
Epigenetic Alterations in Sports-Related Injuries

 , ,
, ,
Abstract
1. Introduction
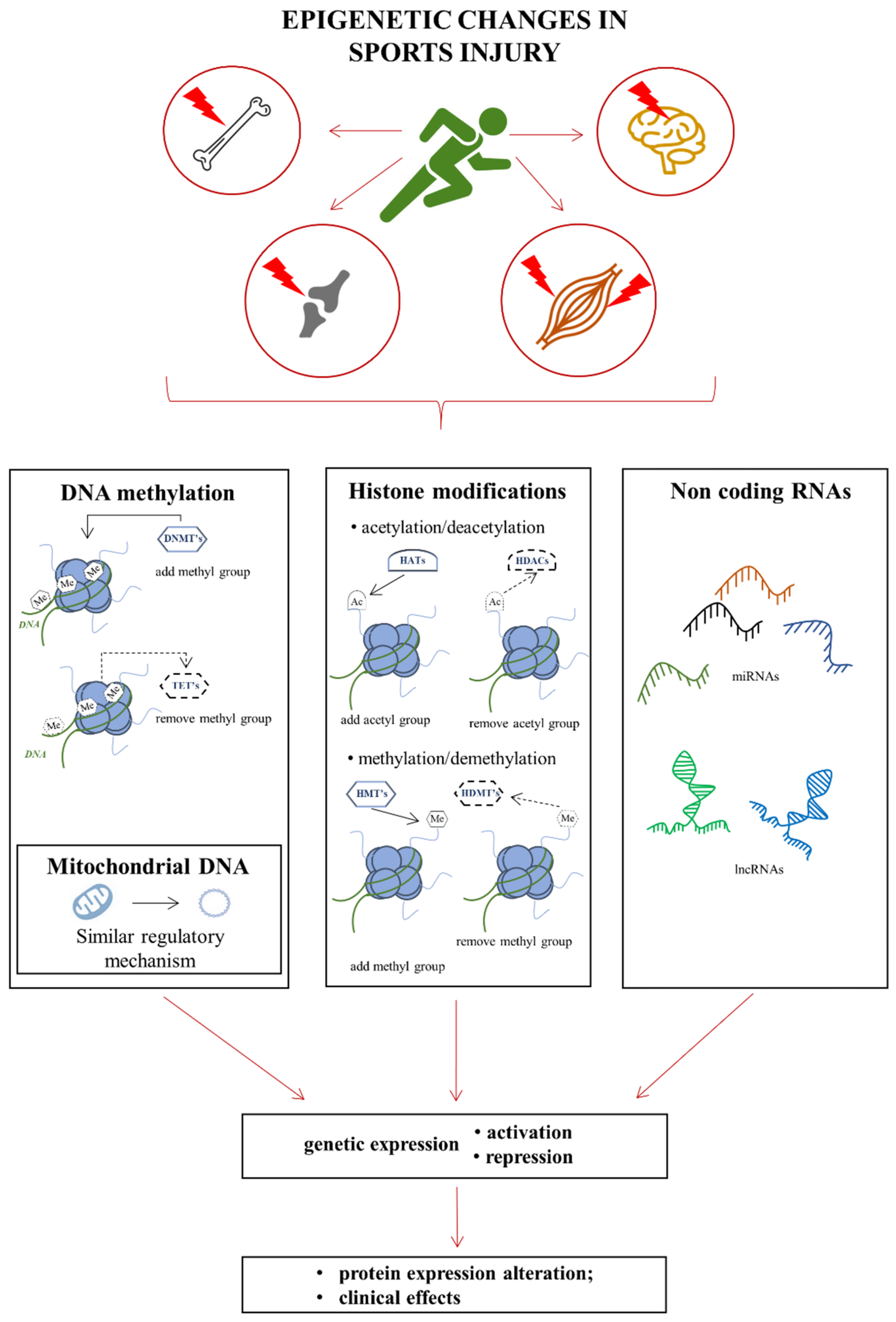
2. Epigenetics
Basics of Epigenetic Modifications
3. Injury to Skeletal Muscles
3.1. Pathophysiology of Skeletal Muscle Injury
3.2. Genetic and Epigenetic Changes Accompanying Skeletal Muscle Injuries
3.3. Epigenetic Mechanisms of Skeletal Muscle Regeneration after Injury
4. Injury to Tendons
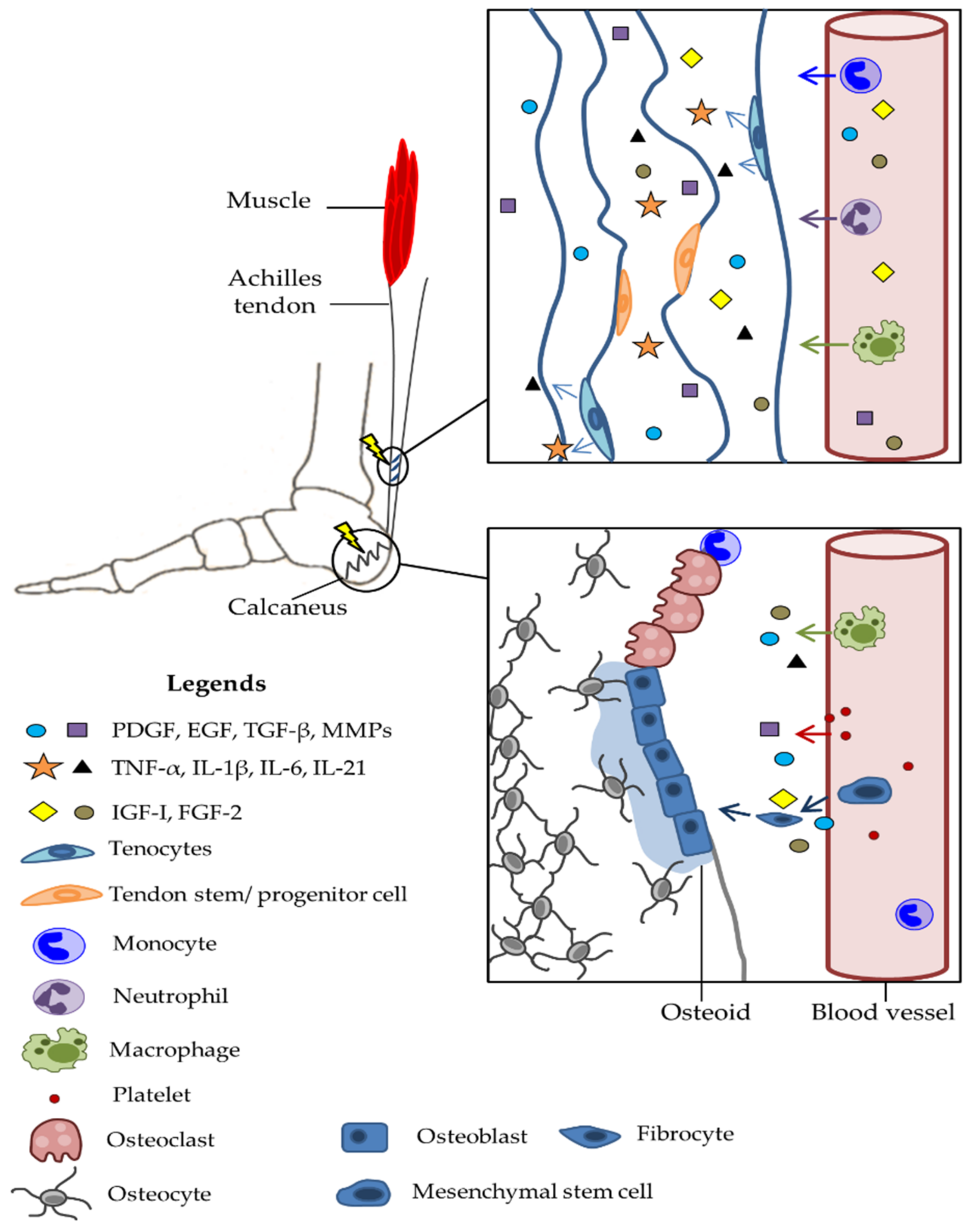
4.1. Pathophysiology of Trauma
4.2. Molecular and Genetic Basis of Damage
4.3. Epigenetic Changes as a Consequence of Damage
4.4. Epigenetic Mechanisms of Tendon Regeneration
5. Bone Injuries
5.1. Pathophysiology of Trauma
5.2. Molecular and Genetic Basis of Damage
5.3. Epigenetic Changes as a Consequence of Damage to the Bone Tissue
5.4. Epigenetic Mechanisms of Bone Regeneration

| Type of miRNA | Functional Activity | Reference |
|---|---|---|
| miR-1, miR-21, miR-135, miR-155, miR-199a, miR-429, miR-675 | Hypoxia-regulated | [161,209] |
| miR-26a, miR-126, miR-143 | Pro-angiogenic | [209,210,211] |
| miR-22, miR-342 | Pro-apoptotic | [161,209,210,211] |
| miR-21 | Inhibit apoptosis | [209,210] |
| miR-21, miR-31a | Bone resorption | [201] |
| miR-128, miR-155, miR-182, miR-222 | Inhibit bone formation | [209,210] |
| miR-21, miR-31a | Osteoclast activity | [201] |
| miR-22 | Suppresses osteoblast viability | [161,209,210,211] |
| miR-21, miR-31a, miR-34c, miR-99, miR-125a, miR-128, miR-142, miR-182, miR-183, miR-218, miR-483 | Osteoclastogenesis | [161,201,209,210,211,212] |
| miR-21, miR-29a, miR-126, miR-128, miR-135b, miR-142, miR-150, miR-218, miR-223, miR-296, miR-451, miR-503 | Increase mineralization | [161,209,210,211] |
