
Split-GFP Reassembly Assay: Strengths and Caveats from a Multiparametric Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
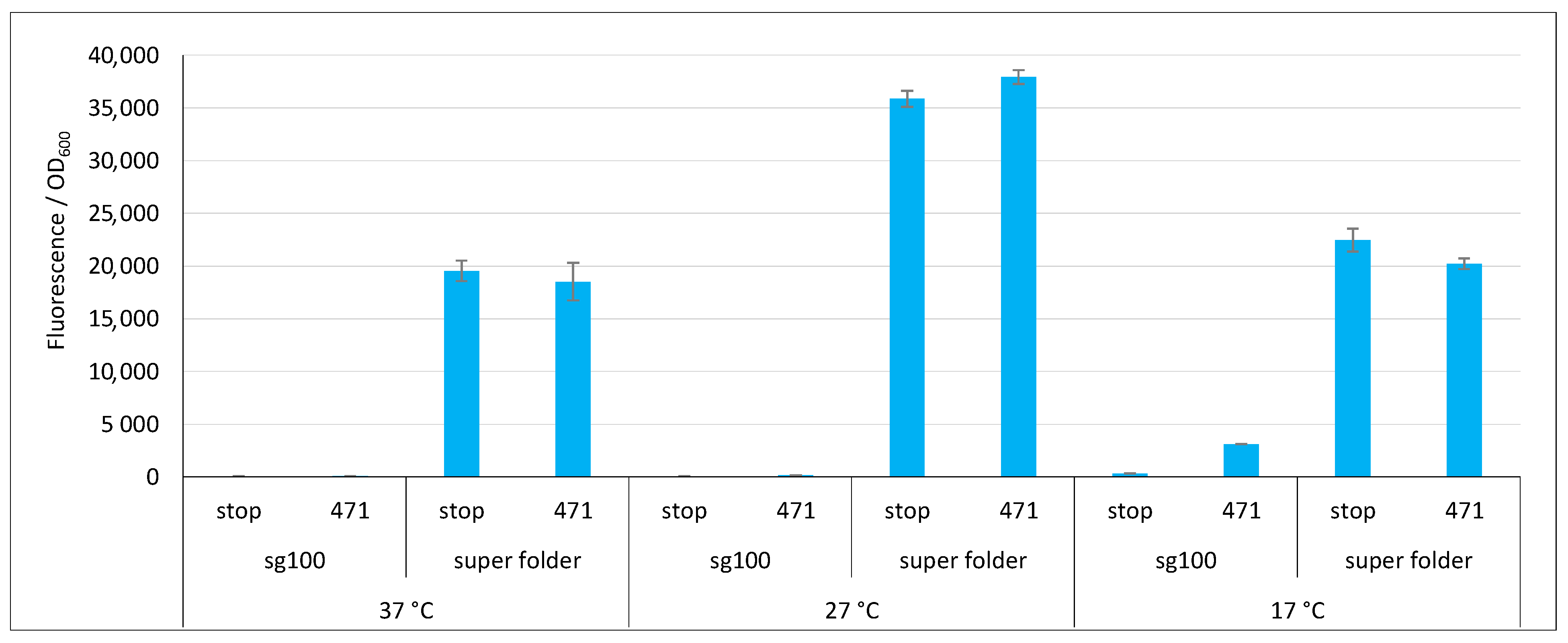{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

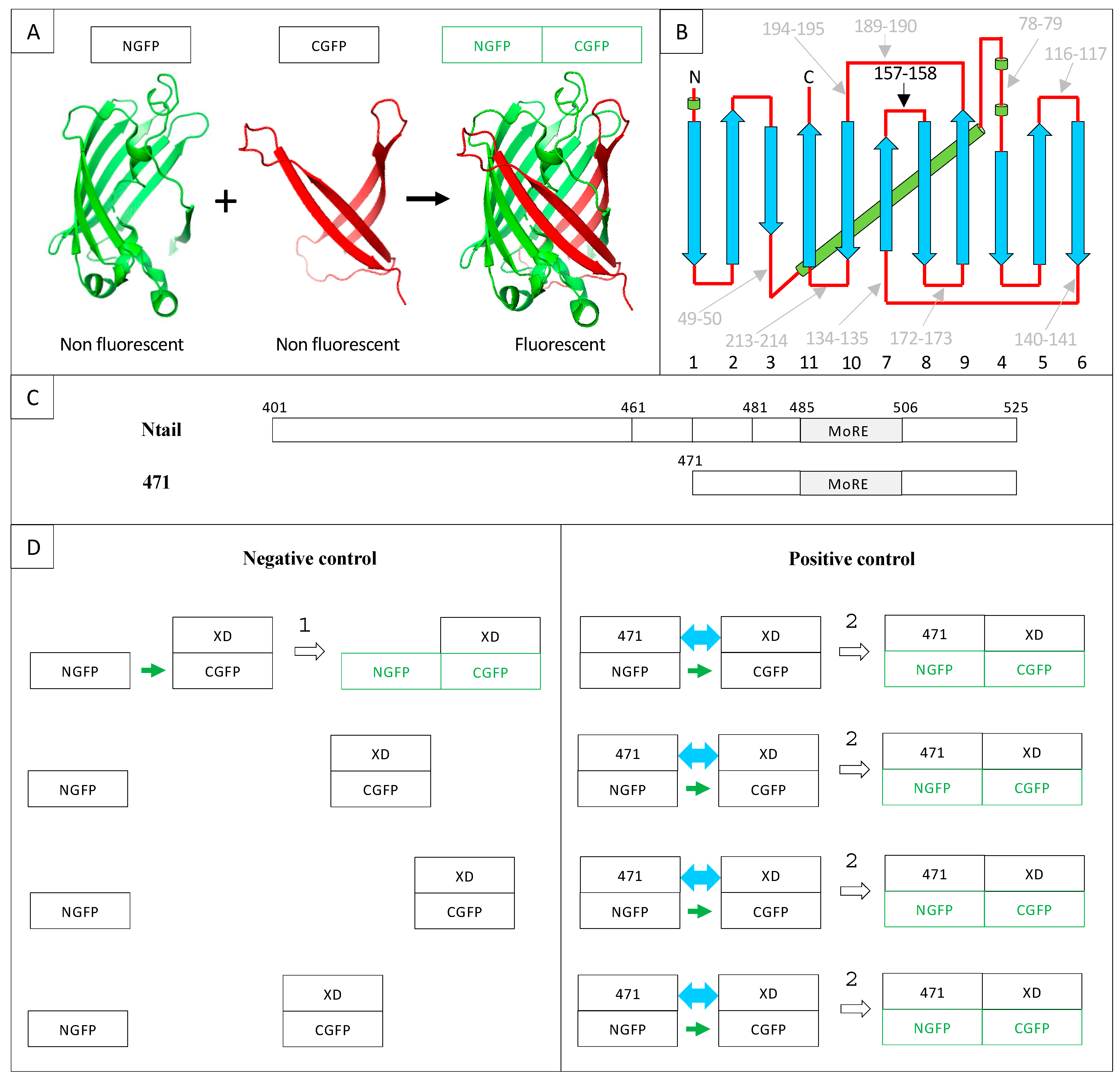
2. Results and Discussion
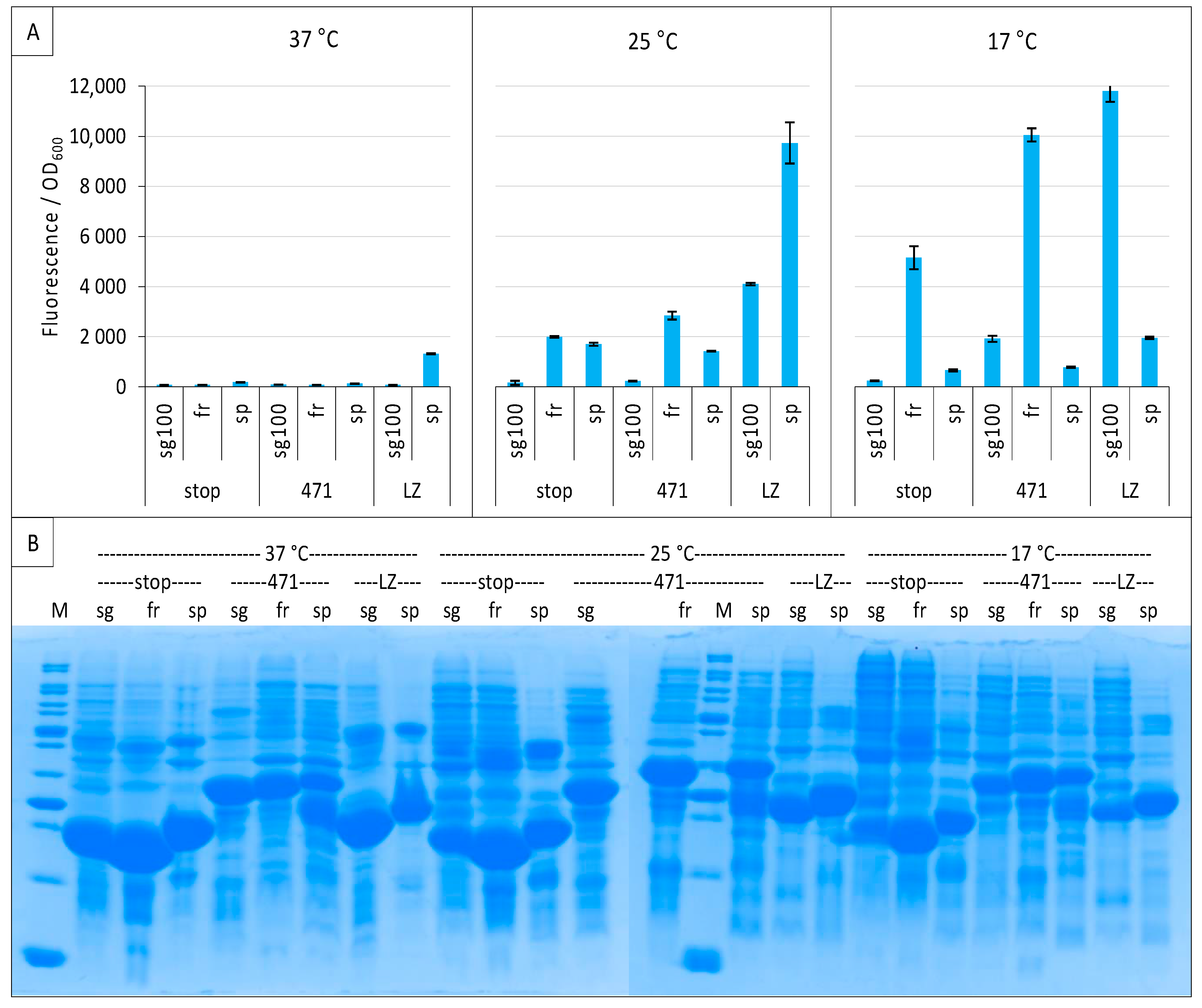
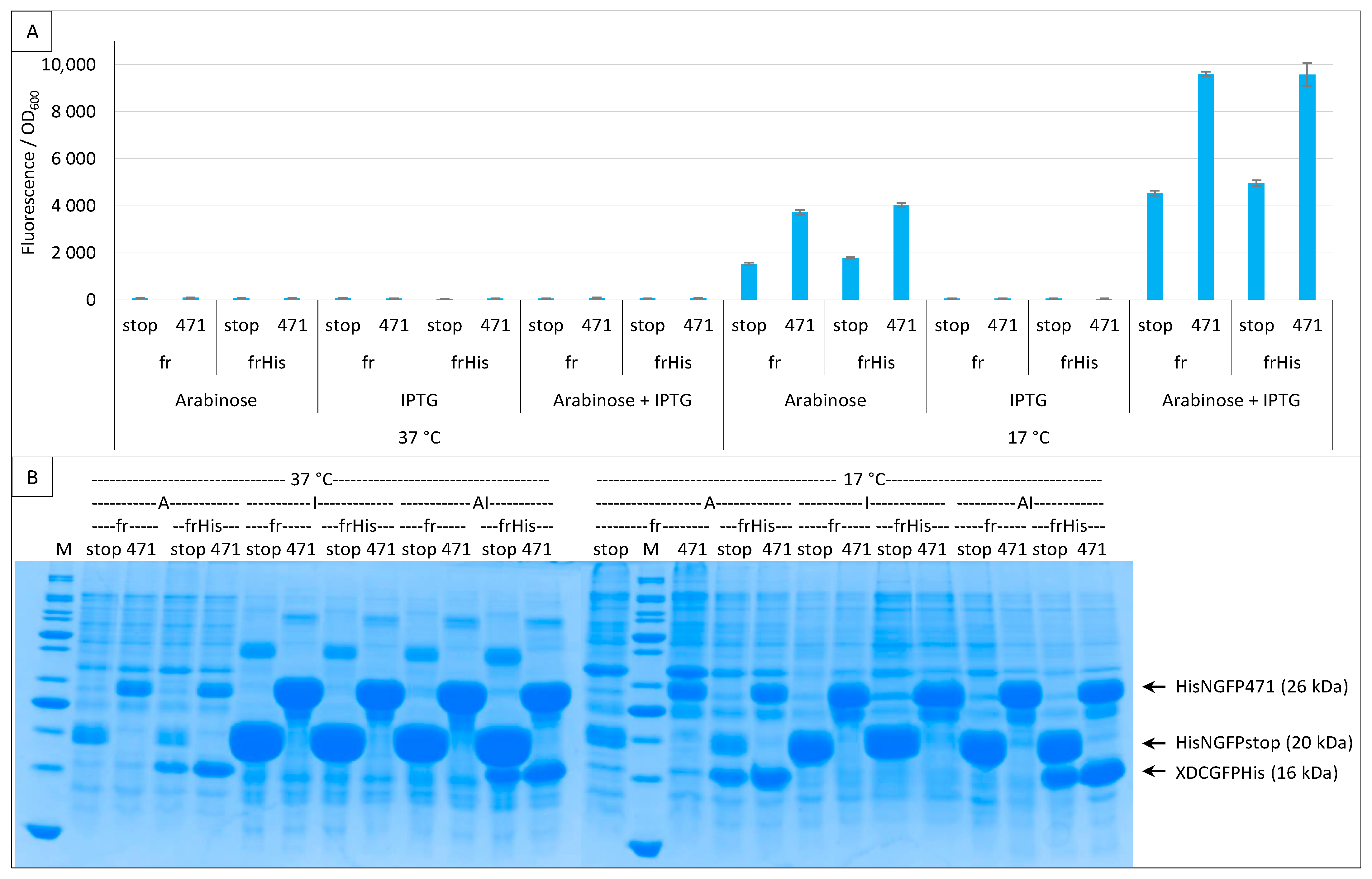
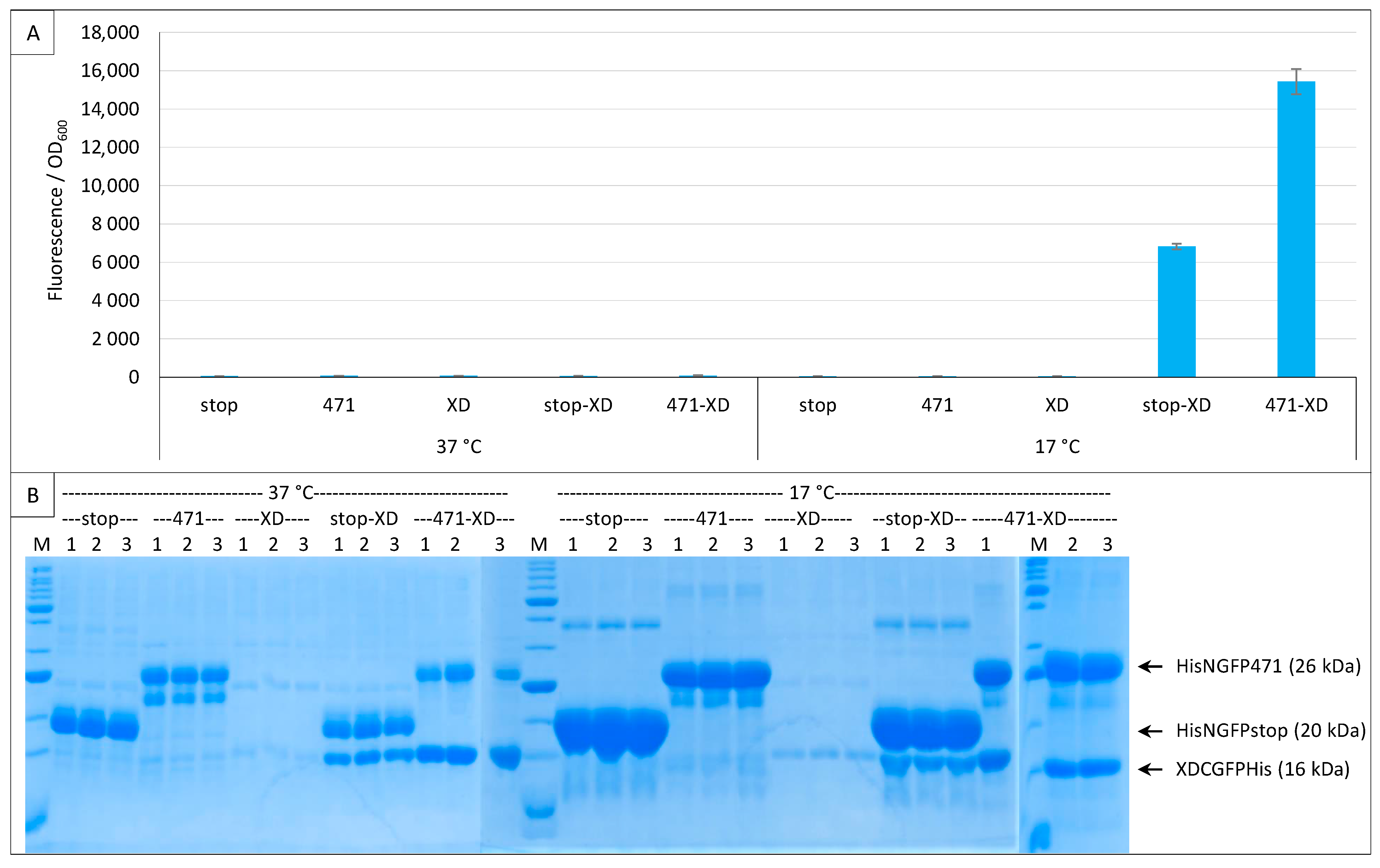
2.1. Experimental Conditions
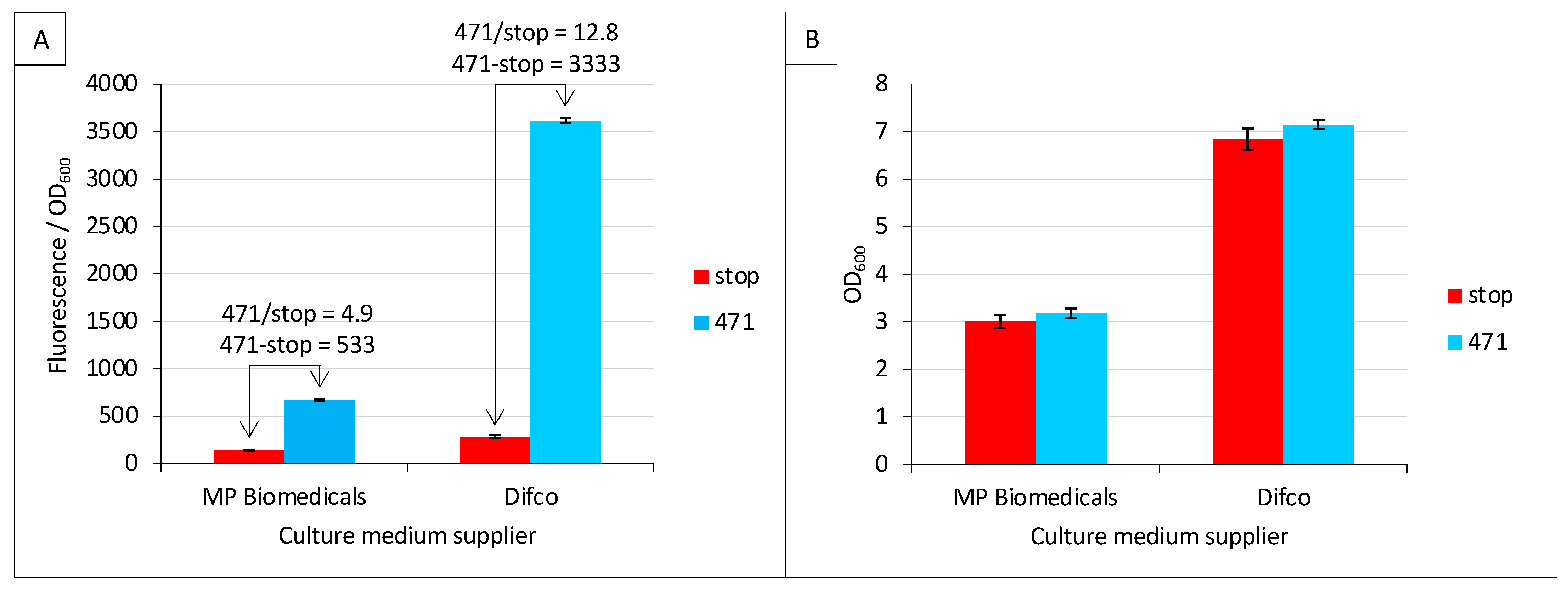
2.2. Effect of Culture Medium Supplier
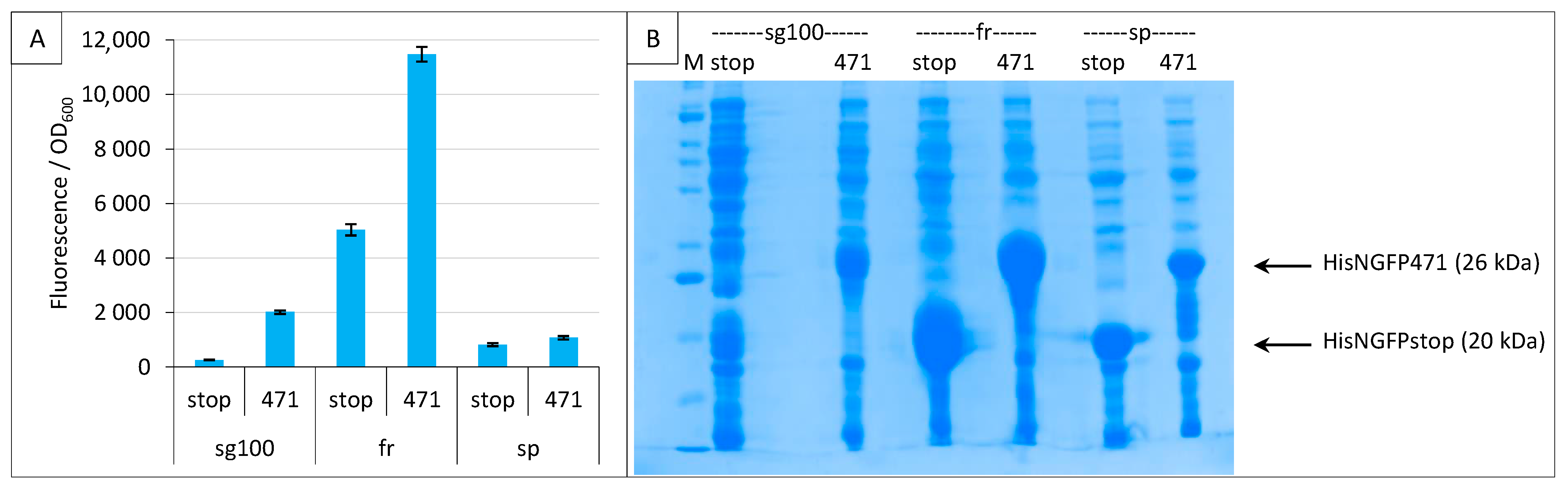
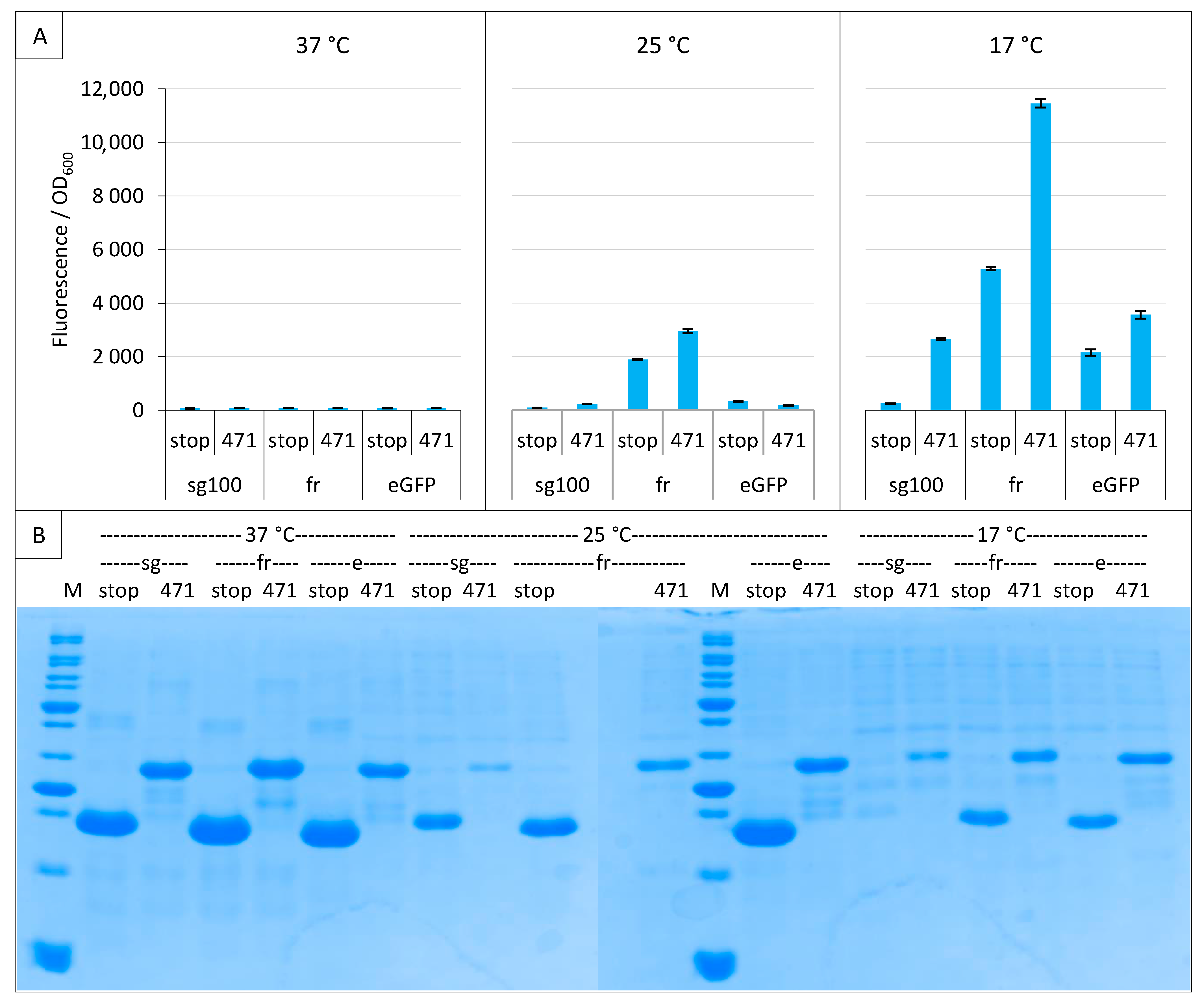
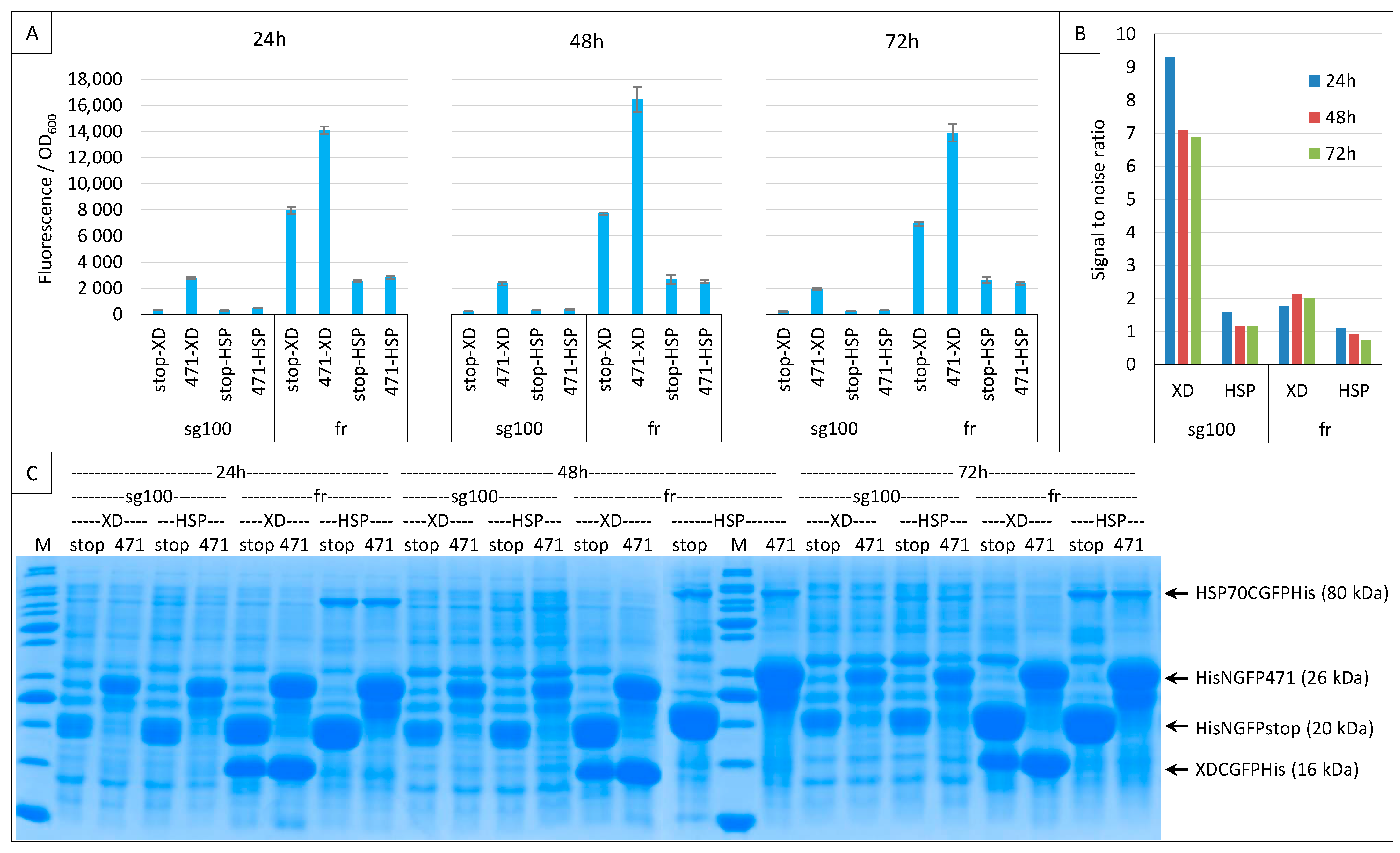
2.3. Impact of GFP Variants
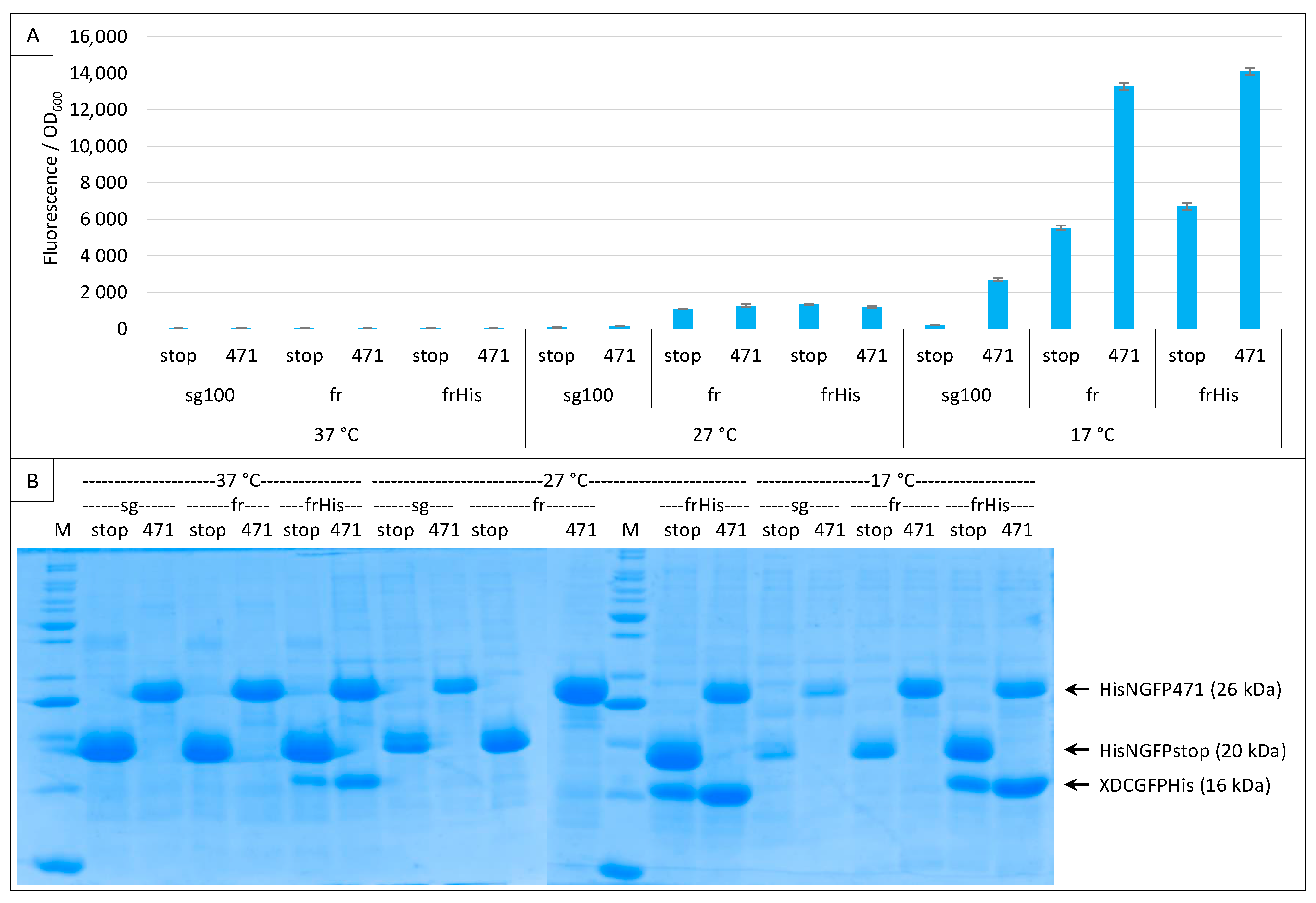
2.4. Addition of a Histidine-Tag to CGFP and Ensuing Analysis of CGFP Stability
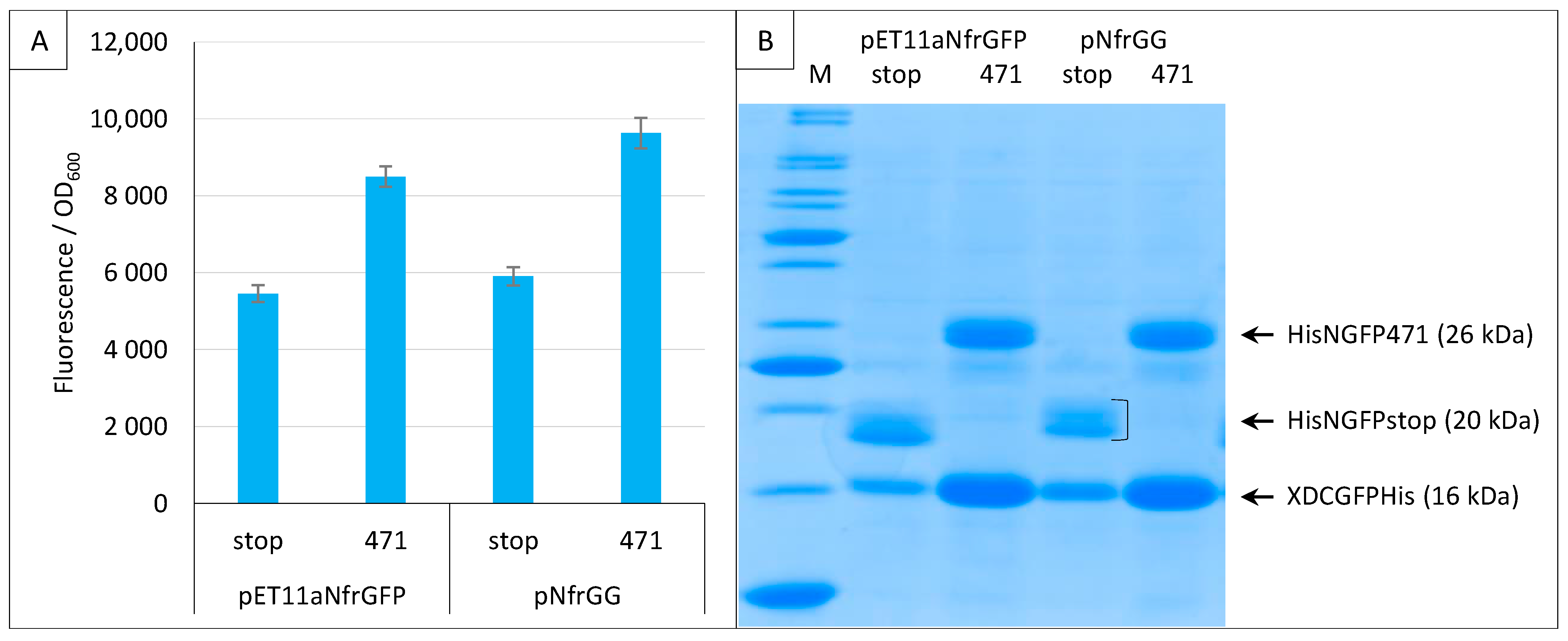
2.5. Generation of a Gateway Vector for NGFP Fusions
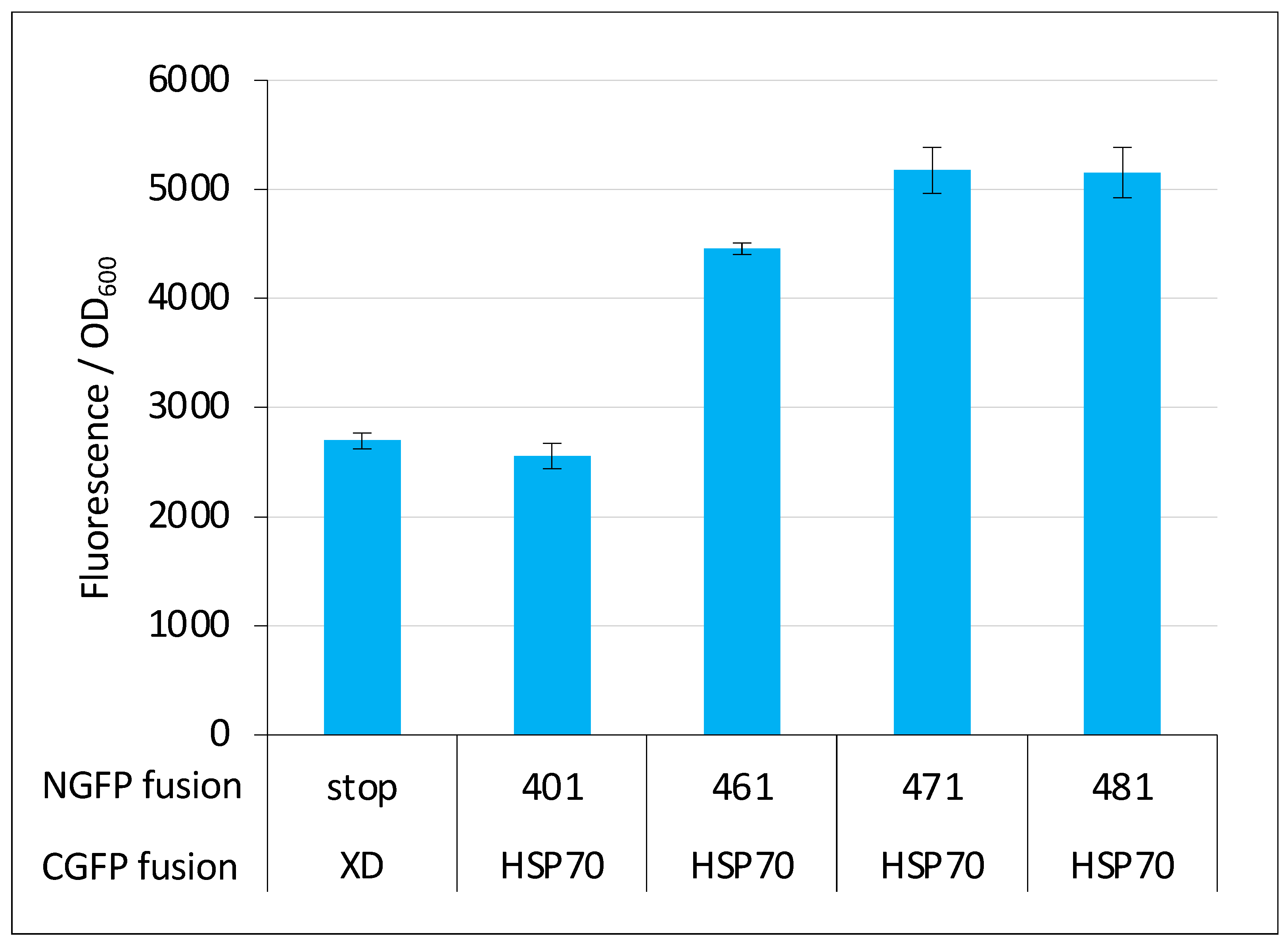
2.6. GFP Variants and Detection of Low-Affinity Interactions
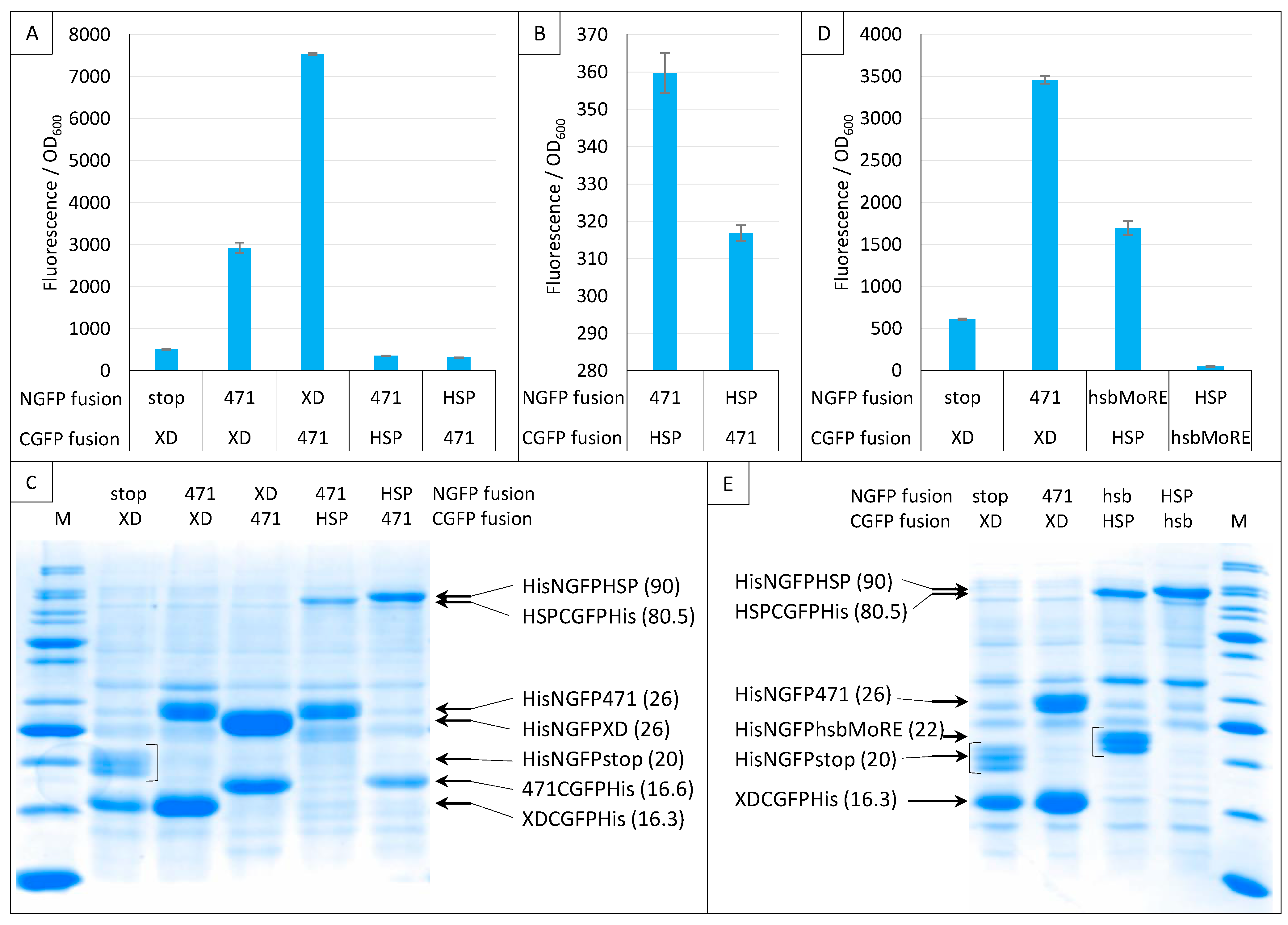
2.7. Impact of NGFP/CGFP Fusion Inversion
2.8. Choice of Negative Controls
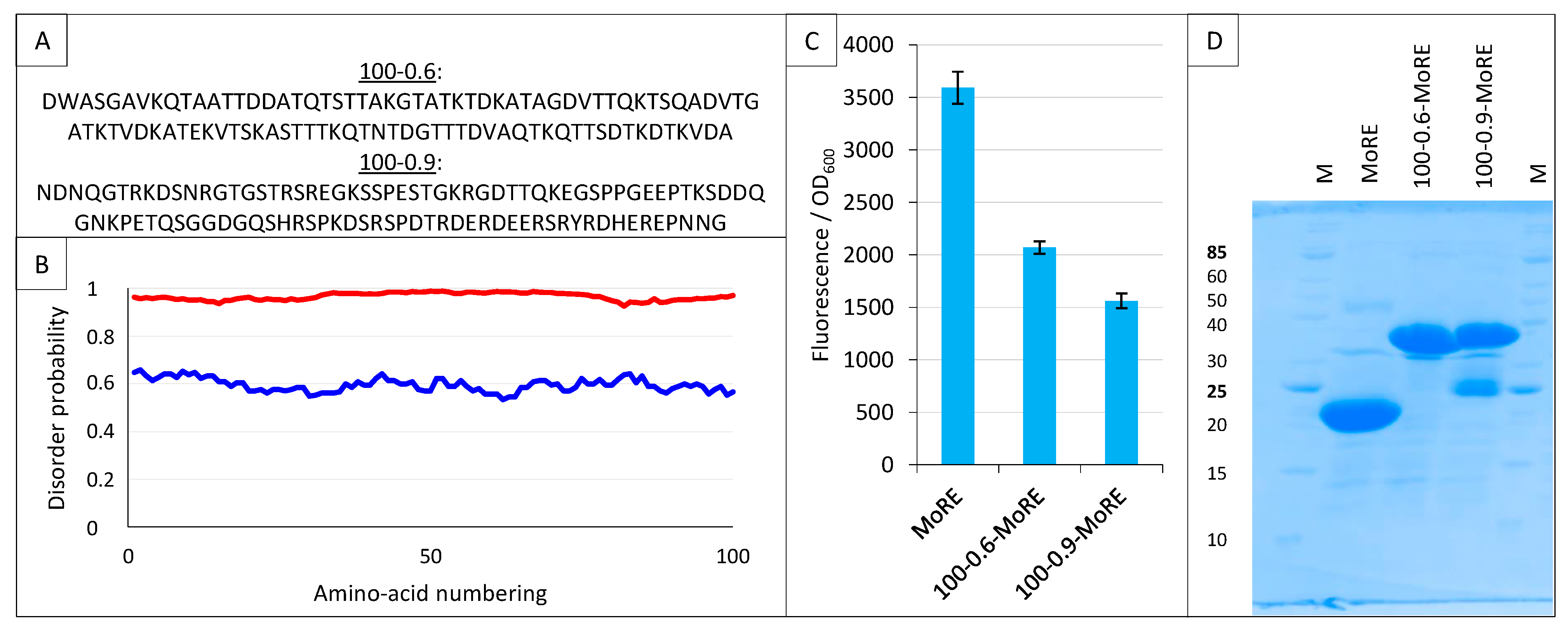
2.9. Split-GFP Reassembly Assay and Artificial IDPs
3. Conclusions
4. Materials and Methods
4.1. DNA Constructs
4.1.1. Folding Reporter (fr) Constructs
4.1.2. Enhanced GFP (eGFP) Constructs
