Molecular Modeling of CO2 and n-Octane in Solubility Process and α-Quartz Nanoslit

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Method
3. Results and Discussions
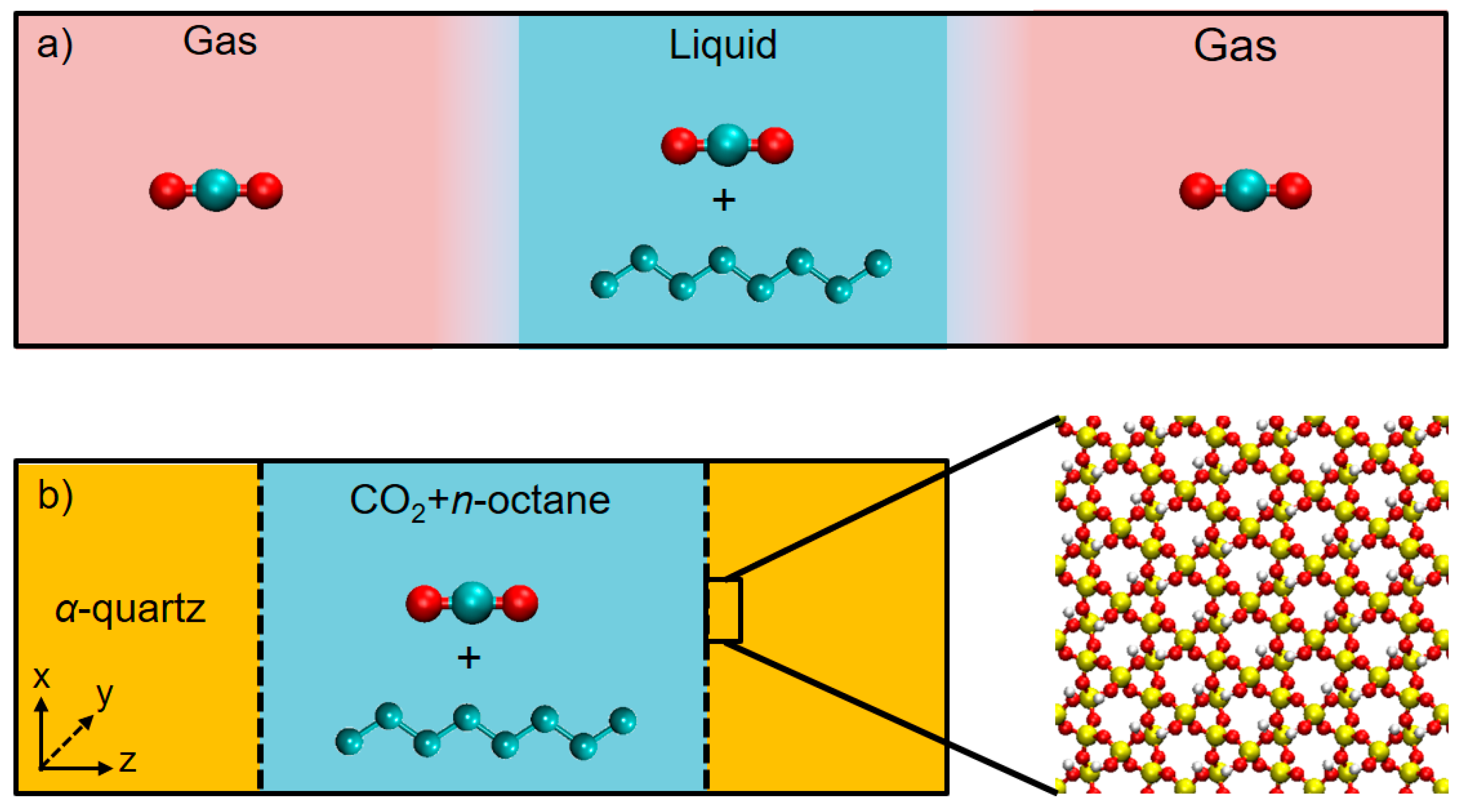
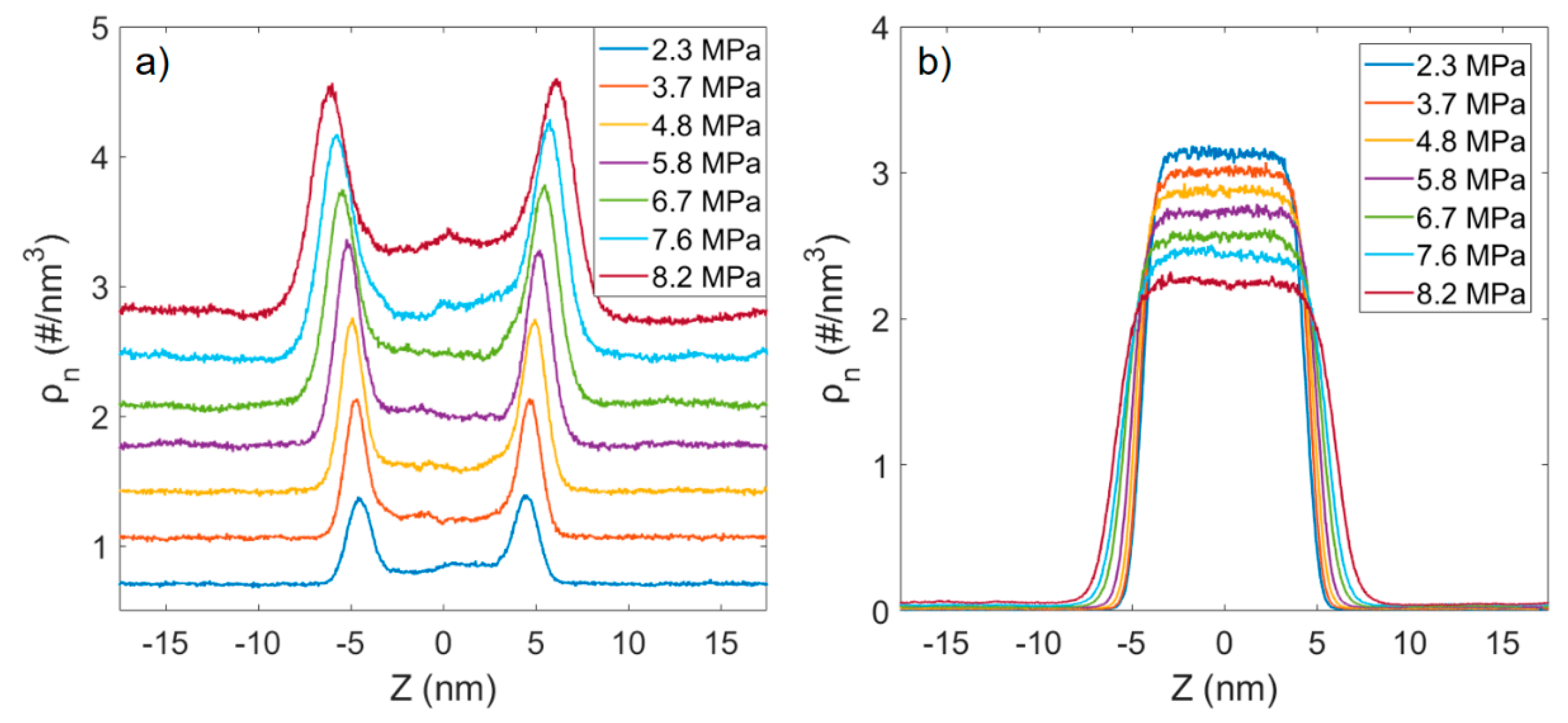
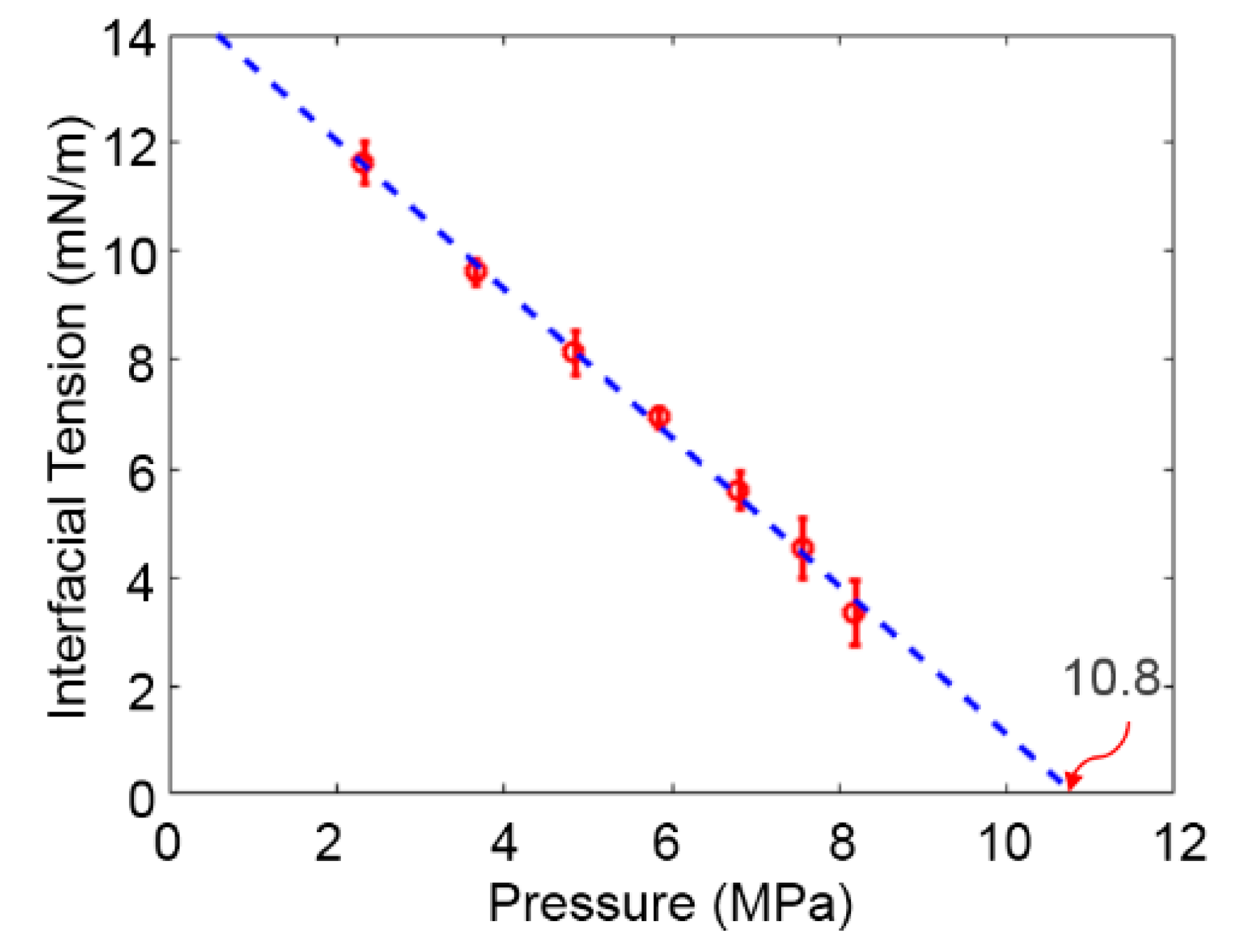
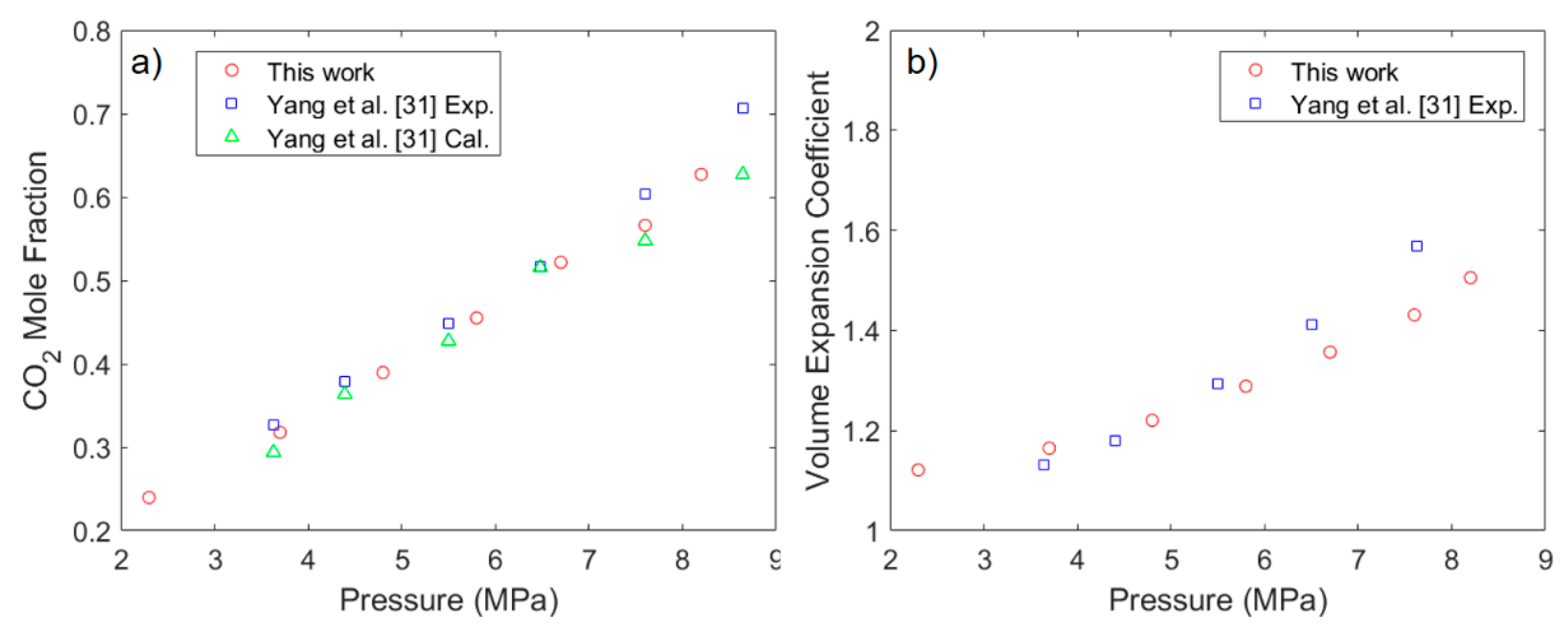
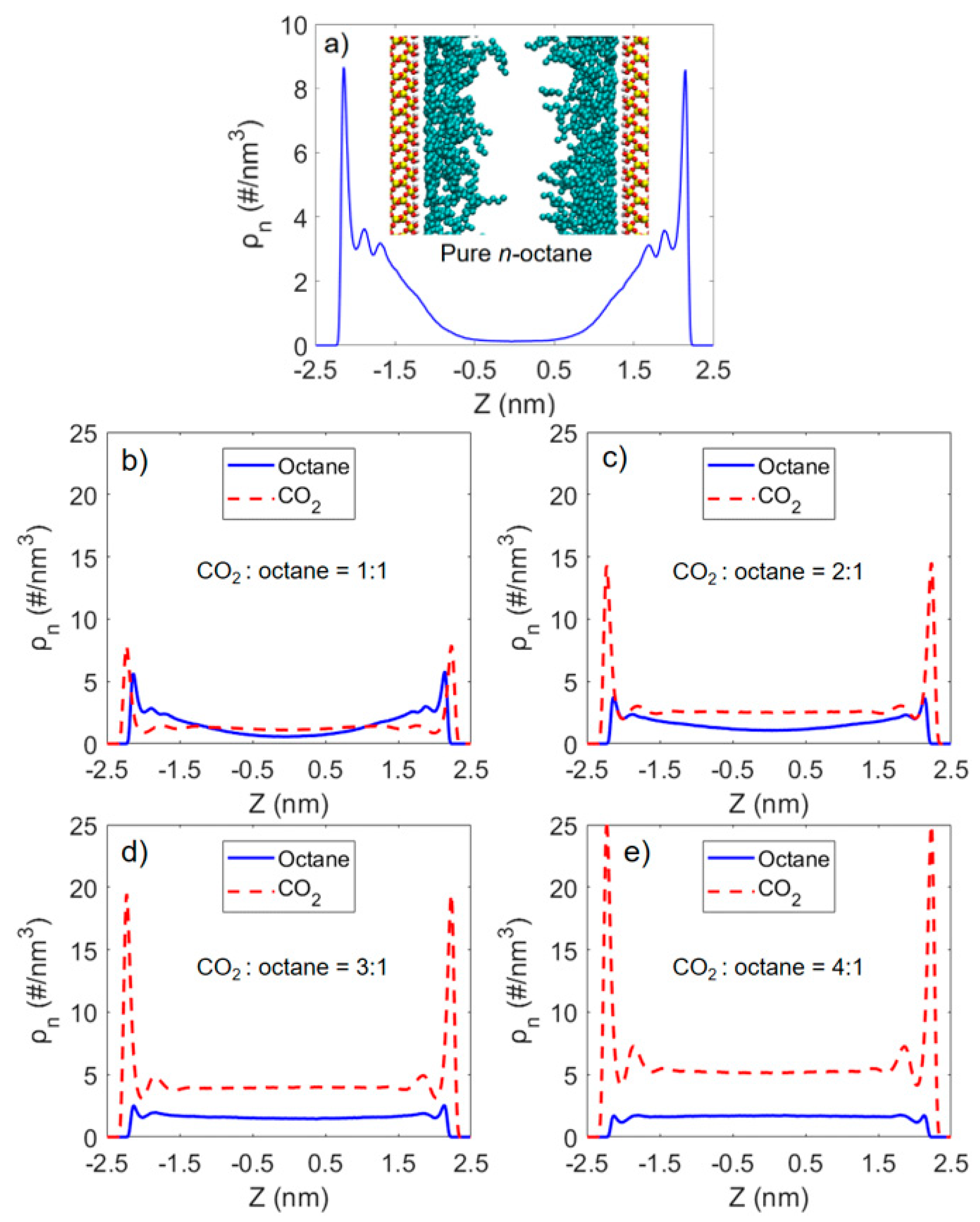
3.1. Solubility Process of CO2 and n-Octane System
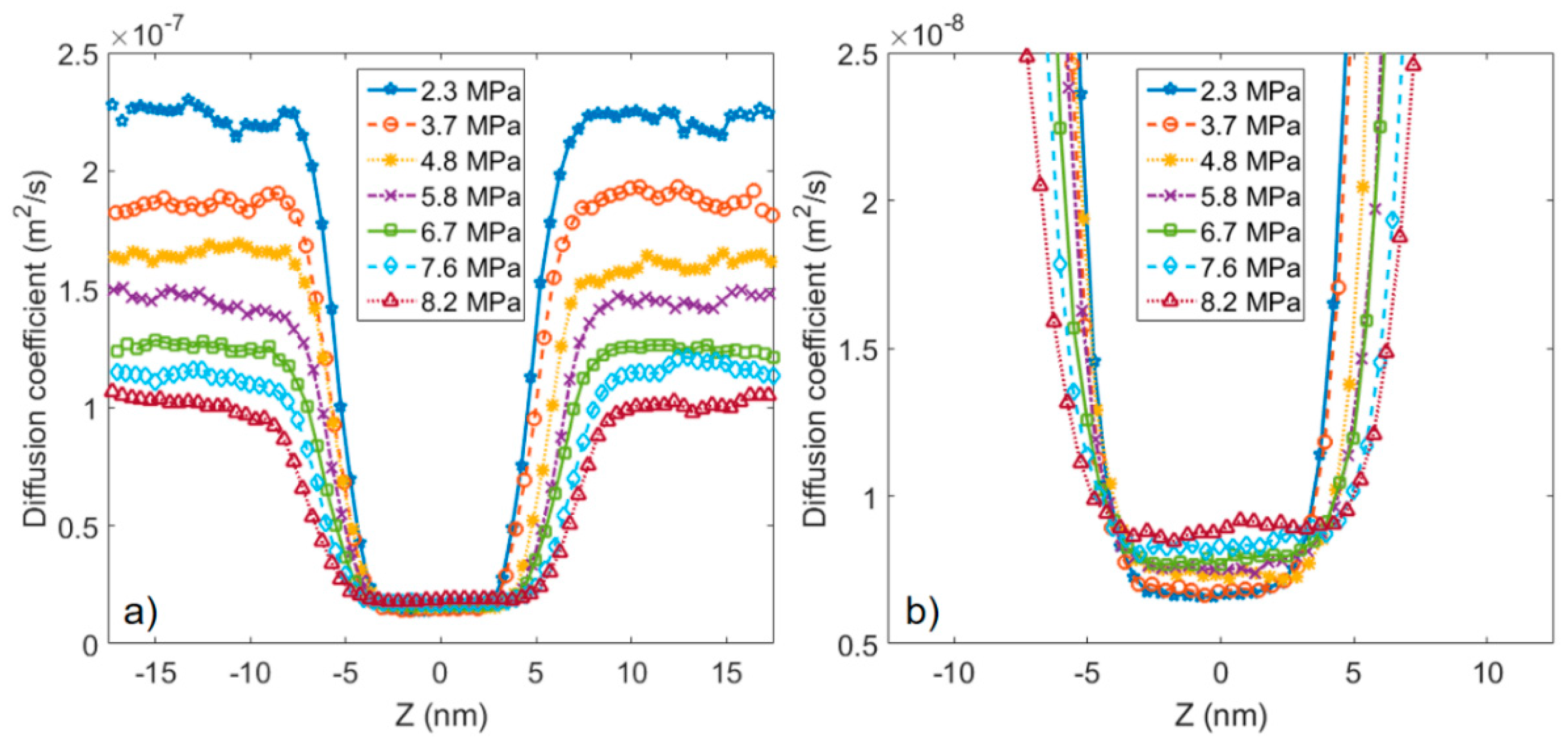
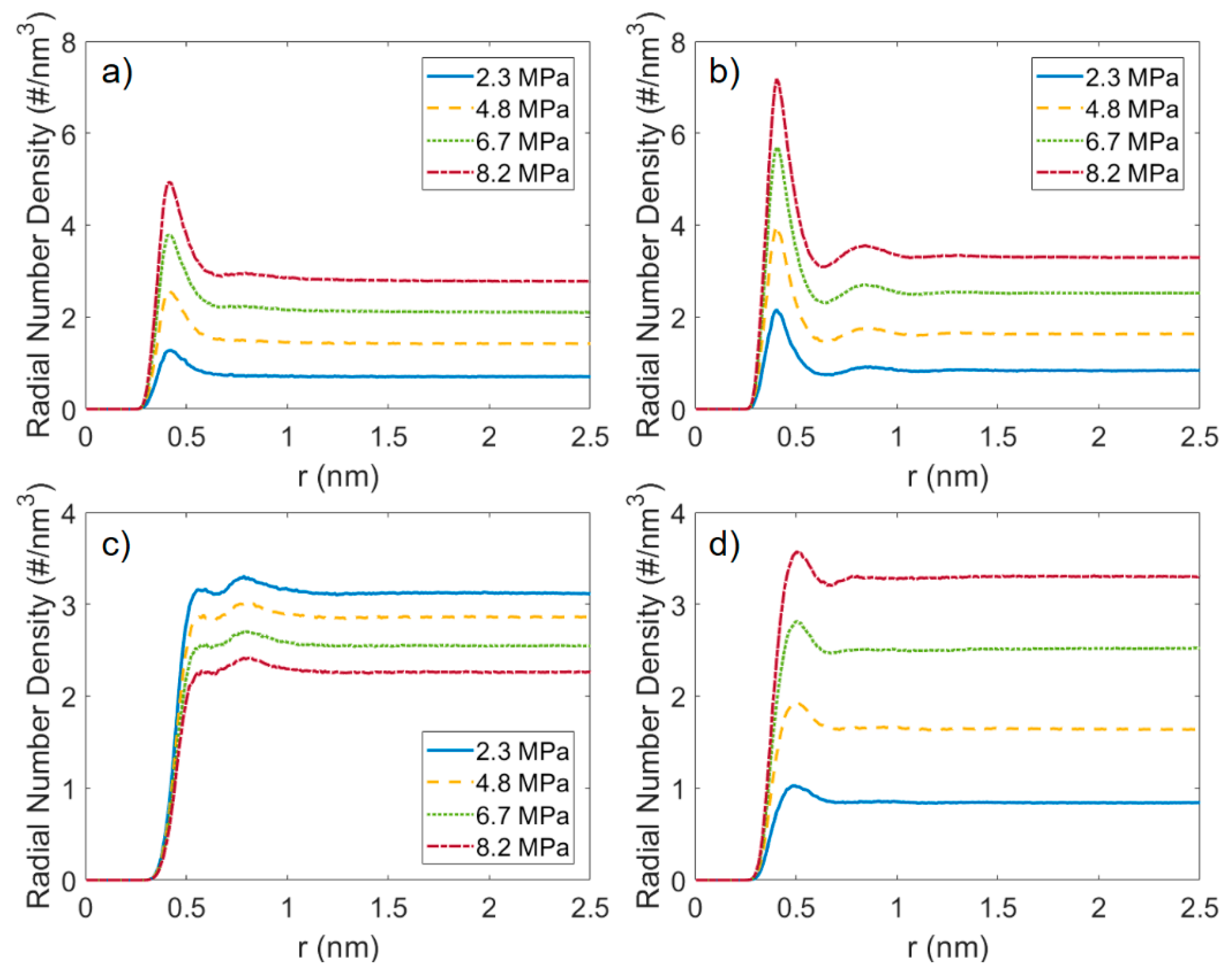
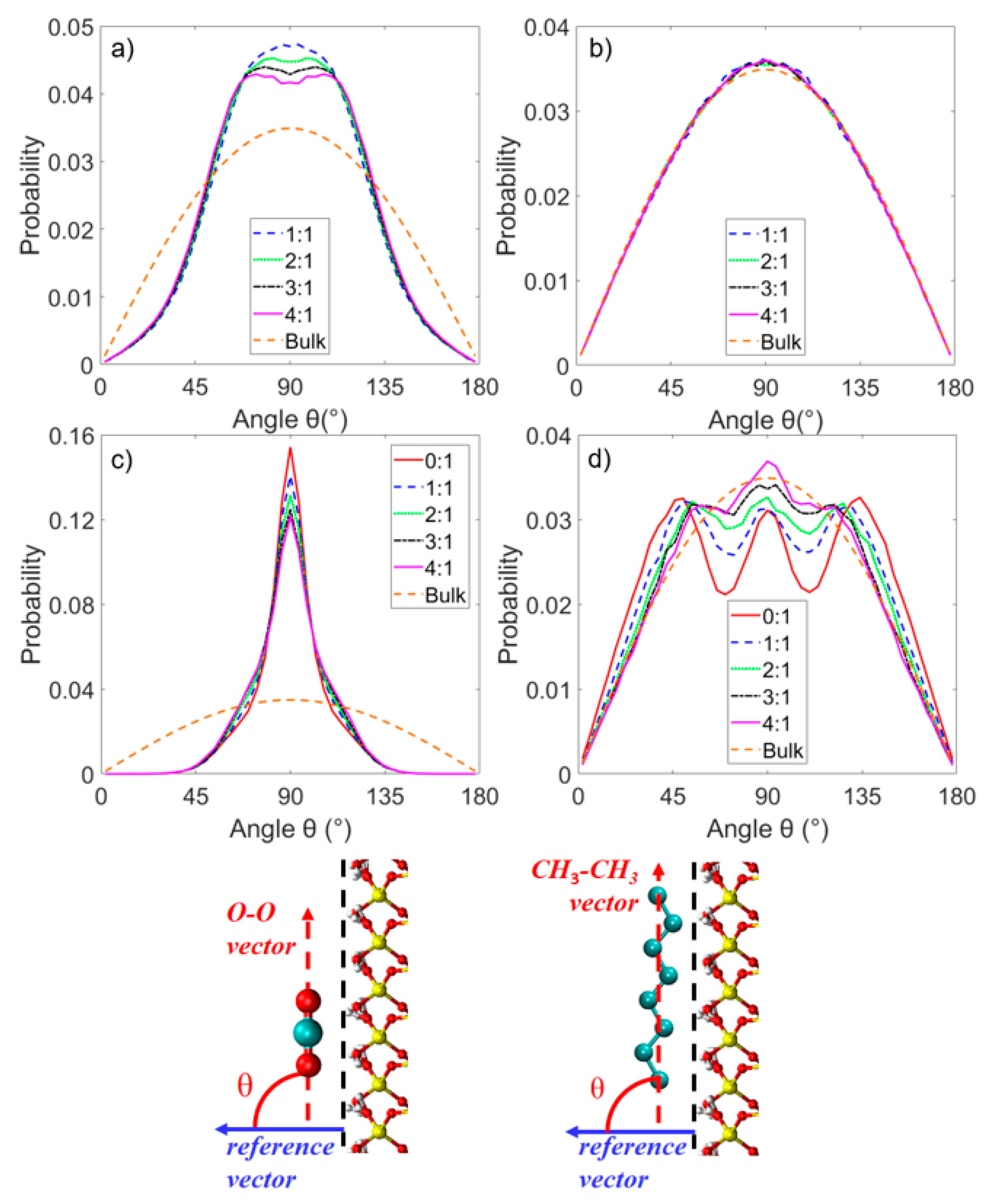
3.2. Microstructure of CO2 and n-Octane in α-Quartz Nanoslit
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babadagli, T. Development of mature oil fields—A review. J. Pet. Sci. Eng. 2007, 57, 221–246. [Google Scholar] [CrossRef]
- Alvarado, V.; Manrique, E. Enhanced oil recovery: An update review. Energies 2010, 3, 1529–1575. [Google Scholar] [CrossRef]
- Zou, C.; Yang, Z.; Tao, S.; Yuan, X.; Zhu, R.; Hou, L.; Wu, S.; Sun, L.; Zhang, G.; Bai, B. Continuous hydrocarbon accumulation over a large area as a distinguishing characteristic of unconventional petroleum: The Ordos Basin, North-Central China. Earth-Sci. Rev. 2013, 126, 358–369. [Google Scholar] [CrossRef]