| miR-10, miR-17, miR-29b, miR-30c, miR-99, miR-124, miR-125b, miR-133a, miR-138, miR-141, miR-148a, miR-186, miR-193a, miR-200a, miR-203, miR-205, miR-214, miR-320a, miR-320b, miR-409, miR-532, miR-542 | Decrease mineralization | [161,200,209,210,211] |
| miR-9, miR-15b, miR-21, miR-23b, miR-27a, miR-98, miR-128, miR-135b, miR-140, miR-143, miR-149, miR-187, miR-194, miR-409, miR-503, miR-664a, miR-877 | Enhance osteogenic differentiation | [161,209,210,211] |
| miR-29a, miR-194, miR-219a, miR-223, miR-296, miR-302a, miR-5106 | Enhance osteoblastic differentiation | [161,209,210,211] |
| miR-146a, miR-346a | Enhance osteogenesis | [161,209,210,211] |
| miR-451 | Enhances osteoblastogenesis | [161,209,210,211] |
| miR-10, miR-17, miR-23, miR-31, miR-34a, miR-34c, miE-103, miR-124, miR-125b, miR-133a, miR-138, miR-139, miR-141, miR-145, miR-150, miR-153, miR-181a, miR-186, miR-200a, miR-203, miR-205, miR-206, miR-214, miR-217, miR-320a, miR-320b, miR-342, miR-363, miR-375, miR-383, miR-449b, miR-505, miR-532, miR-765 | Inhibit osteogenic differentiation | [161,209,210,211] |
| miR-22, miR-144, miR-182, miR-193a, miR-542 | Suppress osteoblastic differentiation | [161,209,210,211] |
| miR-29a, miR-140, miR-181a, miR-218, miR-222, miR-335, miR-337 | Chondrogenesis | [161,209,210,211] |
| miR-1, miR-26b, miR-125b, miR-146a, miR-206, miR-214 | Inhibit chondrogenic differentiation | [161,209,210,211] |
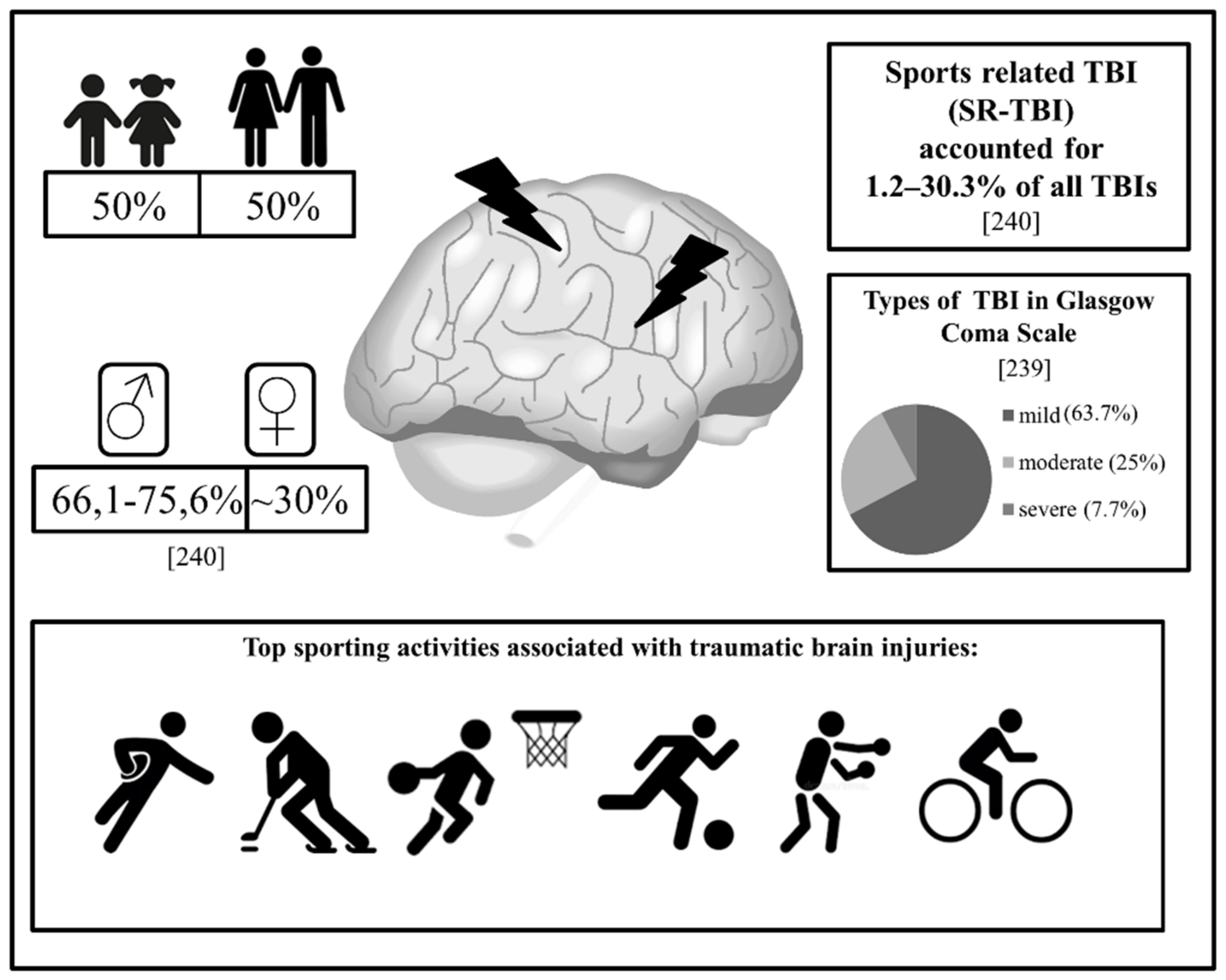
6. Traumatic Brain Injury (TBI)
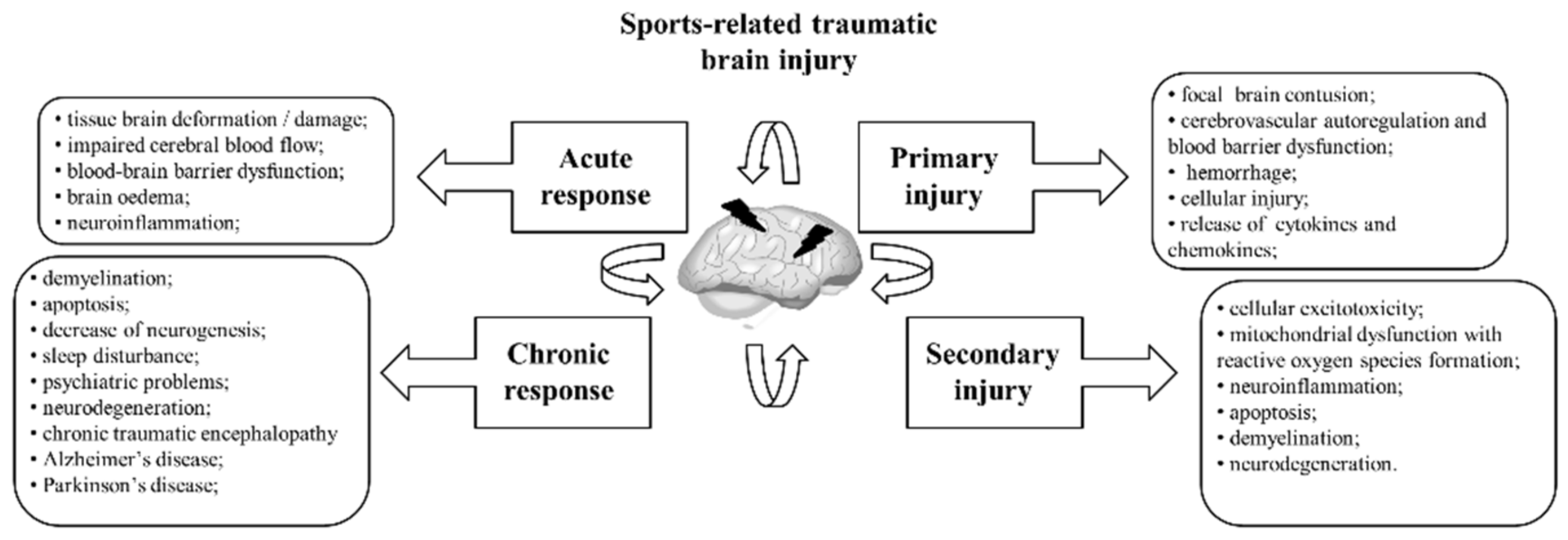
6.1. Pathophysiology of Trauma
6.2. Molecular and Genetic Basis of TBI

{kind=link}
{kind=link}
{kind=link}
{kind=link}
6.3. Epigenetics of TBI
7. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Physical Activity Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs385/en/ (accessed on 20 May 2022).