4.1.3. Super Positive GFP (sp) Constructs
4.1.4. Super Folder (sf) Constructs
4.1.5. sg100 Constructs
4.2. Split-GFP Reassembly Assay
4.3. Analysis of Protein Expression
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Perovic, V.; Sumonja, N.; Marsh, L.A.; Radovanovic, S.; Vukicevic, M.; Roberts, S.G.E.; Veljkovic, N. IDPpi: Protein-Protein Interaction Analyses of Human Intrinsically Disordered Proteins. Sci. Rep. 2018, 8, 10563. [Google Scholar] [CrossRef]
- Davey, N.E.; Seo, M.H.; Yadav, V.K.; Jeon, J.; Nim, S.; Krystkowiak, I.; Blikstad, C.; Dong, D.; Markova, N.; Kim, P.M.; et al. Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J. 2017, 284, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.; Ali, M.; Krystkowiak, I.; Simonetti, L.; Sayadi, A.; Mihalic, F.; Kliche, J.; Andersson, E.; Jemth, P.; Davey, N.E.; et al. Proteome-scale mapping of binding sites in the unstructured regions of the human proteome. Mol. Syst. Biol. 2022, 18, e10584. [Google Scholar] [CrossRef] [PubMed]
- Receveur-Bréchot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins Struct. Funct. Bioinform. 2006, 62, 24–45. [Google Scholar] [CrossRef]
- Schramm, A.; Bignon, C.; Brocca, S.; Grandori, R.; Santambrogio, C.; Longhi, S. An arsenal of methods for the experimental characterization of intrinsically disordered proteins—How to choose and combine them? Arch. Biochem. Biophys. 2019, 676, 108055. [Google Scholar] [CrossRef]