- Orr, F.M., Jr.; Taber, J.J. Use of carbon dioxide in enhanced oil recovery. Science 1984, 224, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Markewitz, P.; Kuckshinrichs, W.; Leitner, W.; Linssen, J.; Zapp, P.; Bongartz, R.; Schreiber, A.; Müller, T.E. Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 2012, 5, 7281–7305. [Google Scholar] [CrossRef]
- Hirasaki, G.J.; Miller, C.A.; Puerto, M. Recent advances in surfactant EOR. SPE J. 2011, 16, 889–907. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, J.; Sun, F.; Zhang, F.; Feng, G.; Yang, J.; Zhang, X.; Xiang, W. A review of polymer EOR on offshore heavy oil field in Bohai Bay, China. In Proceedings of the SPE Enhanced Oil Recovery Conference, Kuala Lumpur, Malaysia, 19–21 July 2011. [Google Scholar] [CrossRef]
- Sen, R. Biotechnology in petroleum recovery: The microbial EOR. Prog. Energy Combust. Sci. 2008, 34, 714–724. [Google Scholar] [CrossRef]
- Van Bergen, F.; Gale, J.; Damen, K.; Wildenborg, A. Worldwide selection of early opportunities for CO2-enhanced oil recovery and CO2-enhanced coal bed methane production. Energy 2004, 29, 1611–1621. [Google Scholar] [CrossRef]
- Jimenez-Gallegos, R.; Galicia-Luna, L.A.; Elizalde-Solis, O. Experimental vapor—Liquid equilibria for the carbon dioxide+ octane and carbon dioxide+ decane systems. J. Chem. Eng. Data 2006, 51, 1624–1628. [Google Scholar] [CrossRef]
- Peng, D.-Y.; Robinson, D.B. A new two-constant equation of state. Ind. Eng. Chem. Fundam. 1976, 15, 59–64. [Google Scholar] [CrossRef]
- Nagarajan, N.; Robinson, R., Jr. Equilibrium phase compositions, phase densities, and interfacial tensions for carbon dioxide+ hydrocarbon systems. 2. Carbon dioxide+ n-decane. J. Chem. Eng. Data 1986, 31, 168–171. [Google Scholar] [CrossRef]
- Shaver, R.; Robinson, R., Jr.; Gasem, K. An automated apparatus for equilibrium phase compositions, densities, and interfacial tensions: Data for carbon dioxide+ decane. Fluid Phase Equilib. 2001, 179, 43–66. [Google Scholar] [CrossRef]
