Lifetime Prediction of a Polymer Electrolyte Membrane Fuel Cell under Automotive Load Cycling Using a Physically-Based Catalyst Degradation Model


Abstract
1. Introduction
2. Simulation Methodology
2.1. Overview of Simulation Framework
2.2. Fuel Cell Car Model
2.3. Single Cell Model
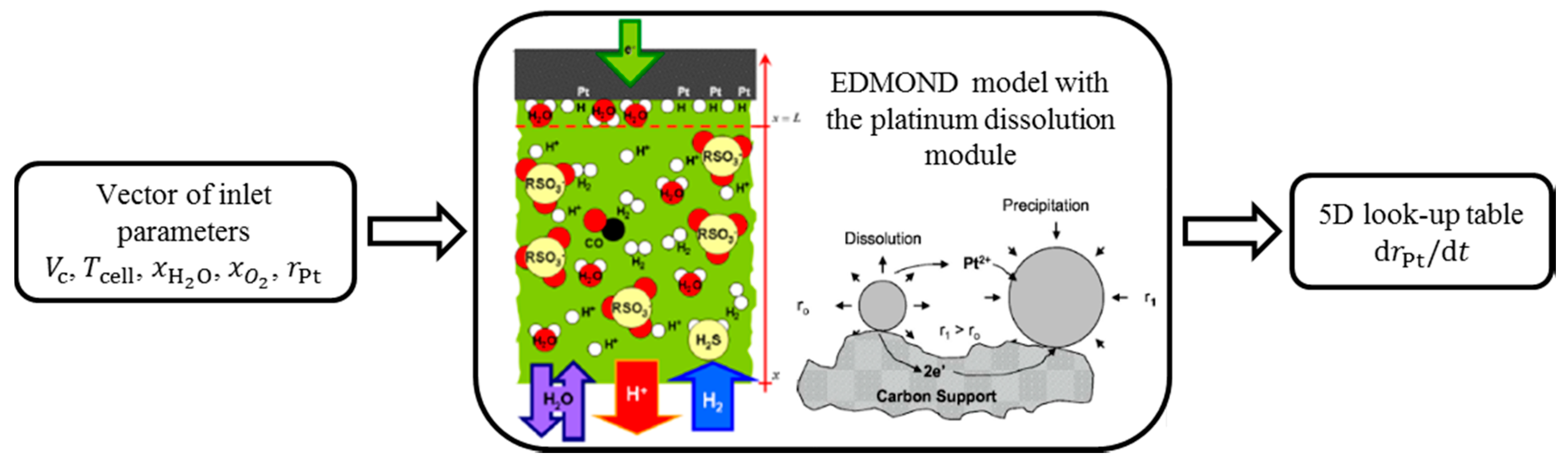
2.4. Degradation Library
2.5. Time Upscaling
3. Pt Dissolution Model
3.1. Pt Dissolution Mechanism
- The free energy of Pt extraction from Pt crystal lattice, : This free energy is calculated from density functional theory (DFT) which takes into account of the coverage of the intermediate species that depend upon the amount of hydration.
- The free energy of Pt oxidation, : This is calculated from the local potential based on transition state theory (TST) as , where is the symmetry factor of the Pt dissolution reaction and is the local potential at the catalyst surface (calculated by the EDMOND model [26]).
- The free energy of desorption, : This is calculated as, from , the transfer coefficient and the Gibbs Thomson energy [35] which depends of the particle radius and is given by:with as the surface energy of Pt[111], the molar mass of , the mass density of , and the particle radius. The dependence of the on the reciprocal of platinum radius shows that small particles are dissolved faster than the larger particles.