- Hechanova, R.L.; Wegler, J.L.; Forest, C.P. Exercise: A vitally important prescription. J. Am. Acad. Pas 2017, 30, 17–22. [Google Scholar] [CrossRef]
- Daskalopoulou, C.; Stubbs, B.; Kralj, C.; Koukounari, A.; Prince, M.; Prina, A.M. Physical activity and healthy ageing: A systematic review and metaanalysis of longitudinal cohort studies. Ageing Res. Rev. 2017, 38, 6. [Google Scholar] [CrossRef]
- Home|Pac Report. Available online: http://www.physicalactivitycouncil.com (accessed on 21 June 2021).
- Ryan, J.L.; Pracht, E.E.; Orban, B.L. Inpatient and emergency department costs from sports injuries among youth aged 5–18 years. BMJ Open Sport Exerc. Med. 2019, 5, e000491. [Google Scholar]
- Trentacosta, N. Pediatric Sports Injuries. Pediatr. Clin. North USA 2020, 67, 205–225. [Google Scholar] [CrossRef]
- Rui, P.; Ashman, J.J.; Akinseye, A. Emergency department visits for injuries sustained during sports and recreational activities by patients aged 5–24 years, 2010–2016. Natl. Health Stat. Rep. 2019, 133, 2010–2016. [Google Scholar]
- Sheu, Y.; Chen, L.-H.; Hedegaard, H. Sports- and recreation-related injury episodes in the United States, 2011–2014. Natl. Health Stat. Rep. 2016, 99, 1–10. [Google Scholar]
- Prieto-González, P.; Martínez-Castillo, J.L.; Fernández-Galván, L.M.; Casado, A.; Soporki, S.; Sánchez-Infante, J. Epidemiology of Sports-Related Injuries and Associated Risk Factors in Adolescent Athletes: An Injury Surveillance. Int. J. Environ. Res. Public Health 2021, 18, 4857. [Google Scholar] [CrossRef]
- Visentini, P.J.; McDowell, A.H.; Pizzari, T. Factors associated with overuse injury in cyclists: A systematic review. J. Sci. Med. Sport 2022, 25, 391–398. [Google Scholar] [CrossRef]
- O’Donovan, G.; Sarmiento, O.L.; Hamer, M. The Rise of the “Week-end Warrior”. J. Orthop. Sports Phys. Ther. 2018, 48, 604–606. [Google Scholar] [CrossRef]
- Gimigliano, F.; Resmini, G.; Moretti, A.; Aulicino, M.; Gargiulo, F.; Gimigliano, A.; Liguori, S.; Paoletta, M.; Iolascon, G. Epidemiology of Musculoskeletal Injuries in Adult Athletes: A Scoping Review. Medicina 2021, 57, 1118. [Google Scholar] [CrossRef]
- Gomes, C.; Almeida, J.A.; Franco, O.L.; Petriz, B. Omics and the molecular exercise physiology. Adv. Clin. Chem. 2020, 96, 55–84. [Google Scholar] [PubMed]
- Georgiades, E.; Klissouras, V.; Baulch, J.; Wang, G.; Pitsiladis, Y. Why nature prevails over nurture in the making of the elite athlete. BMC Genom. 2017, 18, 835. [Google Scholar] [CrossRef] [PubMed]
- De Moor, M.H.M.; Spector, T.D.; Cherkas, L.F.; Falchi, M.; Hottenga, J.J.; Boomsma, D.I.; de Geus, E. Genome-wide linkage scan for athlete status in 700 British female DZ twin pairs. Twin Res. Hum. Genet. 2007, 10, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, J.P.L.F.; Tritto, A.C.C.; North, K.N.; Lancha Junior, A.H.; Artioli, G.G. Genetics and sport performance: Current challenges and directions to the future. Rev. Bras. Educ. Física E Esporte 2014, 28, 177–193. [Google Scholar] [CrossRef]
- Varillas-Delgado, D.; Del Coso, J.; Gutiérrez-Hellín, J.; Aguilar-Navarro, M.; Muñoz, A.; Maestro, A.; Morencos, E. Genetics and sports performance: The present and future in the identification of talent for sports based on DNA testing. Eur. J. Appl. Physiol. 2022, 16, 1–20. [Google Scholar] [CrossRef]
- Ginevičienė, V.; Utkus, A.; Pranckevičienė, E.; Semenova, E.A.; Hall, E.C.R.; Ahmetov, I.I. Perspectives in Sports Genomics. Biomedicines 2022, 10, 298. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef]
- Pandorf, C.E.; Haddad, F.; Wright, C.; Bodell, P.W.; Baldwin, K.M. Differential epigenetic modifications of histones at the myosin heavy chain genes in fast and slow skeletal muscle fibers and in response to muscle unloading. Am. J. Physiol. Cell Physiol. 2009, 297, C6–C16. [Google Scholar] [CrossRef]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef]
- Ecker, S.; Pancaldi, V.; Valencia, A.; Beck, S.; Paul, D.S. Epigenetic and transcriptional variability shape phenotypic plasticity. Bioessays 2018, 40, 1700148. [Google Scholar] [CrossRef]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Deans, C.; Maggert, K.A. What do you mean, “epigenetic”? Genetics 2015, 199, 887–896. [Google Scholar] [CrossRef]
- Woo, H.; Dam Ha, S.; Lee, S.B.; Buratowski, S.; Kim, T. Modulation of gene expression dynamics by co-transcriptional histone methylations. Exp. Mol. Med. 2017, 49, e326. [Google Scholar] [CrossRef]
- Thompson, R.F.; Einstein, F.H. Epigenetic basis for fetal origins of age-related disease. J. Womens Health 2010, 19, 581–587. [Google Scholar] [CrossRef]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2021, 70, 29–49. [Google Scholar] [CrossRef]
- Tarnowski, M.; Kopytko, P.; Piotrowska, K. Epigenetic Regulation of Inflammatory Responses in the Context of Physical Activity. Genes 2021, 12, 1313. [Google Scholar] [CrossRef]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef]
- Bell, O.; Tiwari, V.K.; Thoma, N.H.; Schubeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011, 12, 554–564. [Google Scholar] [CrossRef]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorf, N. The interplay of histone modifcations—writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Amin, N.; Mcgrath, A.; Chen, Y.P. Evaluation of deep learning in non-coding RNA classification. Nat. Mach. Intell. 2019, 1, 246–256. [Google Scholar] [CrossRef]
- Huumonen, K.; Korkalainen, M.; Viluksela, M.; Lahtinen, T.; Naarala, J.; Juutilainen, J. Role of microRNAs and DNA methyltransferases in transmitting induced genomic instability between cell generations. Front. Public Health 2014, 2, 139. [Google Scholar] [CrossRef]
- Shivram, H.; Le, S.V.; Iyer, V.R. MicroRNAs reinforce repression of PRC2 transcriptional targets independently and through a feed-forward regulatory network. Genome Res. 2019, 29, 184–192. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar] [CrossRef]
- Dozmorov, M.G.; Giles, C.B.; Koelsch, K.A.; Wren, J.D. Systematic classification of non-coding RNAs by epigenomic similarity. BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Koscianska, E.; Starega-Roslan, J.; Krzyzosiak, W.J. The role of dicer protein partners in the processing of microRNA precursors. PLoS ONE 2011, 6, e28548. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hamo, R.; Efroni, S. MicroRNA regulation of molecular pathways as a generic mechanism and as a core disease phenotype. Oncotarget 2015, 6, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef]
- Li, X.; Wu, Z.; Fu, X.; Han, W. lncRNAs: Insights into their function and mechanics in underlying disorders. Mutat. Res. Rev. Mutat. Res. 2014, 762, 1–21. [Google Scholar]
- Angrand, P.O.; Vennin, C.; Le Bourhis, X.; Adriaenssens, E. The role of long non-coding RNAs in genome formatting and expression. Front. Genet. 2015, 6, 165. [Google Scholar] [CrossRef]
- Rion, N.; Ruegg, M.A. LncRNA-encoded peptides: More than translational noise? Cell Res. 2017, 27, 604–605. [Google Scholar]
- Wang, Z.; Zheng, Y. lncRNAs Regulate Innate Immune Responses and Their Roles in Macrophage Polarization. Mediat. Inflamm 2018, 2018, 8050956. [Google Scholar] [CrossRef]
- Saini, A.; Mastana, S.; Myers, F.; Lewis, M.P. ‘From death, lead me to immortality’-mantra of ageing skeletal muscle. Curr. Genom. 2013, 14, 256–267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shadrach, J.L.; Wagers, A.J. Stem cells for skeletal muscle repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Epigenetic control of adult skeletal muscle stem cell functions. FEBS J. 2015, 282, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Lee, M.; Akimoto, T. Conditional Deletion of Dicer in Adult Mice Impairs Skeletal Muscle Regeneration. Int. J. Mol. Sci. 2019, 13, 5686. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Gillespie, M.A.; Rudnicki, M.A. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem. Cell 2008, 2, 22–31. [Google Scholar] [CrossRef]