- Rommerskirchen, R.; Bilgili, H.; Fischer, J.; Sottmann, T. Impact of Miscibility Enhancing Additives on the Flooding Scheme in CO2 EOR Processes. In Proceedings of the SPE Improved Oil Recovery Conference, Tulsa, OH, USA, 14–18 April 2018. [Google Scholar] [CrossRef]
- Pathak, M.; Cho, H.; Deo, M. Experimental and Molecular Modeling Study of Bubble Points of Hydrocarbon Mixtures in Nanoporous Media. Energy Fuels 2017, 31, 3427–3435. [Google Scholar] [CrossRef]
- Alfarge, D.; Wei, M.; Bai, B. Data analysis for CO2-EOR in shale-oil reservoirs based on a laboratory database. J. Pet. Sci. Eng. 2018, 162, 697–711. [Google Scholar] [CrossRef]
- Majumder, M.; Chopra, N.; Andrews, R.; Hinds, B.J. Nanoscale hydrodynamics: Enhanced flow in carbon nanotubes. Nature 2005, 438, 44. [Google Scholar] [CrossRef] [PubMed]
- Whitby, M.; Cagnon, L.; Thanou, M.; Quirke, N. Enhanced fluid flow through nanoscale carbon pipes. Nano Lett. 2008, 8, 2632–2637. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Javadpour, F.; Feng, Q. Fast mass transport of oil and supercritical carbon dioxide through organic nanopores in shale. Fuel 2016, 181, 741–758. [Google Scholar] [CrossRef]