3.2. Integration of Pt Dissolution into Degradation Library
3.3. Fuel Cell Durability Estimation under Pt Dissolution
4. Results and Discussion
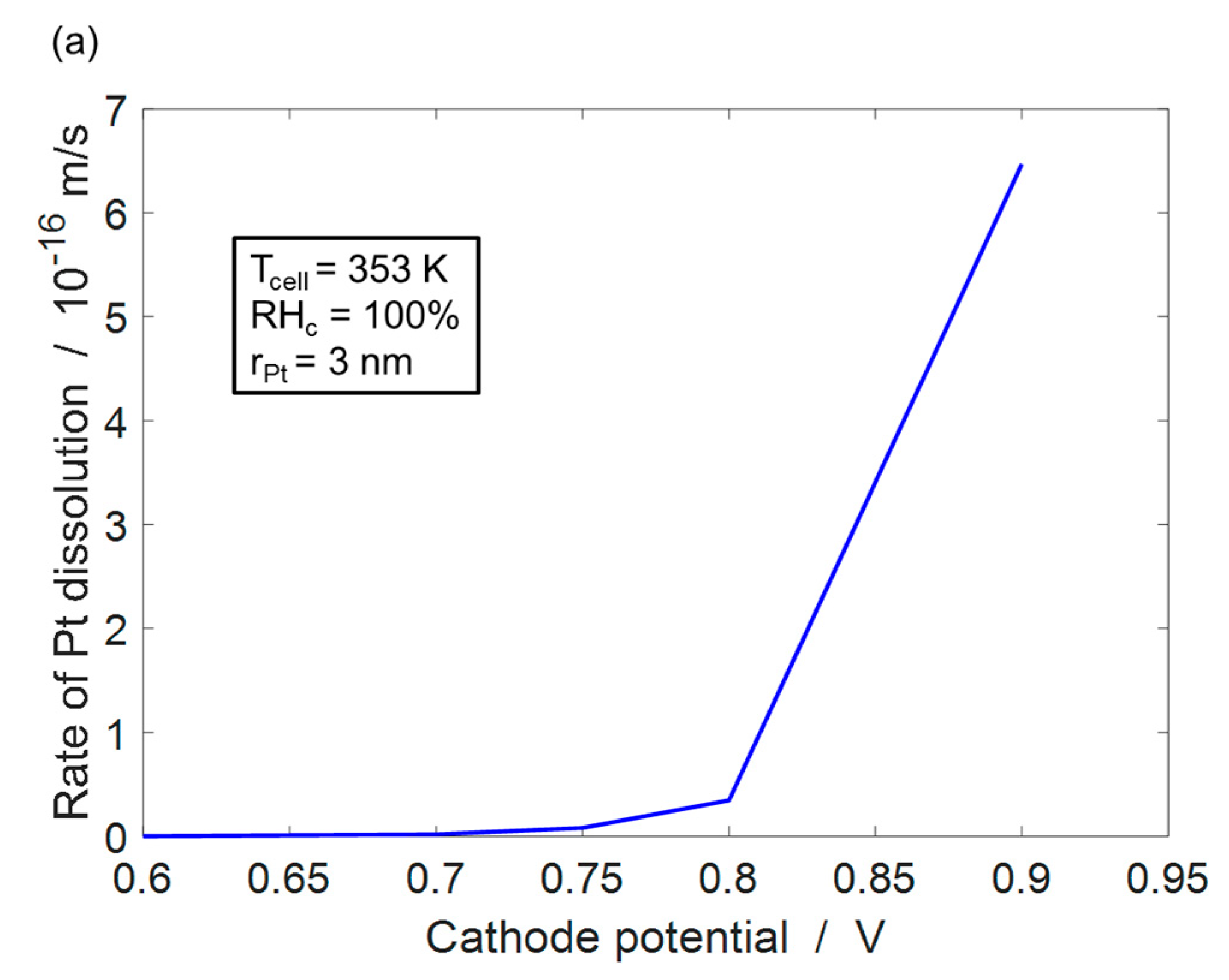
4.1. Pt Dissolution Model
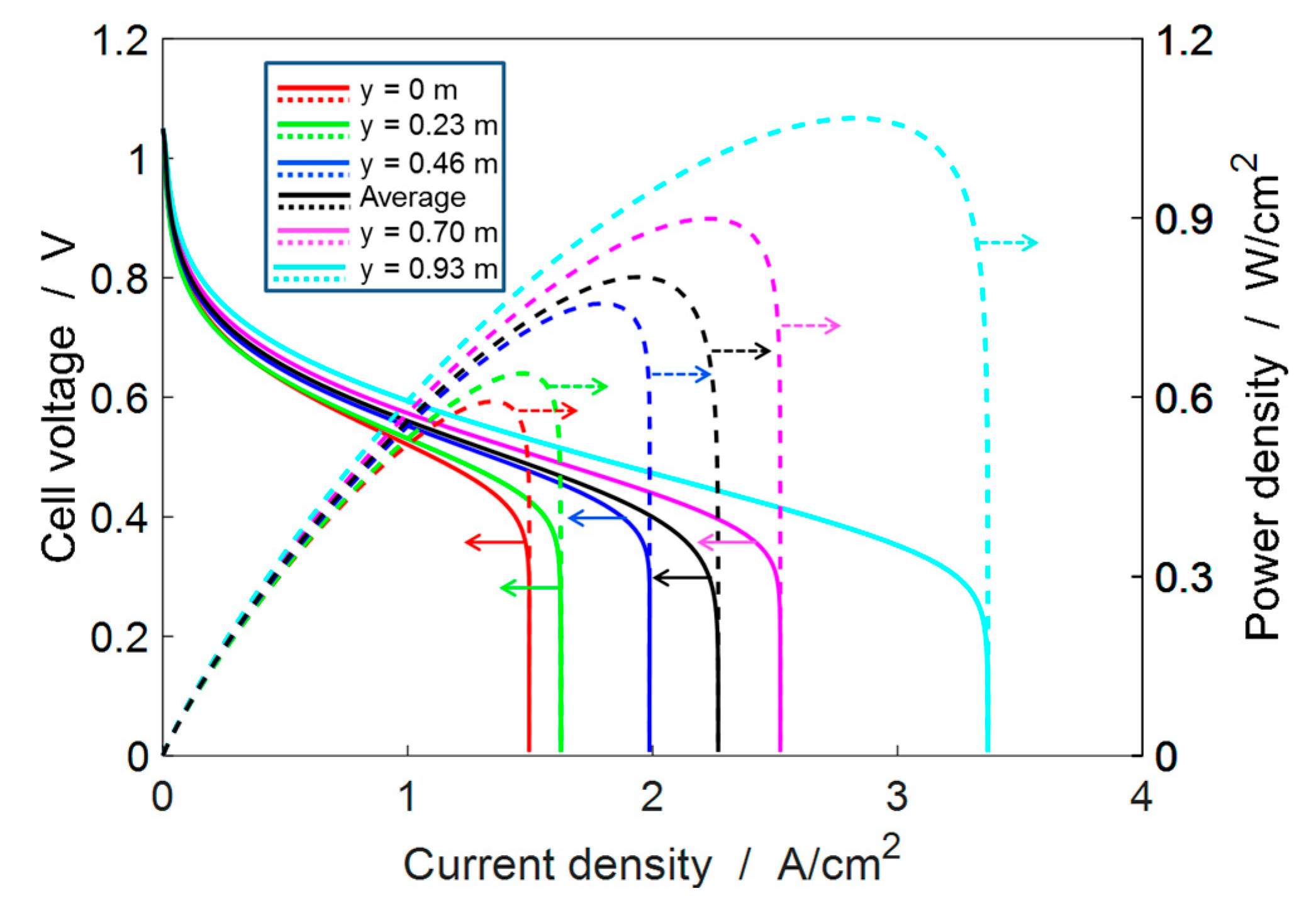
4.2. Single-Cell Performance
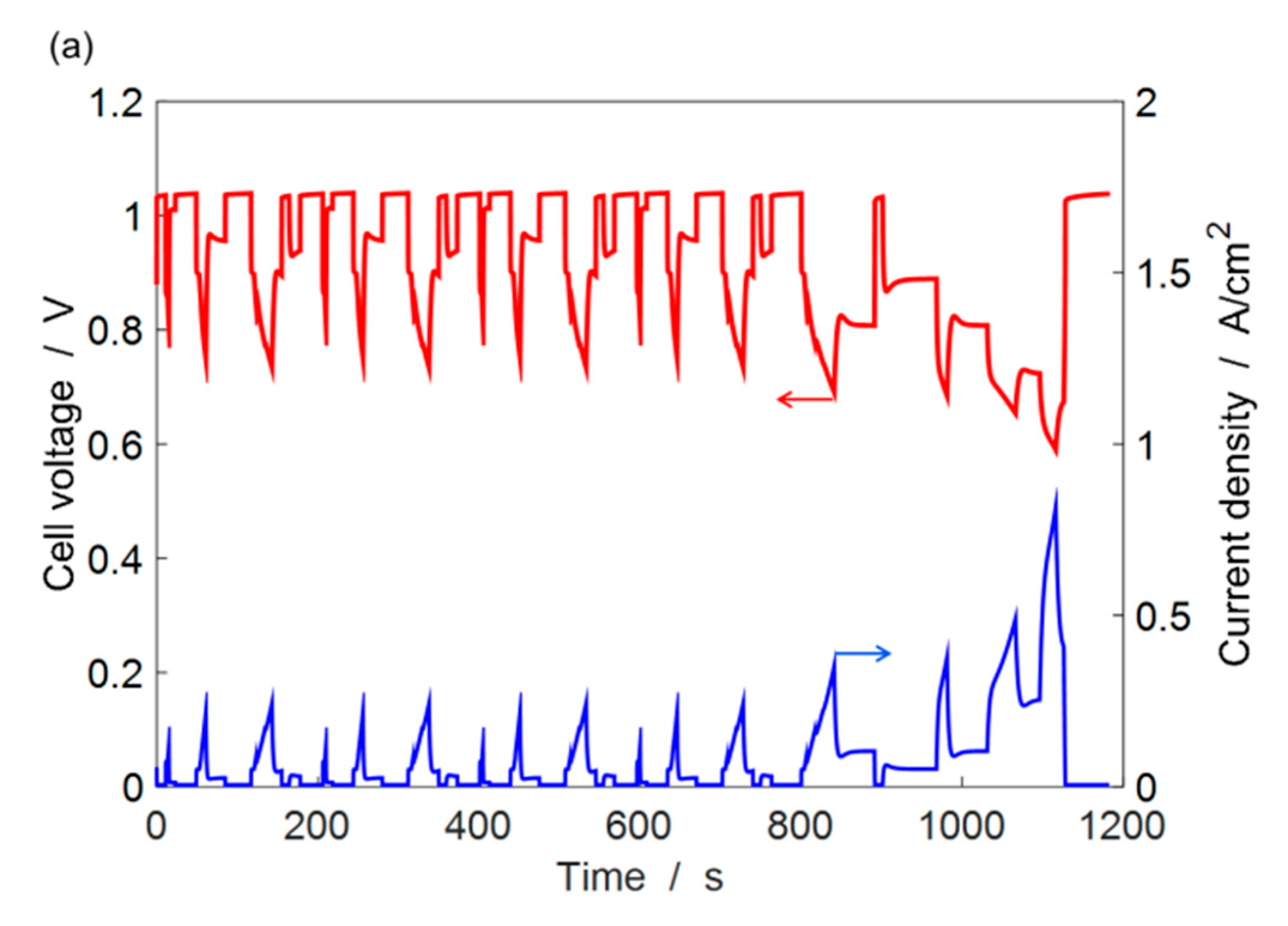
4.3. ECSA Loss under Automotive Drive Cycle
4.4. Durability Estimation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, X.; Ye, S. Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC. J. Power Sources 2007, 172, 145–154. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Shao-Horn, Y.; Sheng, W.C.; Chen, S.; Ferreira, P.J.; Holby, E.F.; Morgan, D. Instability of Supported Platinum Nanoparticles in Low-Temperature Fuel Cells. Top. Catal. 2007, 46, 285–305. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Arisetty, S.; Wang, X.; Subbaraman, R.; Ball, S.C.; DeCrane, S.; Myers, D.J. Thermodynamics and Kinetics of Platinum Dissolution from Carbon-Supported Electrocatalysts in Aqueous Media under Potentiostatic and Potentiodynamic Conditions. J. Electrochem. Soc. 2013, 160, F447–F455. [Google Scholar] [CrossRef]
- Virkar, A.V.; Zhou, Y. Mechanism of Catalyst Degradation in Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2007, 154, B540. [Google Scholar] [CrossRef]
- Shao, Y.; Kou, R.; Wang, J.; Viswanathan, V.V.; Kwak, J.H.; Liu, J.; Wang, Y.; Lin, Y. The influence of the electrochemical stressing (potential step and potential-static holding) on the degradation of polymer electrolyte membrane fuel cell electrocatalysts. J. Power Sources 2008, 185, 280–286. [Google Scholar] [CrossRef]
- Luo, Z.; Li, D.X.; Tang, H.; Pan, M.; Ruan, R. Degradation behavior of membrane-electrode-assembly materials in 10-cell PEMFC stack. Int. J. Hydrog. Energy 2006, 31, 1831–1837. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, X.Z.; Hin, J.N.; Wang, H.; Friedrich, K.A.; Schulze, M. A review of platinum-based catalyst layer degradation in proton exchange membrane fuel cells. J. Power Sources 2009, 194, 588–600. [Google Scholar] [CrossRef]