- Ekstrand, J.; Hägglund, M.; Waldén, M. Epidemiology of muscle injuries in professional football (soccer). Am. J. Sports Med. 2011, 39, 1226–1232. [Google Scholar] [CrossRef]
- Guermazi, A.; Roemer, F.W.; Robinson, P.; Tol, J.L.; Regatte, R.R.; Crema, M.D. Imaging of Muscle Injuries in Sports Medicine: Sports Imaging Series. Radiology 2017, 282, 646–663, Erratum in: Radiology 2017, 285, 1063. [Google Scholar] [CrossRef]
- Järvinen, T.A.; Järvinen, T.L.; Kääriäinen, M.; Kalimo, H.; Järvinen, M. Muscle injuries: Biology and treatment. Am. J. Sports Med. 2005, 33, 745–764. [Google Scholar] [CrossRef]
- Barroso, G.C.; Thiele, E.S. Muscle injuries in athletes. Rev. Bras. Ortop. 2015, 46, 354–358. [Google Scholar] [CrossRef]
- Malliaropoulos, N.; Isinkaye, T.; Tsitas, K.; Maffulli, N. Reinjury after acute posterior thigh muscle injuries in elite track and field athletes. Am. J. Sports Med. 2011, 39, 304–310. [Google Scholar] [CrossRef]
- Peake, J.M.; Neubauer, O.; Della Gatta, P.A.; Nosaka, K. Muscle damage and inflammation during recovery from exercise. J. Appl. Physiol. 2017, 122, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Hyldahl, R.D.; Hubal, M.J. Lengthening our perspective: Morphological, cellular, and molecular responses to eccentric exercise. Muscle Nerve 2014, 49, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Delos, D.; Maak, T.G.; Rodeo, S.A. Muscle injuries in athletes: Enhancing recovery through scientific understanding and novel therapies. Sports Health 2013, 5, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Carazzato, J.G. Lesões musculotendineas e seu tratamento. Rev. Bras. Ortop. 1994, 29, 723–728. [Google Scholar]
- Brukner, P.; Khan, K. Clinical Sports Medicine, 3rd ed.; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- O’Donoghue, D.O. Treatment of Injuries to Athletes; Saunders: Philadelphia, PA, USA, 1894; pp. 51–56. [Google Scholar]
- SantAnna, J.P.C.; Pedrinelli, A.; Hernandez, A.J.; Fernandes, T.L. Muscle Injury: Pathophysiology, Diagnosis, and Treatment. Rev. Bras. Ortop. 2022, 57, 1–13. [Google Scholar]
- Koelwyn, G.J.; Wennerberg, E.; Demaria, S.; Jones, L.W. Exercise in regulation of inflammation-immune axis function in cancer initiation and progression. Oncology 2015, 29, 908–922. [Google Scholar]
- Hollander, J.; Fiebig, R.; Gore, M.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide dismutase gene expression is activated by a single bout of exercise in rat skeletal muscle. Pflugers Arch. Eur. J. Physiol. 2001, 442, 426–434. [Google Scholar] [CrossRef]
- Kramer, H.F.; Goodyear, L.J. Exercise, MAPK, and NF-kappaB signaling in skeletal muscle. J. Appl. Physiol. 2007, 103, 388–395. [Google Scholar] [CrossRef]
- Proske, U.; Allen, T.J. Damage to skeletal muscle from eccentric exercise. Exerc. Sport Sci. Rev. 2005, 33, 98–104. [Google Scholar] [CrossRef]
- Peake, J.; Nosaka, K.; Suzuki, K. Characterization of inflammatory responses to eccentric exercise in humans. Exerc. Immunol. Rev. 2005, 11, 64–85. [Google Scholar]
- Toumi, H.; Best, T.M. The inflammatory response: Friend or enemy for muscle injury? Br. J. Sports Med. 2003, 37, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Windelinckx, A.; Nielens, H.; Ramaekers, M.; Van Leemputte, M.; Hespel, P.; Thomis, M.A. Protective role of α-actinin-3 in the response to an acute eccentric exercise bout. J. Appl. Physiol. 2010, 109, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Yamin, C.; Duarte, J.A.R.; Oliveira, J.M.F.; Amir, O.; Sagiv, M.; Eynon, N.; Sagiv, M.; Amir, R.E. IL6 (−174) and TNFA (−308) promoter polymorphisms are associated with systemic creatine kinase response to eccentric exercise. Eur. J. Appl. Physiol. 2008, 104, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Devaney, J.M.; Hoffman, E.P.; Gordish-Dressman, H.; Kearns, A.; Zambraski, E.; Clarkson, P.M. IGF-II gene region polymorphisms related to exertional muscle damage. J. Appl. Physiol. 2007, 102, 1815–1823. [Google Scholar] [CrossRef]
- Giordani, L.; Puri, P.L. Epigenetic control of skeletal muscle regeneration: Integrating genetic determinants and environmental changes. FEBS J. 2013, 280, 4014–4025. [Google Scholar] [CrossRef]
- Lluis, F.; Ballestar, E.; Suelves, M.; Esteller, M.; Munoz-Canoves, P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005, 24, 974–984. [Google Scholar] [CrossRef]
- Serra, C.; Palacios, D.; Mozzetta, C.; Forcales, S.V.; Morantte, I.; Ripani, M.; Jones, D.R.; Du, K.; Jhala, U.S.; Simone, C.; et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol. Cell 2007, 28, 200–213. [Google Scholar] [CrossRef]
- Yu, X.; Zuo, Q. MicroRNAs in the regeneration of skeletal muscle. Front. Biosci. (Landmark Ed.) 2013, 18, 608–615. [Google Scholar] [CrossRef]
- Kirby, T.J.; McCarthy, J.J. MicroRNAs in skeletal muscle biology and exercise adaptation. Free Radic. Biol. Med. 2013, 64, 95–105. [Google Scholar] [CrossRef]
- Pegoraro, V.; Merico, A.; Angelini, C. MyomiRNAs Dysregulation in ALS Rehabilitation. Brain Sci. 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J. MicroRNA-206: The skeletal muscle- specific myomiR. Biochim. Biophys. Acta 2008, 1779, 682–691. [Google Scholar] [CrossRef]
- Winbanks, C.E.; Wang, B.; Beyer, C.; Koh, P.; White, L.; Kantharidis, P.; Gregorevic, P. TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of histone deacetylase 4 (HDAC4). J. Biol. Chem. 2011, 286, 13805–13814. [Google Scholar] [CrossRef]
- Liu, H.; Chen, S.E.; Jin, B.; Carson, J.A.; Niu, A.; Durham, W.; Lai, J.Y.; Li, Y.P. TIMP3: A physiological regulator of adult myogenesis. J. Cell Sci. 2010, 123, 2914–2921. [Google Scholar] [CrossRef]
- Nakasa, T.; Ishikawa, M.; Shi, M.; Shibuya, H.; Adachi, N.; Ochi, M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J. Cell Mol. Med. 2010, 14, 2495–2505. [Google Scholar] [CrossRef]
- Naguibneva, I.; Ameyar-Zazoua, M.; Polesskaya, A.; Ait-Si-Ali, S.; Groisman, R.; Souidi, M.; Cuvellierm, S.; Harel-Bellan, A. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat. Cell Biol. 2006, 8, 278–284. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, Y.; Drnevich, J.; Zhao, Y.; Band, M.; Chen, J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 2010, 189, 1157–1169. [Google Scholar] [CrossRef]
- Chen, Y.; Melton, D.W.; Gelfond, J.A.; McManus, L.M.; Shireman, P.K. MiR-351 transiently increases during muscle regeneration and promotes progenitor cell proliferation and survival upon differentiation. Physiol. Genom. 2012, 44, 1042–1051. [Google Scholar] [CrossRef]
- Dey, B.K.; Gagan, J.; Yan, Z.; Dutta, A. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes. Dev. 2012, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.P.C.; Oliveira-Jr, G.P.; Madrid, B.; Almeida, J.A.; Franco, O.L.; Pereira, R.W. Circulating miR-1, miR-133a, and miR-206 levels are increased after a half-marathon run. Biomarkers 2014, 19, 585–589. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, R.F.; Markworth, J.F.; Aasen, K.M.M.; Zeng, N.; Cameron-Smith, D.; Mitchell, C.J. Acute resistance exercise modulates microRNA expression profiles: Combined tissue and circulatory targeted analyses. PLoS ONE 2017, 12, e0181594. [Google Scholar] [CrossRef]
- Baggish, A.L.; Park, J.; Min, P.K.; Isaacs, S.; Parker, B.A.; Thompson, P.D.; Troyanos, C.; D’Hemecourt, P.; Dyer, S.; Thiel, M.; et al. Rapid upregulation and clearance of distinct circulating microRNAs after prolonged aerobic exercise. J. Appl. Physiol. 2014, 116, 522–531. [Google Scholar] [CrossRef]