- Morell, M.; Ventura, S.; Avilés, F.X. Protein complementation assays: Approaches for the in vivo analysis of protein interactions. FEBS Lett. 2009, 583, 1684–1691. [Google Scholar] [CrossRef]
- Magliery, T.J.; Wilson, C.G.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar] [CrossRef]
- Wilson, C.G.; Magliery, T.J.; Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Hashimoto, J.; Watanabe, T.; Seki, T.; Karasawa, S.; Izumikawa, M.; Seki, T.; Iemura, S.; Natsume, T.; Nomura, N.; Goshima, N.; et al. Novel in vitro protein fragment complementation assay applicable to high-throughput screening in a 1536-well format. J. Biomol. Screen. 2009, 14, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Ohmuro-Matsuyama, Y.; Chung, C.I.; Ueda, H. Demonstration of protein-fragment complementation assay using purified firefly luciferase fragments. BMC Biotechnol. 2013, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Hamilton, A.D.; Regan, L. Antiparallel Leucine Zipper-Directed Protein Reassembly: Application to the Green Fluorescent Protein. J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar] [CrossRef]
- Waldo, G.S.; Standish, B.M.; Berendzen, J.; Terwilliger, T.C. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999, 17, 691–695. [Google Scholar] [CrossRef]
- Sarkar, M.; Magliery, T.J. Re-engineering a split-GFP reassembly screen to examine RING-domain interactions between BARD1 and BRCA1 mutants observed in cancer patients. Mol. Biosyst. 2008, 4, 599–605. [Google Scholar] [CrossRef]
- Blakeley, B.D.; Chapman, A.M.; McNaughton, B.R. Split-superpositive GFP reassembly is a fast, efficient, and robust method for detecting protein-protein interactions in vivo. Mol. Biosyst. 2012, 8, 2036–2040. [Google Scholar] [CrossRef]
- Roggo, C.; Carraro, N.; van der Meer, J.R. Probing chemotaxis activity in Escherichia coli using fluorescent protein fusions. Sci. Rep. 2019, 9, 3845. [Google Scholar] [CrossRef]
- Hu, C.D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar] [CrossRef]
- Abedi, M.R.; Caponigro, G.; Kamb, A. Green fluorescent protein as a scaffold for intracellular presentation of peptides. Nucleic Acids Res. 1998, 26, 623–630. [Google Scholar] [CrossRef]
- Santner, A.A.; Croy, C.H.; Vasanwala, F.H.; Uversky, V.N.; Van, Y.Y.; Dunker, A.K. Sweeping away protein aggregation with entropic bristles: Intrinsically disordered protein fusions enhance soluble expression. Biochemistry 2012, 51, 7250–7262. [Google Scholar] [CrossRef]