- Bi, W.; Sun, Q.; Deng, Y.; Fuller, T.F. The effect of humidity and oxygen partial pressure on degradation of Pt/C catalyst in PEM fuel cell. Electrochim. Acta 2009, 54, 1826–1833. [Google Scholar] [CrossRef]
- Bussian, D.A.; O’Dea, J.R.; Metiu, H.; Buratto, S.K. Nanoscale current imaging of the conducting channels in proton exchange membrane fuel cells. Nano Lett. 2007, 7, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Cleghorn, S.J.C.; Derouin, C.R.; Wilson, M.S.; Gottesfeld, S. A printed circuit board approach to measuring current distribution in a fuel cell. J. Appl. Electrochem. 1998, 28, 663–672. [Google Scholar] [CrossRef]
- Lilavivat, V.; Shimpalee, S.; van Zee, J.W.; Xu, H.; Mittelsteadt, C.K. Current Distribution Mapping for PEMFCs. Electrochim. Acta 2015, 174, 1253–1260. [Google Scholar] [CrossRef]
- Durst, J.; Lamibrac, A.; Charlot, F.; Dillet, J.; Castanheira, L.F.; Maranzana, G.; Dubau, L.; Maillard, F.; Chatenet, M.; Lottin, O. Degradation heterogeneities induced by repetitive start/stop events in proton exchange membrane fuel cell: Inlet vs. outlet and channel vs. land. Appl. Catal. B Environ. 2013, 138–139, 416–426. [Google Scholar] [CrossRef]
- Rajalakshmi, N.; Raja, M.; Dhathathreyan, K.S. Evaluation of current distribution in a proton exchange membrane fuel cell by segmented cell approach. J. Power Sources 2002, 112, 331–336. [Google Scholar] [CrossRef]
- Schulze, M.; Gülzow, E.; Schönbauer, S.; Knöri, T.; Reissner, R. Segmented Cells as Tool for Development of Fuel Cells and Error Prevention/Prediagnostic in Fuel Cell Stacks. J. Power Sources 2007, 173, 19–27. [Google Scholar] [CrossRef]
- Pérez, L.C.; Brandão, L.; Sousa, J.M.; Mendes, A. Segmented polymer electrolyte membrane fuel cells—A review. Renew. Sustain. Energy Rev. 2011, 15, 169–185. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Wu, J.; Zhang, J. PEM Fuel Cell Testing and Diagnosis; Elsevier Science: Oxford, UK, 2013. [Google Scholar]