- Mooren, F.C.; Viereck, J.; Krüger, K.; Thum, T. Circulating microRNAs as potential biomarkers of aerobic exercise capacity. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H557–H563. [Google Scholar] [CrossRef] [PubMed]
- Clauss, S.; Wakili, R.; Hildebrand, B.; Kääb, S.; Hoster, E.; Klier, I.; Martens, E.; Hanley, A.; Hanssen, H.; Halle, M.; et al. MicroRNAs as Biomarkers for Acute Atrial Remodeling in Marathon Runners (The miRathon Study--A Sub-Study of the Munich Marathon Study). PLoS ONE 2016, 11, e0148599. [Google Scholar] [CrossRef]
- Ramos, A.E.; Lo, C.; Estephan, L.E.; Tai, Y.Y.; Tang, Y.; Zhao, J.; Sugahara, M.; Gorcsan, J., 3rd; Brown, M.G.; Lieberman, D.E.; et al. Specific circulating microRNAs display dose-dependent responses to variable intensity and duration of endurance exercise. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H273–H283. [Google Scholar] [CrossRef] [PubMed]
- Chalchat, E.; Charlot, K.; Garcia-Vicencio, S.; Hertert, P.; Baugé, S.; Bourdon, S.; Bompard, J.; Farges, C.; Martin, V.; Bourrilhon, C.; et al. Circulating microRNAs after a 24-h ultramarathon run in relation to muscle damage markers in elite athletes. Scand. J. Med. Sci. Sports 2021, 31, 1782–1795. [Google Scholar] [CrossRef]
- Fernández-Sanjurjo, M.; Úbeda, N.; Fernández-García, B.; Del Valle, M.; Ramírez de Molina, A.; Crespo, M.C.; Martín-Hernández, R.; Casas-Agustench, P.; Martínez-Camblor, P.; de Gonzalo-Calvo, D.; et al. Exercise dose affects the circulating microRNA profile in response to acute endurance exercise in male amateur runners. Scand. J. Med. Sci. Sports 2020, 30, 1896–1907. [Google Scholar] [CrossRef]
- Uhlemann, M.; Möbius-Winkler, S.; Fikenzer, S.; Adam, J.; Redlich, M.; Möhlenkamp, S.; Hilberg, T.; Schuler, G.C.; Adams, V. Circulating microRNA-126 increases after different forms of endurance exercise in healthy adults. Eur. J. Prev. Cardiol. 2014, 21, 484–491. [Google Scholar] [CrossRef]
- Eyileten, C.; Wicik, Z.; Fitas, A.; Marszalek, M.; Simon, J.E.; De Rosa, S.; Wiecha, S.; Palatini, J.; Postula, M.; Malek, L.A. Altered Circulating MicroRNA Profiles After Endurance Training: A Cohort Study of Ultramarathon Runners. Front. Physiol. 2022, 12, 792931. [Google Scholar] [PubMed]
- Baggish, A.L.; Hale, A.; Weiner, R.B.; Lewis, G.D.; Systrom, D.; Wang, F.; Wang, T.J.; Chan, S.Y. Dynamic regulation of circulating microRNA during acute exhaustive exercise and sustained aerobic exercise training. J. Physiol. 2011, 589, 3983–3994. [Google Scholar] [CrossRef] [PubMed]
- Danese, E.; Benati, M.; Sanchis-Gomar, F.; Tarperi, C.; Salvagno, G.L.; Paviati, E.; Montagnana, M.; Schena, F.; Lippi, G. Influence of middle-distance running on muscular micro RNAs. Scan. J. Clin. Lab. Invest. 2018, 78, 165–170. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; Dávalos, A.; Fernández-Sanjurjo, M.; Amado-Rodríguez, L.; Díaz-Coto, S.; Tomás-Zapico, C.; Montero, A.; García-González, Á.; Llorente-Cortés, V.; Heras, M.E.; et al. Circulating microRNAs as emerging cardiac biomarkers responsive to acute exercise. Int. J. Cardiol. 2018, 264, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Karvinen, S.; Sievänen, T.; Karppinen, J.E.; Hautasaari, P.; Bart, G.; Samoylenko, A.; Vainio, S.J.; Ahtiainen, J.P.; Laakkonen, E.K.; Kujala, U.M. MicroRNAs in Extracellular Vesicles in Sweat Change in Response to Endurance Exercise. Front. Physiol. 2020, 11, 676. [Google Scholar] [CrossRef]
- Radom-Azik, S.; Zaldivar, F., Jr.; Oliver, S.; Galassetti, P.; Cooper, D.M. Evidence for microRNA involvement in exercise-associated neutrophil gene expression changes. J. Appl. Physiol. 2010, 109, 252–261. [Google Scholar] [CrossRef]
- Radom-Aizik, S.; Zaldivar, F., Jr.; Leu, S.Y.; Adams, G.R.; Oliver, S.; Cooper, D.M. Effects of exercise on microRNA expression in young males peripheral blood mononuclear cells. Clin. Transl. Sci. 2012, 5, 32–38. [Google Scholar] [CrossRef]
- Radom-Aizik, S.; Zaldivar, F.; Haddad, F.; Cooper, D.M. Impact of brief exercise on peripheral blood NK cell gene and microRNA expression in young adults. J. Appl. Physiol. 2013, 114, 628–636. [Google Scholar] [CrossRef]
- Yuan, W.; Condorelli, G.; Caruso, M.; Felsani, A.; Giordano, A. Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem. 1996, 271, 9009–9013. [Google Scholar] [CrossRef]
- Polesskaya, A.; Duquet, A.; Naguibneva, I.; Weise, C.; Vervisch, A.; Bengal, E.; Hucho, F.; Robin, P.; Harel-Bellan, A. CREB-binding protein/p300 activates MyoD by acetylation. J. Biol. Chem. 2000, 275, 34359–34364. [Google Scholar] [CrossRef]
- Palacios, D.; Puri, P.L. The epigenetic network regulating muscle development and regeneration. J. Cell Physiol. 2006, 207, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, F.J.; Blais, A. Epigenetic regulation of satellite cell activation during muscle regeneration. Stem. Cell Res. Ther. 2011, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Renzini, A.; Marroncelli, N.; Noviello, C.; Moresi, V.; Adamo, S. HDAC4 Regulates Skeletal Muscle Regeneration via Soluble Factors. Front. Physiol. 2018, 9, 1387. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ji, S.; Li, W.; Yi, B.; Li, H.; Zhang, H.; Ma, W. LncRNA H19 promotes the differentiation of bovine skeletal muscle satellite cells by suppressing Sirt1/FoxO1. Cell Mol. Biol. Lett. 2017, 22, 102017. [Google Scholar] [CrossRef]
- Chen, X.; He, L.; Zhao, Y.; Li, Y.; Zhang, S.; Sun, K.; So, K.; Chen, F.; Zhou, L.; Lu, L.; et al. Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov. 2017, 3, 170022017. [Google Scholar] [CrossRef]
- Wang, G.Q.; Wang, Y.; Xiong, Y.; Chen, X.C.; Ma, M.L.; Cai, R.; Gao, Y.; Sun, Y.M.; Yang, G.S.; Pang, W.J. Sirt1 AS lncRNA interacts with its mRNA to inhibit muscle formation by attenuating function of miR-34a. Sci. Rep. 2016, 6, 218652016. [Google Scholar] [CrossRef]
- Zheng, L.; Liu, X.; Chen, P.; Xiao, W. Expression and role of lncRNAs in the regeneration of skeletal muscle following contusion injury [published correction appears in Exp. Ther. Med. 2020, 19, 797]. Exp. Ther. Med. 2019, 18, 2617–2627. [Google Scholar]
- Sharma, P.; Maffulli, N. Tendon injury and tendinopathy: Healing and repair. J. Bone Jt. Surg. Am. 2005, 87, 187–202. [Google Scholar]
- Cięszczyk, P.; Willard, K.; Gronek, P.; Zmijewski, P.; Trybek, G.; Gronek, J.; Weber-Rajek, M.; Stastny, P.; Petr, M.; Lulińska-Kuklik, E.; et al. Are genes encoding proteoglycans really associated with the risk of anterior cruciate ligament rupture? Biol. Sport 2017, 34, 97–103. [Google Scholar] [CrossRef]
- Abate, M.; Silbernagel, K.G.; Siljeholm, C.; Di Iorio, A.; De Amicis, D.; Salini, V.; Werner, S.; Paganelli, R. Pathogenesis of tendinopathies: Inflammation or degeneration? Arthritis Res. Ther. 2009, 11, 235. [Google Scholar] [CrossRef]
- Thankam, F.G. Epigenetic mechanisms and implications in tendon inflammation (Review). Int. J. Mol. Med. 2019, 43, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fredberg, U.; Bolvig, L.; Andersen, N.T. Prophylactic training in asymptomatic soccer players with ultrasonographic abnormalities in Achilles and patellar tendons: The Danish super league study. Am. J. Sports Med. 2008, 36, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Maffulli, N. Biology of tendon injury: Healing, modeling and remodeling. J. Musculoskelet Neuronal Interact. 2006, 6, 181–190. [Google Scholar] [PubMed]
- Kannus, P.; Natri, A. Etiology and pathophysiology of tendon ruptures in sports. Scand. J. Med. Sci. Sports 1997, 7, 107–112. [Google Scholar] [CrossRef] [PubMed]
- September, A.V.; Schwellnus, M.P.; Collins, M. Tendon and ligament injuries: The genetic component. Br. J. Sports Med. 2007, 41, 241–246. [Google Scholar] [CrossRef]