- Santos, M.S.; Franco, L.F.; Castier, M.; Economou, I.G. Molecular dynamics simulation of n-alkanes and CO2 confined by calcite nanopores. Energy Fuels 2018, 32, 1934–1941. [Google Scholar] [CrossRef]
- Wang, R.; Peng, F.; Song, K.; Feng, G.; Guo, Z. Molecular dynamics study of interfacial properties in CO2 enhanced oil recovery. Fluid Phase Equilib. 2018, 467, 25–32. [Google Scholar] [CrossRef]
- Le, T.; Striolo, A.; Cole, D.R. CO2–C4H10 mixtures simulated in silica slit pores: Relation between structure and dynamics. J. Phys. Chem. C 2015, 119, 15274–15284. [Google Scholar] [CrossRef]
- Yan, Y.; Dong, Z.; Zhang, Y.; Wang, P.; Fang, T.; Zhang, J. CO2 activating hydrocarbon transport across nanopore throat: Insights from molecular dynamics simulation. PCCP 2017, 19, 30439–30444. [Google Scholar] [CrossRef] [PubMed]
- Mejía, A.S.; Cartes, M.; Segura, H.; Müller, E.A. Use of equations of state and coarse grained simulations to complement experiments: Describing the interfacial properties of carbon dioxide+ decane and carbon dioxide+ eicosane mixtures. J. Chem. Eng. Data 2014, 59, 2928–2941. [Google Scholar] [CrossRef]