- Wang, H.H.; Yuan, X.-Z.; Li, H. PEM Fuel Cell Diagnostic Tools; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Uchimura, M.; Kocha, S.S. The Impact of Cycle Profile on PEMFC Durability. ECS Trans. 2007, 11, 1215–1226. [Google Scholar]
- Borup, R.; Davey, J.; Garzon, F.; Wood, D.; Welch, P.; More, K. PEM Fuel Cell Durability with Transportation Transient Operation. ECS Trans. 2006, 3, 879–886. [Google Scholar]
- Schmittinger, W.; Vahidi, A. A review of the main parameters influencing long-term performance and durability of PEM fuel cells. J. Power Sources 2008, 180, 1–14. [Google Scholar] [CrossRef]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shi, Z.; Glass, N.; Zhang, L.; Zhang, J.; Song, D.; Liu, Z.-S.; Wang, H.; Shen, J. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar] [CrossRef]
- Jahnke, T.; Futter, G.; Latz, A.; Malkow, T.; Papakonstantinou, G.; Tsotridis, G.; Schott, P.; Gérard, M.; Quinaud, M.; Quiroga, M.; et al. Performance and degradation of Proton Exchange Membrane Fuel Cells: State of the art in modeling from atomistic to system scale. J. Power Sources 2016, 304, 207–233. [Google Scholar] [CrossRef]
- Mayur, M.; Strahl, S.; Husar, A.; Bessler, W.G. A Multi-Timescale Modeling Methodology for PEMFC Performance and Durability in a Virtual Fuel Cell Car. Int. J. Hydrog. Energy 2015, 40, 16466–16476. [Google Scholar] [CrossRef]
- Robin, C.; Gérard, M.; Quinaud, M.; d’Arbigny, J.; Bultel, Y. Proton exchange membrane fuel cell model for aging predictions. Simulated equivalent active surface area loss and comparisons with durability tests. J. Power Sources 2016, 326, 417–427. [Google Scholar] [CrossRef]
- Gerard, M.; Robin, C.; Chandesris, M.; Schott, P. (Invited) Polymer Electrolyte Fuel Cells Lifetime Prediction by a Full Multi-Scale Modeling Approach. ECS Trans. 2016, 75, 35–43. [Google Scholar] [CrossRef]
- Bao, C.; Bessler, W.G. Two-Dimensional Modeling of a Polymer Electrolyte Membrane Fuel Cell with Long Flow Channel. Part I. Model development. J. Power Sources 2015, 275, 922–934. [Google Scholar] [CrossRef]