- Vaughn, N.H.; Stepanyan, H.; Gallo, R.A.; Dhawan, A. Genetic Factors in Tendon Injury: A Systematic Review of the Literature. Orthop. J. Sports Med. 2017, 5, 2325967117724416. [Google Scholar] [CrossRef]
- Gaida, J.E.; Ashe, M.; Bass, S.L.; Cook, J.L. Is adiposity an under-recognized risk factor for tendinopathy? A systematic review. Arthritis Care. Res. 2009, 61, 840–849. [Google Scholar] [CrossRef]
- Ilfeld, F.W. Can stroke modification relieve tennis elbow? Clin. Orthop. 1992, 276, 182–186. [Google Scholar] [CrossRef]
- Cottrell, J.A.; Turner, J.C.; Livingston Arinzeh, T.; O’Connor, J.P. The Biology of Bone and Ligament Healing. Foot. Ankle Clin. 2016, 21, 739–761. [Google Scholar] [CrossRef]
- Jobe, F.W.; Ciccotti, M.G. Lateral and Medial Epicondylitis of the Elbow. J. Am. Acad. Orthop. Surg. 1994, 2, 1–8. [Google Scholar] [CrossRef]
- Mokone, G.G.; Gajjar, M.; September, A.V.; Schwellnus, M.P.; Greenberg, J.; Noakes, T.D.; Collins, M. The guanine-thymine dinucleotide repeat polymorphism within the tenascin-C-gene is associated with Achilles tendon injuries. Am. J. Sports Med. 2005, 33, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Jozsa, L.; Balint, J.B.; Kannus, P.; Reffy, A.; Barzo, M. Distribution of blood groups in patients with tendon rupture. An analysis of 832 cases. J. Bone Joint. Surg. Br. 1989, 71, 272–274. [Google Scholar] [CrossRef]
- Kujala, U.M.; Järvinen, M.; Natri, A.; Lehto, M.; Nelimarkka, O.; Hurme, M.; Virta, L.; Finne, J. ABO blood groups and musculoskeletal injuries. Injury 1992, 23, 131–133. [Google Scholar] [CrossRef]
- Corps, A.N.; Robinson, A.H.; Harrall, R.L.; Avery, N.C.; Curry, V.A.; Hazleman, B.L.; Riley, G.P. Changes in matrix protein biochemistry and the expression of mRNA encoding matrix proteins and metalloproteinases in posterior tibialis tendinopathy. Ann. Rheum. Dis. 2012, 71, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Corps, A.N.; Robinson, A.H.; Movin, T.; Costa, M.L.; Ireland, D.C.; Hazleman, B.L.; Riley, G.P. Versican splice variant messenger RNA expression in normal human Achilles tendon and tendinopathies. Rheumatology 2004, 43, 969–972. [Google Scholar] [CrossRef]
- Archambault, J.M.; Jelinsky, S.A.; Lake, S.P.; Hill, A.A.; Glaser, D.L.; Soslowsky, L.J. Rat supraspinatus tendon expresses cartilage markers with overuse. J. Orthop. Res. 2007, 25, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Mohmara, Y.A.; Cook, J.; Benítez-Martínez, J.C. Influence of genetic factors in elbow tendon pathology: A case-control study. Sci. Rep. 2020, 10, 6503. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.C.; Corps, A.N.; Pennington, C.J.; Clark, I.M.; Edwards, D.R.; Bradley, M.M.; Hazleman, B.L.; Riley, G.P. Expression profiling of metalloproteinases and tissue inhibitors of metalloproteinases in normal and degenerate human achilles tendon. Arthritis Rheum. 2006, 54, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Peffers, M.J.; Fang, Y.; Cheung, K.; Wei, T.K.; Clegg, P.D.; Birch, H.L. Transcriptome analysis of ageing in uninjured human Achilles tendon. Arthritis Res. Ther. 2015, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Kambouris, M.; Ntalouka, F.; Ziogas, G.; Maffulli, N. Predictive genomics DNA profiling for athletic performance. Recent Pat. DNA Gene. Seq. 2012, 6, 229–239. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stastny, P.; Lehnert, M.; De Ste Croix, M.; Petr, M.; Svoboda, Z.; Maixnerova, E.; Varekova, R.; Botek, M.; Petrek, M.; Kocourkova, L.; et al. Effect of COL5A1, GDF5, and PPARA Genes on a Movement Screen and Neuromuscular Performance in Adolescent Team Sport Athletes. J. Strength Cond. Res. 2019, 8, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Page, P. Shoulder muscle imbalance and subacromial impingement syndrome in overhead athletes. Int. J. Sports Phys. Ther. 2011, 6, 51–58. [Google Scholar] [PubMed]
- Guo, J.; Zhang, J.F.; Li, G.; Chan, K.M. A Mini-review: MicroRNA in tendon injuries. J. Stem. Cell Res. Ther. 2015, 5, 303. [Google Scholar]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Porta, G.D.; Peretti, G.M.; Maffulli, N. Therapeutic potential of microRNA in tendon injuries. Br. Med. Bull. 2020, 133, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Nakasa, T.; Nagata, Y.; Yamasaki, K.; Ochi, M. A mini-review: microRNA in arthritis. Physiol. Genom. 2011, 43, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.L.; Gilchrist, D.S.; Akbar, M.; Reilly, J.H.; Kerr, S.C.; Campbell, A.L.; Murrell, G.A.C.; Liew, F.Y.; Kurowska-Stolarska, M.; McInnes, I.B. MicroRNA29a regulates IL-33-mediated tissue remodelling in tendon disease. Nat. Commun. 2015, 6, 6774. [Google Scholar] [CrossRef] [PubMed]
- Plachel, F.; Heuberer, P.; Gehwolf, R.; Frank, J. MicroRNA Profiling Reveals Distinct Signatures in Degenerative Rotator Cuff Pathologies. J. Orthop. Res. 2020, 38, 202–211. [Google Scholar] [CrossRef]
- Takata, A.; Otsuka, M.; Kojima, K.; Yoshikawa, T.; Kishikawa, T.; Yoshida, H.; Koike, K. MicroRNA-22 and microRNA-140 suppress NF-κB activity by regulating the expression of NF-κB coactivators. Biochem. Biophys. Res. Commun. 2011, 411, 826–831. [Google Scholar] [CrossRef]
- Thankam, F.G.; Boosani, C.S.; Dilisio, M.F.; Gross, R.M.; Agrawal, D.K. Genes interconnecting AMPK and TREM-1 and associated microRNAs in rotator cuff tendon injury. Mol. Cell Biochem. 2019, 454, 97–109. [Google Scholar] [CrossRef]
- Ge, H.; Shrestha, A.; Liu, C.; Wu, P.; Cheng, B. MicroRNA 148a-3p promotes Thrombospondin-4 expression and enhances angiogenesis during tendinopathy development by inhibiting Krüppel-like factor 6. Biochem. Biophys. Res. Commun. 2018, 502, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gai, F.; Chen, H. MiR-30b-5p Influences Chronic Exercise Arthritic Injury by Targeting Hoxa1. Int. J. Sports Med. 2021, 42, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Peffers, M.J.; Goljanek-Whysall, K.; Collins, J. Decoding the Regulatory Landscape of Ageing in Musculoskeletal Engineered Tissues Using Genome-Wide DNA Methylation and RNASeq. PLoS ONE 2016, 11, e0160517. [Google Scholar] [CrossRef] [PubMed]
- Waki, T.; Lee, S.Y.; Niikura, T.; Iwakura, T.; Dogaki, Y.; Okumachi, E.; Oe, K.; Kuroda, R.; Kurosaka, M. Profiling microRNA expression during fracture healing. BMC Musculoskelet. Disord. 2016, 17, 83. [Google Scholar] [CrossRef]
- Donati, S.; Ciuffi, S.; Palmini, G.; Brandi, M.L. Circulating miRNAs: A New Opportunity in Bone Fragility. Biomolecules 2020, 10, 927. [Google Scholar] [CrossRef]
- Bellavia, D.; Salamanna, F.; Raimondi, L.; De Luca, A.; Carina, V.; Costa, V.; Alessandro, R.; Fini, M.; Giavaresi, G. Deregulated miRNAs in osteoporosis: Effects in bone metastasis. Cell Mol. Life Sci. 2019, 76, 3723–3744. [Google Scholar] [CrossRef]
- Michou, L. Epigenetics in bone diseases. Jt. Bone Spine 2018, 85, 701–707. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Houchen, C.W.; Kennedy, S.; Dieckgraefe, B.K.; Anant, S. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol. Cell 2003, 11, 113–126. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Tang, J. MiR-30a: A novel biomarker and potential therapeutic target for cancer. J. Oncol. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Nakagawa, R.; Muroyama, R.; Saeki, C.; Goto, K.; Kaise, Y.; Koike, K.; Nakano, M.; Matsubara, Y.; Takano, K.; Ito, S.; et al. miR-425 regulates inflammatory cytokine production in CD4(+) T cells via N-Ras upregulation in primary biliary cholangitis. J. Hepatol. 2017, 66, 1223–1230. [Google Scholar] [CrossRef]
- Mendias, C.L.; Gumucio, J.P.; Bakhurin, K.I.; Lynch, E.B.; Brooks, S.V. Physiological loading of tendons induces scleraxis expression in epitenon fibroblasts. J. Orthop. Res. 2012, 30, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Rickaby, R.; El Khoury, L.Y.; Samiric, T.; Raleigh, S.M. Epigenetic Status of The Human MMP11 Gene Promoter is Altered in Patellar Tendinopathy. J. Sports Sci. Med. 2019, 18, 155–159. [Google Scholar] [PubMed]