- Bresme, F.; Chacón, E.; Tarazona, P. Force-field dependence on the interfacial structure of oil–water interfaces. Mol. Phys. 2010, 108, 1887–1898. [Google Scholar] [CrossRef]
- Zhu, A.; Zhang, X.; Liu, Q.; Zhang, Q. A fully flexible potential model for carbon dioxide. Chin. J. Chem. Eng. 2009, 17, 268–272. [Google Scholar] [CrossRef]
- Nath, S.K.; Escobedo, F.A.; de Pablo, J.J. On the simulation of vapor–liquid equilibria for alkanes. J. Chem. Phys. 1998, 108, 9905–9911. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hockney, R.; Goel, S.; Eastwood, J. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Yang, Z.; Li, M.; Peng, B.; Lin, M.; Dong, Z. Dispersion property of CO2 in oil. 1. Volume expansion of CO2+ alkane at near critical and supercritical condition of CO2. J. Chem. Eng. Data 2012, 57, 882–889. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The nose–hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Dong, T.; Harris, N.B. 8 pore size distribution and morphology in the Horn River Shale, Middle and Upper Devonian, Northeastern British Columbia, Canada. In Electron Microscopy of Shale Hydrocarbon Reservoirs; AAPG: Houston, TX, USA, 2013. [Google Scholar]