- United Nations UNECE. Agreement Concerning the Adoption of Uniform Technical Prescriptions for Wheeled Vehicles, Equipment and Parts Which Can Be Fitted and/or Be Used on Wheeled Vehicles and the Conditions for Reciprocal Recognition of Approvals Granted on the Basis of These Prescriptions. Available online: http://digitallibrary.un.org/record/492895 (accessed on 17 September 2015).
- Tutuianu, M.; Marotta, A.; Steven, H.; Ericsson, E.; Haniu, T.; Ichikawa, N.; Ishii, H. Development of a World-Wide Worldwide Harmonized Light Duty Driving Test Cycle (WLTC). Available online: https://www.unece.org/fileadmin/DAM/trans/doc/2014/wp29grpe/GRPE-68-03e.pdf (accessed on 17 September 2015).
- Martin, A.; Joerissen, L. Auto-Stack—Implementing a European Automotive Fuel Cell Stack Cluster. ECS Trans. 2012, 42, 31–38. [Google Scholar]
- Owejan, J.P.; Owejan, J.E.; Gu, W. Impact of Platinum Loading and Catalyst Layer Structure on PEMFC Performance. J. Electrochem. Soc. 2013, 160, F824–F833. [Google Scholar] [CrossRef]
- Li, Y.; Moriyama, K.; Gu, W.; Arisetty, S.; Wang, C.Y. A One-Dimensional Pt Degradation Model for Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2015, 162, F834–F842. [Google Scholar] [CrossRef]
- Robin, C.; Gerard, M.; Franco, A.A.; Schott, P. Multi-scale coupling between two dynamical models for PEMFC aging prediction. Int. J. Hydrog. Energy 2013, 38, 4675–4688. [Google Scholar] [CrossRef]
- Holby, E.F.; Sheng, W.; Shao-Horn, Y.; Morgan, D. Pt nanoparticle stability in PEM fuel cells. Influence of particle size distribution and crossover hydrogen. Energy Environ. Sci. 2009, 2, 865. [Google Scholar] [CrossRef]
- Franco, A.A.; Schott, P.; Jallut, C.; Maschke, B. A Multi-Scale Dynamic Mechanistic Model for the Transient Analysis of PEFCs. Fuel Cells 2007, 7, 99–117. [Google Scholar] [CrossRef]
- Franco, A.A.; Passot, S.; Fugier, P.; Anglade, C.; Billy, E.; Guétaz, L.; Guillet, N.; de Vito, E.; Mailley, S. PtxCoy Catalysts Degradation in PEFC Environments: Mechanistic Insights. J. Electrochem. Soc. 2009, 156, B410. [Google Scholar] [CrossRef]