- El Khoury, L.Y.; Rickaby, R.; Samiric, T.; Raleigh, S.M. Promoter methylation status of the TIMP2 and ADAMTS4 genes and patellar tendinopathy. J. Sci. Med. Sport 2018, 21, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Chen, Z.-Y.; Lin, Y.-H.; Chen, S.-H.; Chou, P.-Y.; Kao, H.-K.; Lin, F.-H. Extracellular Vesicles of Adipose-Derived Stem Cells Promote the Healing of Traumatized Achilles Tendons. Int. J. Mol. Sci. 2021, 22, 12373. [Google Scholar] [CrossRef] [PubMed]
- Trumble, T.E.; Budoff, J.E.; Cornwall, R. Hand, Elbow & Shoulder: Core Knowledge in Orthopaedics. Chapter 12 Tendon Trauma. Edra Urban Partn. 2017, 210–211. [Google Scholar]
- Dai, G.C.; Li, Y.J.; Chen, M.H.; Lu, P.P.; Rui, Y.F. Tendon stem/progenitor cell ageing: Modulation and rejuvenation. World J. Stem. Cells 2019, 11, 677–692. [Google Scholar] [CrossRef]
- Ding, L.; Wang, M.; Qin, S.; Xu, L. The Roles of MicroRNAs in Tendon Healing and Regeneration. Front. Cell Dev. Biol. 2021, 9, 687117. [Google Scholar] [CrossRef]
- Riasat, K.; Bardell, D.; Goljanek-Whysall, K.; Clegg, P.D.; Peffers, M.J. Epigenetic mechanisms in Tendon Ageing. Br. Med. Bull. 2020, 135, 90–107. [Google Scholar] [CrossRef]
- Lu, Y.F.; Liu, Y.; Fu, W.M.; Xu, J.; Wang, B.; Sun, Y.X.; Wu, T.Y.; Xu, L.L.; Chan, K.M.; Zhang, J.F.; et al. Long noncoding RNA H19 accelerates tenogenic differentiation and promotes tendon healing through targeting miR-29b-3p and activating TGF-β1 signaling. FASEB J. 2017, 31, 954–964. [Google Scholar] [CrossRef]
- Wada, S.; Ideno, H.; Shimada, A.; Kamiunten, T.; Nakamura, Y.; Nakashima, K.; Kimura, H.; Shinkai, Y.; Tachibana, M.; Nifuji, A. H3K9MTase G9a is essential for the differentiation and growth of tenocytes in vitro. Histochem. Cell Biol. 2015, 144, 13–20. [Google Scholar] [CrossRef]
- Webb, S.; Gabrelow, C.; Pierce, J.; Gibb, E.; Elliott, J. Retinoic acid receptor signaling preserves tendon stem cell characteristics and prevents spontaneous differentiation in vitrox. Stem. Cell Res. Ther. 2016, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Oryan, A.; Monazzah, S.; Bigham-Sadegh, A. Bone Injury and Fracture Healing Biology. Biomed. Environ. Sci. 2015, 28, 57–71. [Google Scholar] [PubMed]
- Ackerman, K.E.; Nazem, T.; Chapko, D.; Russell, M.; Mendes, N.; Taylor, A.P.; Bouxsein, M.L.; Misra, M. Bone microarchitecture is impaired in adolescent amenorrheic athletes compared with eumenorrheic athletes and nonathletic controls. J. Clin. Endocrinol. Metab. 2011, 96, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Bennell, K.L.; Malcolm, S.A.; Thomas, S.A.; Wark, J.D.; Brukner, P.D. The incidence and distribution of stress fractures in competitive track and field athletes: A twelve-month prospective study. Am. J. Sports Med. 1996, 24, 211–217. [Google Scholar] [CrossRef]
- Changstrom, B.G.; Brou, L.; Khodaee, M.; Braund, C.; Comstock, R.D. Epidemiology of stress fracture injuries among us high school athletes, 2005–2006, through 2012–2013. Am. J. Sports Med. 2015, 43, 26–33. [Google Scholar] [CrossRef]
- Goolsby, M.A.; Barrack, M.T.; Nattiv, A. A displaced femoral neck stress fracture in an amenorrheic adolescent female runner. Sports Health 2012, 4, 352–356. [Google Scholar] [CrossRef]
- Fredericson, M.; Jennings, F.; Beaulieu, C.; Matheson, G.O. Stress fractures in athletes. Top. Magn. Reson. Imaging 2006, 17, 309–325. [Google Scholar] [CrossRef]
- Greaser, M.C. Foot and Ankle Stress Fractures in Athletes. Orthop. Clin. N. Am. 2016, 47, 809–822. [Google Scholar] [CrossRef]
- Harrast, M.A.; Colonno, D. Stress fractures in runners. Clin. Sports Med. 2010, 29, 399–416. [Google Scholar] [CrossRef]
- MacKnight, J.M. Osteopenia and Osteoporosis in Female Athletes. Clin. Sports Med. 2017, 36, 687–702. [Google Scholar] [CrossRef]
- Herbert, A.J.; Williams, A.G.; Hennis, P.J.; Erskine, R.M.; Sale, C.; Day, S.H.; Stebbings, G.K. The interactions of physical activity, exercise and genetics and their associations with bone mineral density: Implications for injury risk in elite athletes. Eur. J. Appl. Physiol. 2019, 119, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Hornberger, T.A.; Robling, A.G. Bone and skeletal muscle: Key players in mechanotransduction and potential overlapping mechanisms. Bone 2015, 80, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Elliott-Sale, J.K.; Sale, C. Exercise and bone health across the lifespan. Biogerontology 2017, 18, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Zhong, Z.; Williams, B.O.; Yang, Y. Wnt signaling in bone development and disease: Making stronger bone with wnts. Cold Spring Harb. Perspect Biol. 2012, 4, a007997. [Google Scholar] [CrossRef] [PubMed]
- Sibonga, J.D. Spaceflight-induced bone loss: Is there an osteoporosis risk? Curr. Osteoporos. Rep. 2013, 11, 92–98. [Google Scholar] [CrossRef]
- Grimes, R.; Jepsen, K.J.; Fitch, J.L.; Einhorn, T.A.; Gerstenfeld, L.C. The transcriptome of fracture healing defines mechanisms of coordination of skeletal and vascular development during endochondral bone formation. J. Bone Miner. Res. 2011, 26, 2597–2609. [Google Scholar] [CrossRef]
- Botor, M.; Fus-Kujawa, A.; Uroczynska, M.; Stepien, K.L.; Galicka, A.; Gawron, K.; Sieron, A.L. Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules 2021, 11, 1493. [Google Scholar] [CrossRef]
- Friedman, E.; Moran, D.S.; Ben-Avraham, D.; Yanovich, R.; Atzmon, G. Novel candidate genes putatively involved in stress fracture predisposition detected by whole-exome sequencing. Genet. Res. (Camb.) 2014, 96, e004. [Google Scholar] [CrossRef]
- Varley, I.; Hughes, D.C.; Greeves, J.P.; Stellingwerff, T.; Ranson, C.; Fraser, W.D.; Sale, C. The association of novel polymorphisms with stress fracture injury in Elite Athletes: Further insights from the SFEA cohort. J. Sci. Med. Sport 2018, 21, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Varley, I.; Greeves, J.P.; Sale, C.; Friedman, E.; Moran, D.S.; Yanovich, R.; Wilson, P.J.; Gartland, A.; Hughes, D.C.; Stellingwerff, T.; et al. Functional polymorphisms in the P2X7 receptor gene are associated with stress fracture injury. Purinergic Signal 2016, 12, 103–113. [Google Scholar] [CrossRef]
- Brennan-Olsen, S.L.; Page, R.S.; Berk, M.; Riancho, J.A.; Leslie, W.D.; Wilson, S.G.; Saban, K.L.; Janusek, L.; Pasco, J.A.; Hodge, J.M.; et al. DNA methylation and the social gradient of osteoporotic fracture: A conceptual model. Bone 2016, 84, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.I.; Kim, H.S.; Jung, Y.C.; Kim, Y.H.; Hong, S.J.; Kim, M.K.; Baek, K.H.; Kim, C.C.; Rhyu, M.G. Transitional CpG methylation between promoters and retroelements of tissue-specific genes during human mesenchymal cell differentiation. J. Cell Biochem. 2007, 102, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Yasui, T.; Hirose, J.; Aburatani, H.; Tanaka, S. Epigenetic regulation of osteoclast differentiation. Ann. N. Y. Acad. Sci. 2011, 1240, 7–13. [Google Scholar] [CrossRef]
- Steeve, K.T.; Marc, P.; Sandrine, T.; Dominique, H.; Yannick, F. IL-6, RANKL, TNFalpha/IL-1: Interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004, 15, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Ito, H.; Yoshitomi, H.; Yamamoto, K.; Fukuda, A.; Yoshikawa, J.; Furu, M.; Ishikawa, M.; Shibuya, H.; Matsuda, S. Inhibition of miR-92a enhances fracture healing via promoting angiogenesis in a model of stabilized fracture in young mice. J. Bone Miner. Res. 2014, 29, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Meng, F.Y.; Zhao, Y.H.; Jin, C.L.; Tian, J.; Yi, X.J. Inhibition of microRNA-214-5p promotes cell survival and extracellular matrix formation by targeting collagen type IV alpha 1 in osteoblastic MC3T3-E1 cells. Bone Jt. Res. 2017, 6, 464–471. [Google Scholar] [CrossRef]