- Gruet, A.; Longhi, S.; Bignon, C. One-step generation of error-prone PCR libraries using Gateway® technology. Microb. Cell Fact. 2012, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Bourhis, J.M.; Campanacci, V.; Cambillau, C.; Canard, B.; Longhi, S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 2003, 278, 44567–44573. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.L.; Hamel, D.J.; Gay, L.S.; Dahlquist, F.W.; Matthews, B.W. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. USA 2004, 101, 8301–8306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bourhis, J.M.; Longhi, S.; Carsillo, T.; Buccellato, M.; Morin, B.; Canard, B.; Oglesbee, M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 2005, 337, 162–174. [Google Scholar] [CrossRef]
- Zhang, X.; Glendening, C.; Linke, H.; Parks, C.L.; Brooks, C.; Udem, S.A.; Oglesbee, M. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 2002, 76, 8737–8746. [Google Scholar] [CrossRef] [PubMed]
- Couturier, M.; Buccellato, M.; Costanzo, S.; Bourhis, J.M.; Shu, Y.; Nicaise, M.; Desmadril, M.; Flaudrops, C.; Longhi, S.; Oglesbee, M. High Affinity Binding between Hsp70 and the C-Terminal Domain of the Measles Virus Nucleoprotein Requires an Hsp40 Co-Chaperone. J. Mol. Recognit. 2010, 23, 301–315. [Google Scholar] [CrossRef]