- Ferreira, P.J.; Shao-Horn, Y.; Morgan, D.; Makharia, R.; Kocha, S.; Gasteiger, H.A. Instability of Pt/C electrocatalysts in proton exchange membrane fuel cells—A mechanistic investigation. J. Electrochem. Soc. 2005, 152, A2256–A2271. [Google Scholar] [CrossRef]
- Redmond, E.L.; Setzler, B.P.; Juhas, P.; Billinge, S.J.L.; Fuller, T.F. In-Situ Monitoring of Particle Growth at PEMFC Cathode under Accelerated Cycling Conditions. Electrochem. Solid-State Lett. 2012, 15, B72. [Google Scholar] [CrossRef]
- Mitsushima, S.; Kawahara, S.; Ota, K.-I.; Kamiya, N. Consumption Rate of Pt under Potential Cycling. J. Electrochem. Soc. 2007, 154, B153. [Google Scholar] [CrossRef]
- Cherevko, S.; Zeradjanin, A.R.; Keeley, G.P.; Mayrhofer, K.J.J. A Comparative Study on Gold and Platinum Dissolution in Acidic and Alkaline Media. J. Electrochem. Soc. 2014, 161, H822–H830. [Google Scholar] [CrossRef]
- Jovanovič, P.; Pavlišič, A.; Šelih, V.S.; Šala, M.; Hodnik, N.; Bele, M.; Hočevar, S.; Gaberšček, M. New Insight into Platinum Dissolution from Nanoparticulate Platinum-Based Electrocatalysts Using Highly Sensitive In Situ Concentration Measurements. ChemCatChem 2014, 6, 449–453. [Google Scholar] [CrossRef]
- Lopes, J.A.P.; Moreira, C.L.; Madureira, A.G. Defining Control Strategies for MicroGrids Islanded Operation. IEEE Trans. Power Syst. 2006, 21, 916–924. [Google Scholar] [CrossRef]
- Parthasarathy, P.; Virkar, A.V. Electrochemical Ostwald ripening of Pt and Ag catalysts supported on carbon. J. Power Sources 2013, 234, 82–90. [Google Scholar] [CrossRef]
- Weber, A.Z.; Kusoglu, A. Unexplained transport resistances for low-loaded fuel-cell catalyst layers. J. Mater. Chem. A 2014, 2, 17207–17211. [Google Scholar] [CrossRef]
- Bose, A.; Babburi, P.; Kumar, R.; Myers, D.; Mawdsley, J.; Milhuff, J. Performance of individual cells in polymer electrolyte membrane fuel cell stack under-load cycling conditions. J. Power Sources 2013, 243, 964–972. [Google Scholar] [CrossRef]
