mae, S.; Matoto, R. Raman spectroscopic determination of hydration number of methane hydrates. AIChE J. 1999, 45, 2641–2645. [Google Scholar] [CrossRef]
- Sum, A.K. Measurements of clathrate hydrates properties via Raman spectroscopy. In Proceedings of the 2nd International Conference on Natural Gas Hydrates, Toulouse, France, 2–6 June 1996; pp. 51–58. [Google Scholar]
- Bozzo, A.T.; Hsiao-Sheng, C.; Kass, J.R.; Barduhn, A.J. The properties of the hydrates of chlorine and carbon dioxide. Desalination 1975, 16, 303–320. [Google Scholar] [CrossRef]
- Uchida, T.; Takagi, A.; Kawabata, J.; Mae, S.; Hondoh, T. Raman spectroscopic analyses of the growth process of CO2 hydrates. Energy Convers. Manag. 1995, 36, 547–550. [Google Scholar] [CrossRef]
- Hammerschmidt, E. Formation of gas hydrates in natural gas transmission lines. Ind. Eng. Chem. 1934, 26, 851–855. [Google Scholar] [CrossRef]
- Kvamme, B.; Aromada, S.A.; Kuznetsova, T.; Gjerstad, P.B.; Canonge, P.C.; Zarifi, M. Maximum tolerance for water content at various stages of a natuna production. Heat Mass Transf. 2019, 55, 1059–1079. [Google Scholar] [CrossRef]
- Aromada, S.A.; Kvamme, B. Impacts of CO2 and H2S on the risk of hydrate formation during pipeline transport of natural gas. Front. Chem. Sci. Eng. 2019, 13, 616–627. [Google Scholar] [CrossRef]
- Aromada, S.A.; Kvamme, B. Simulation of Hydrate Plug Prevention in Natural Gas Pipeline from Bohai Bay to Onshore Facilities in China. In Proceedings of the 10th EUROSIM2019 Congress, Logroño (La Rioja), Spain, 1–5 July 2019. [Google Scholar]
- Sloan, E.D. Fundamental principles and applications of natural gas hydrates. Nature 2003, 426, 353. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, B. Environmentally Friendly Production of Methane from Natural Gas Hydrate Using Carbon Dioxide. Sustainability 2019, 11, 1964. [Google Scholar] [CrossRef]
- Ohgaki, K.; Takano, K.; Sangawa, H.; Matsubara, T.; Nakano, S. Methane Exploitation by Carbon Dioxide from Gas Hydrates—Phase Equilibria for CO2-CH4 Mixed Hydrate System. J. Chem. Eng. Jpn. 1996, 29, 478–483. [Google Scholar] [CrossRef]
- Lee, H.; Seo, Y.; Seo, Y.T.; Moudrakovski, I.L.; Ripmeester, J.A. Recovering methane from solid methane hydrate with carbon dioxide. Angew. Chem. Int. Ed. 2003, 42, 5048–5051. [Google Scholar] [CrossRef]
- Falenty, A.; Salamatin, A.; Kuhs, W. Kinetics of CO2-hydrate formation from ice powders: Data summary and modeling extended to low temperatures. J. Phys. Chem. C 2013, 117, 8443–8457. [Google Scholar] [CrossRef]
- Buanes, T.; Kvamme, B.; Svandal, A. Computer simulation of CO2 hydrate growth. J. Cryst. Growth 2006, 287, 491–494. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water Phase, m | a0 | a1 |
|---|---|---|
| Empty structure I | −3.087 | −18.246 |
| Empty structure II | −3.188 | −18.186 |
| Ice (T < 273.15 K) | −2.639 | −19.051 |
| Liquid water (T > 273.15 K) | −5.610 | −16.080 |
| Temperature | Methane (CH4) | Carbon Dioxide (CO2) | ||||
|---|---|---|---|---|---|---|
| Pressure | Hydration Number (n) | Pressure | Hydration Number (n) | |||
| (K) | (kPa) | (kJ/mol Guest) | (kPa) | (kJ/mol Guest) | ||
| 273.16 | 25.19 | 6.46 | 57.07 | 14.19 | 7.26 | 67.79 |
| 274.17 | 27.87 | 6.43 | 56.63 | 15.73 | 7.24 | 67.24 |
| 275.13 | 31.01 | 6.41 | 56.19 | 17.59 | 7.22 | 66.67 |
| 276.15 | 34.53 | 6.38 | 55.75 | 19.73 | 7.20 | 66.08 |
| 277.16 | 38.45 | 6.37 | 55.31 | 22.21 | 7.18 | 65.50 |
| 278.17 | 42.83 | 6.35 | 54.88 | 25.06 | 7.16 | 64.91 |
| 279.13 | 47.43 | 6.34 | 54.47 | 28.17 | 7.14 | 64.36 |
| 280.14 | 52.87 | 6.32 | 54.03 | 31.96 | 7.11 | 63.77 |
| 281.16 | 58.97 | 6.31 | 53.57 | 36.34 | 7.09 | 63.18 |
| 282.17 | 65.80 | 6.30 | 53.11 | 41.42 | 7.07 | 62.59 |
| 283.13 | 73.03 | 6.30 | 52.66 | 46.95 | 7.05 | 62.03 |
| 284.14 | 81.64 | 6.29 | 52.17 | 109.88 | 6.69 | 60.96 |
| 285.15 | 91.41 | 6.28 | 51.65 | 128.38 | 6.67 | 60.40 |
| 286.17 | 102.55 | 6.28 | 51.11 | 152.24 | 6.64 | 59.86 |
| 287.13 | 114.65 | 6.27 | 50.57 | 183.38 | 6.62 | 59.39 |
| 288.14 | 129.55 | 6.27 | 49.97 | 233.44 | 6.59 | 58.96 |
| 289.16 | 147.28 | 6.26 | 49.33 | 313.22 | 6.57 | 58.68 |
| 290.00 | 164.94 | 6.26 | 48.76 | 404.15 | 6.55 | 58.55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aromada, S.A.; Kvamme, B.; Wei, N.; Saeidi, N. Enthalpies of Hydrate Formation and Dissociation from Residual Thermodynamics. Energies 2019, 12, 4726. https://doi.org/10.3390/en12244726
Aromada SA, Kvamme B, Wei N, Saeidi N. Enthalpies of Hydrate Formation and Dissociation from Residual Thermodynamics. Energies. 2019; 12(24):4726. https://doi.org/10.3390/en12244726
Chicago/Turabian StyleAromada, Solomon Aforkoghene, Bjørn Kvamme, Na Wei, and Navid Saeidi. 2019. "Enthalpies of Hydrate Formation and Dissociation from Residual Thermodynamics" Energies 12, no. 24: 4726. https://doi.org/10.3390/en12244726
APA StyleAromada, S. A., Kvamme, B., Wei, N., & Saeidi, N. (2019). Enthalpies of Hydrate Formation and Dissociation from Residual Thermodynamics. Energies, 12(24), 4726. https://doi.org/10.3390/en12244726