- Gruet, A.; Dosnon, M.; Vassena, A.; Lombard, V.; Gerlier, D.; Bignon, C.; Longhi, S. Dissecting partner recognition by an intrinsically disordered protein using descriptive random mutagenesis. J. Mol. Biol. 2013, 425, 3495–3509. [Google Scholar] [CrossRef]
- Gruet, A.; Dosnon, M.; Blocquel, D.; Brunel, J.; Gerlier, D.; Das, R.K.; Bonetti, D.; Gianni, S.; Fuxreiter, M.; Longhi, S.; et al. Fuzzy regions in an intrinsically disordered protein impair protein-protein interactions. FEBS J. 2016, 283, 576–594. [Google Scholar] [CrossRef]
- Bignon, C.; Troilo, F.; Gianni, S.; Longhi, S. Partner-Mediated Polymorphism of an Intrinsically Disordered Protein. J. Mol. Biol. 2018, 430, 2493–2507. [Google Scholar] [CrossRef]
- Bignon, C.; Troilo, F.; Gianni, S.; Longhi, S. Modulation of Measles Virus N(TAIL) Interactions through Fuzziness and Sequence Features of Disordered Binding Sites. Biomolecules 2018, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Benoit, I.; Coutard, B.; Oubelaid, R.; Asther, M.; Bignon, C. Expression in Escherichia coli, refolding and crystallization of Aspergillus niger feruloyl esterase A using a serial factorial approach. Protein Expr. Purif. 2007, 55, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Pédelacq, J.D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef] [PubMed]