- Yang, Z.; Ball, S.; Condit, D.; Gummalla, M. Systematic Study on the Impact of Pt Particle Size and Operating Conditions on PEMFC Cathode Catalyst Durability. J. Electrochem. Soc. 2011, 158, B1439. [Google Scholar] [CrossRef]
- Coms, F.D. The Chemistry of Fuel Cell Membrane Chemical Degradation. ECS Trans. 2008, 16, 235–255. [Google Scholar]
- Coulon, R.; Bessler, W.; Franco, A.A. Modeling Chemical Degradation of a Polymer Electrolyte Membrane and its Impact on Fuel Cell Performance. ECS Trans. 2010, 25, 259–273. [Google Scholar]
- Ghelichi, M.; Melchy, P.-É.A.; Eikerling, M.H. Radically coarse-grained approach to the modeling of chemical degradation in fuel cell ionomers. J. Phys. Chem. B 2014, 118, 11375–11386. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, J.; Tang, Y.; Song, C.; Shen, J.; Song, D.; Zhang, J. Hydrogen crossover in high-temperature PEM fuel cells. J. Power Sources 2007, 167, 25–31. [Google Scholar] [CrossRef]
- Park, J.; Oh, H.; Ha, T.; Lee, Y.I.; Min, K. A review of the gas diffusion layer in proton exchange membrane fuel cells: Durability and degradation. Appl. Energy 2015, 155, 866–880. [Google Scholar] [CrossRef]
- Lapicque, F.; Belhadj, M.; Bonnet, C.; Pauchet, J.; Thomas, Y. A critical review on gas diffusion micro and macroporous layers degradations for improved membrane fuel cell durability. J. Power Sources 2016, 336, 40–53. [Google Scholar] [CrossRef]
- Papadias, D.D.; Ahluwalia, R.K.; Thomson, J.K.; Meyer, H.M.; Brady, M.P.; Wang, H.; Turner, J.A.; Mukundan, R.; Borup, R. Degradation of SS316L bipolar plates in simulated fuel cell environment: Corrosion rate, barrier film formation kinetics and contact resistance. J. Power Sources 2015, 273, 1237–1249. [Google Scholar] [CrossRef]
- Eikerling, M. Water management in cathode catalyst layers of PEM fuel cells. J. Electrochem. Soc. 2006, 153, E58–E70. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Mass of car + H2 tank (m) | 1100 kg + 99.7 kg a |
| Coefficient of rolling resistance (Cr) | 0.0085 a |
| Drag coefficient (Cw) | 0.3 a |