- Cygan, R.T.; Liang, J.; Kalinichev, A.G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Yeh, I.; Berkowitz, M.L. Ewald summation for systems with slab geometry. J. Chem. Phys. 1999, 111, 3155–3162. [Google Scholar] [CrossRef]
- Jones, J.E. On the determination of molecular fields.—II. From the equation of state of a gas. Proc. R. Soc. Lond. A 1924, 106, 463–477. [Google Scholar] [CrossRef]
- Zhang, Y.; Feller, S.E.; Brooks, B.R.; Pastor, R.W. Computer simulation of liquid/liquid interfaces. I. Theory and application to octane/water. J. Chem. Phys. 1995, 103, 10252–10266. [Google Scholar] [CrossRef]
- Rao, D.N. A new technique of vanishing interfacial tension for miscibility determination. Fluid Phase Equilib. 1997, 139, 311–324. [Google Scholar] [CrossRef]
- Jianyi, L.; Guangdong, Z.; Yanli, L.; Fan, Z. Study on the Methods of Reducing the Miscible Pressure of CO2 Flooding. In Proceedings of the The Second China Energy Scientist Forum, Xuzhou, China, 18–19 October 2010. [Google Scholar]
- Yang, Z.; Li, M.; Peng, B.; Lin, M.; Dong, Z.; Ling, Y. Interfacial tension of CO2 and organic liquid under high pressure and temperature. Chin. J. Chem. Eng. 2014, 22, 1302–1306. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 2017. [Google Scholar]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, J.; Qin, X.; Gou, F.; Fang, W.; Peng, F.; Wang, R.; Guo, Z. Molecular Modeling of CO2 and n-Octane in Solubility Process and α-Quartz Nanoslit. Energies 2018, 11, 3045. https://doi.org/10.3390/en11113045
Pu J, Qin X, Gou F, Fang W, Peng F, Wang R, Guo Z. Molecular Modeling of CO2 and n-Octane in Solubility Process and α-Quartz Nanoslit. Energies. 2018; 11(11):3045. https://doi.org/10.3390/en11113045
Chicago/Turabian StylePu, Jun, Xuejie Qin, Feifei Gou, Wenchao Fang, Fengjie Peng, Runxi Wang, and Zhaoli Guo. 2018. "Molecular Modeling of CO2 and n-Octane in Solubility Process and α-Quartz Nanoslit" Energies 11, no. 11: 3045. https://doi.org/10.3390/en11113045
APA StylePu, J., Qin, X., Gou, F., Fang, W., Peng, F., Wang, R., & Guo, Z. (2018). Molecular Modeling of CO2 and n-Octane in Solubility Process and α-Quartz Nanoslit. Energies, 11(11), 3045. https://doi.org/10.3390/en11113045