- Dosnon, M.; Bonetti, D.; Morrone, A.; Erales, J.; di Silvio, E.; Longhi, S.; Gianni, S. Demonstration of a folding after binding mechanism in the recognition between the measles virus NTAIL and X domains. ACS Chem. Biol. 2015, 10, 795–802. [Google Scholar] [CrossRef]
- Bloyet, L.M.; Brunel, J.; Dosnon, M.; Hamon, V.; Erales, J.; Gruet, A.; Lazert, C.; Bignon, C.; Roche, P.; Longhi, S.; et al. Modulation of Re-initiation of Measles Virus Transcription at Intergenic Regions by PXD to NTAIL Binding Strength. PLoS Pathog. 2016, 12, e1006058. [Google Scholar] [CrossRef]
- Caleca, L.; Colombo, M.; van Overeem Hansen, T.; Lázaro, C.; Manoukian, S.; Parsons, M.T.; Spurdle, A.B.; Radice, P. GFP-Fragment Reassembly Screens for the Functional Characterization of Variants of Uncertain Significance in Protein Interaction Domains of the BRCA1 and BRCA2 Genes. Cancers 2019, 11, 151. [Google Scholar] [CrossRef]
- Flechtner, J.B.; Cohane, K.P.; Mehta, S.; Slusarewicz, P.; Leonard, A.K.; Barber, B.H.; Levey, D.L.; Andjelic, S. High-affinity interactions between peptides and heat shock protein 70 augment CD8+ T lymphocyte immune responses. J. Immunol. 2006, 177, 1017–1027. [Google Scholar] [CrossRef]