| Shadow area (A) | 1.91 m2 a |
| Final drive ratio (ηgear) | 6.066 a |
| Powertrain efficiency (ηPT) | 0.8 a |
| Wheel radius (Rw) | 0.291 m a |
| Auxiliary power consumption | 400 W a |
| Fuel cell stack power | 75 kW c |
| Number of cells in the stack (Nstack) | 315 b |
| Active cell area | 0.025 m2 |
| Total stack weight | 153.8 kg b |
| Stack power density | 0.49 kW/kg c |
| Proportional gain (Kp) | 5 c |
| Integrator gain (KI) | 0.6 c |
| Characteristics | NEDC | WLTC |
|---|---|---|
| Duration | 1180 s | 1800 s |
| Idling duration | 280 s | 242 s |
| Theoretical distance | 11,023 m | 23,262 m |
| Maximum speed | 120 km/h | 131.3 km/h |
| Maximum stack power | 37.9 kW | 44.9 kW |
| Parameters | Value |
|---|---|
| Pt dissolution rate constant, | |
| Free energy for Pt extraction, | |
| Surface energy of Pt, | |
| Symmetry factor, | |
| Molar mass of Pt, | |
| Density of Pt, |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayur, M.; Gerard, M.; Schott, P.; Bessler, W.G. Lifetime Prediction of a Polymer Electrolyte Membrane Fuel Cell under Automotive Load Cycling Using a Physically-Based Catalyst Degradation Model. Energies 2018, 11, 2054. https://doi.org/10.3390/en11082054
Mayur M, Gerard M, Schott P, Bessler WG. Lifetime Prediction of a Polymer Electrolyte Membrane Fuel Cell under Automotive Load Cycling Using a Physically-Based Catalyst Degradation Model. Energies. 2018; 11(8):2054. https://doi.org/10.3390/en11082054
Chicago/Turabian StyleMayur, Manik, Mathias Gerard, Pascal Schott, and Wolfgang G. Bessler. 2018. "Lifetime Prediction of a Polymer Electrolyte Membrane Fuel Cell under Automotive Load Cycling Using a Physically-Based Catalyst Degradation Model" Energies 11, no. 8: 2054. https://doi.org/10.3390/en11082054
APA StyleMayur, M., Gerard, M., Schott, P., & Bessler, W. G. (2018). Lifetime Prediction of a Polymer Electrolyte Membrane Fuel Cell under Automotive Load Cycling Using a Physically-Based Catalyst Degradation Model. Energies, 11(8), 2054. https://doi.org/10.3390/en11082054