- Schramm, A.; Lieutaud, P.; Gianni, S.; Longhi, S.; Bignon, C. InSiDDe: A Server for Designing Artificial Disordered Proteins. Int. J. Mol. Sci. 2018, 19, 91. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Bourhis, J.M.; Receveur-Bréchot, V.; Oglesbee, M.; Zhang, X.; Buccellato, M.; Darbon, H.; Canard, B.; Finet, S.; Longhi, S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005, 14, 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, S.; Lofdahl, P.A.; Hard, T.; Berglund, H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006, 121, 291–298. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bignon, C.; Gruet, A.; Longhi, S. Split-GFP Reassembly Assay: Strengths and Caveats from a Multiparametric Analysis. Int. J. Mol. Sci. 2022, 23, 13167. https://doi.org/10.3390/ijms232113167
Bignon C, Gruet A, Longhi S. Split-GFP Reassembly Assay: Strengths and Caveats from a Multiparametric Analysis. International Journal of Molecular Sciences. 2022; 23(21):13167. https://doi.org/10.3390/ijms232113167
Chicago/Turabian StyleBignon, Christophe, Antoine Gruet, and Sonia Longhi. 2022. "Split-GFP Reassembly Assay: Strengths and Caveats from a Multiparametric Analysis" International Journal of Molecular Sciences 23, no. 21: 13167. https://doi.org/10.3390/ijms232113167
APA StyleBignon, C., Gruet, A., & Longhi, S. (2022). Split-GFP Reassembly Assay: Strengths and Caveats from a Multiparametric Analysis. International Journal of Molecular Sciences, 23(21), 13167. https://doi.org/10.3390/ijms232113167