- Huang, M.; Xu, S.; Liu, L.; Zhang, M. m6A Methylation Regulates Osteoblastic Differentiation and Bone Remodeling. Front. Cell Dev. Biol. 2021, 9, 783322. [Google Scholar] [CrossRef]
- del Real, A.; Pérez-Campo, F.M.; Fernández, A.F.; Sañudo, C.; Ibarbia, C.G.; Pérez-Núñez, M.I.; Criekinge, W.V.; Braspenning, M.; Alonso, M.A.; Fraga, M.F.; et al. Differential analysis of genome-wide methylation and gene expression in mesenchymal stem cells of patients with fractures and osteoarthritis. Epigenetics 2017, 12, 113–122. [Google Scholar] [CrossRef]
- Hadjiargyrou, M.; Zhi, J.; Komatsu, D.E. Identification of the microRNA transcriptome during the early phases of mammalian fracture repair. Bone 2016, 87, 78–88. [Google Scholar] [CrossRef]
- Lin, H.-N.; Cottrell, J.; O’Connor, J.P. Variation in lipid mediator and cytokine levels during mouse femur fracture healing. J. Orthop. Res. 2016, 34, 1883–1893. [Google Scholar] [CrossRef]
- Farr, J.N.; Khosla, S. Cellular senescence in bone. Bone 2019, 121, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Deng, Z.; Liu, J. The Mechanism of Bone Remodeling After Bone Aging. Clin. Interv. Aging 2022, 17, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yang, W.; Zhao, H.; Liu, K.; Deng, A.; Zhang, G.; Pan, K. Abnormal expression of miR-135b-5p in bone tissue of patients with osteoporosis and its role and mechanism in osteoporosis progression. Exp. Ther. Med. 2020, 19, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.H.; Miclau, T.; Chow, S.K.-H.; Yang, F.F.; Alt, V. Fracture healing in osteoporotic bone. Injury 2016, 47, S21–S26. [Google Scholar] [CrossRef]
- Groven, R.V.M.; van Koll, J.; Poeze, M.; Blokhuis, T.J.; van Griensven, M. miRNAs Related to Different Processes of Fracture Healing: An Integrative Overview. Front. Surg. 2021, 8, 786564. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M. MicroRNA and fracture healing. Calcif. Tissue Int. 2017, 101, 355–361. [Google Scholar] [CrossRef]
- Xu, F.; Li, W.; Yang, X.; Na, L.; Chen, L.; Liu, G. The Roles of Epigenetics Regulation in Bone Metabolism and Osteoporosis. Front. Cell Dev. Biol. 2021, 8, 619301. [Google Scholar] [CrossRef]
- Lee, W.Y.; Li, N.; Lin, S.; Wang, B.; Lan, H.Y.; Li, G. miRNA-29b improves bone healing in mouse fracture model. Mol. Cell Endocrinol. 2016, 430, 97–107. [Google Scholar] [CrossRef]
- Compton, J.T.; Lee, F.Y. A review of osteocyte function and the emerging importance of sclerostin. J. Bone Jt. Surg. Am. 2014, 96, 1659–1668. [Google Scholar] [CrossRef]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Zhou, G.S.; Zhang, X.L.; Wu, J.P.; Zhang, R.P.; Xiang, L.X.; Dai, L.C.; Shao, J.Z. 5-Azacytidine facilitates osteogenic gene expression and differentiation of mesenchymal stem cells by alteration in DNA methylation. Cytotechnology 2009, 60, 11. [Google Scholar] [CrossRef] [PubMed]
- Farshdousti Hagh, M.; Noruzinia, M.; Mortazavi, Y.; Soleimani, M.; Kaviani, S.; Abroun, S.; Dehghani Fard, A.; Mahmoodinia, M. Different methylation patterns of RUNX2, OSX, DLX5 and BSP in osteoblastic differentiation of mesenchymal stem cells. Cell J. 2015, 17, 71–82. [Google Scholar] [PubMed]
- Thaler, R.; Agsten, M.; Spitzer, S.; Paschalis, E.P.; Karlic, H.; Klaushofer, K.; Varga, F. Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J. Biol. Chem. 2011, 286, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- Wakitani, S.; Yokoi, D.; Hidaka, Y.; Nishino, K. The differentially DNA methylated region responsible for expression of runt-related transcription factor 2. J. Vet. Med. Sci. 2017, 79, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Licini, C.; Vitale-Brovarone, C.; Mattioli-Belmonte, M. Collagen and non-collagenous proteins molecular crosstalk in the pathophysiology of osteoporosis. Cytokine Growth Fact. Rev. 2019, 49, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Westendorf, J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2005, 20, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, W.; Chen, L.; Tang, K. Mechanism of osteogenic and adipogenic differentiation of tendon stem cells induced by sirtuin 1. Mol. Med. Rep. 2016, 14, 1643–1648. [Google Scholar] [CrossRef]
- Yasui, T.; Hirose, J.; Tsutsumi, S.; Nakamura, K.; Aburatani, H.; and Tanaka, S. Epigenetic regulation of osteoclast differentiation: Possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J. Bone Miner. Res. 2011, 26, 2665–2671. [Google Scholar] [CrossRef]
- Fei, Q.; Bai, X.; Lin, J.; Meng, H.; Yang, Y.; Guo, A. Identification of aberrantly expressed long non-coding RNAs in postmenopausal osteoporosis. Int. J. Mol. Med. 2018, 41, 3537–3550. [Google Scholar] [CrossRef]
- Ray, S.K.; Nixon, C.E. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol. Histopathol. 2002, 17, 1137–1152. [Google Scholar]
- Di Pietro, V.; Yakoub, K.M. Antioxidant Therapies in Traumatic Brain Injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Mahon, S. Incidence of Sports-Related Traumatic Brain Injury of All Severities: A Systematic Review. Neuroepidemiology 2020, 54, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, B.; Sagarkar, S. Epigenetic Mechanisms of Traumatic Brain Injuries. Prog. Mol. Biol. Transl. Sci. 2018, 157, 263–298. [Google Scholar] [PubMed]
- Pearn, M.L.; Niesman, I.R. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of sport. Acta Neuropathol. 2014, 127, 29–51. [Google Scholar] [CrossRef]
- Pavlovic, D.; Pekic, S. Traumatic brain injury: Neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 2019, 22, 270–282. [Google Scholar] [CrossRef]
- Brazinova, A.; Rehorcikova, V. Epidemiology of Traumatic Brain Injury in Europe: A Living Systematic Review. J. Neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef]
- Sherer, M.; Struchen, M.A. Comparison of indices of traumatic brain injury severity: Glasgow Coma Scale, length of coma and post-traumatic amnesia. J. Neurol. Neurosurg. Psychiatry 2008, 79, 678–685. [Google Scholar] [CrossRef]
- Selassie, A.W.; Wilson, D.A. Incidence of sport-related traumatic brain injury and risk factors of severity: A population-based epidemiologic study. Ann. Epidemiol. 2013, 23, 750–756. [Google Scholar] [CrossRef]
- VanItallie, T.B. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 2019, 100S, 153943. [Google Scholar] [CrossRef]
- Weiner, M.W.; Crane, P.K. Traumatic brain injury may not increase the risk of Alzheimer disease. Neurology 2017, 89, 1923–1925. [Google Scholar] [CrossRef]
- Sussman, E.S.; Pendharkar, A.V. Mild traumatic brain injury and concussion: Terminology and classification. Handb. Clin. Neurol. 2018, 158, 21–24. [Google Scholar]
- Ladak, A.A.; Enam, S.A. A Review of the Molecular Mechanisms of Traumatic Brain Injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bao, Y. The Association Between Apolipoprotein E and Functional Outcome After Traumatic Brain Injury: A Meta-Analysis. Medicine 2015, 46, e2028. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, C.A.; Zeiler, F.A. Apolipoprotein E4 Polymorphism and Outcomes from Traumatic Brain Injury: A Living Systematic Review and Meta-Analysis. J. Neurotrauma 2021, 38, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Maiti, T.K.; Konar, S. Role of apolipoprotein E polymorphism as a prognostic marker in traumatic brain injury and neurodegenerative disease: A critical review. Neurosurg. Focus 2015, 39, E3. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.R.; Reuter-Rice, K. Genetic Influences in Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: Oxfordshire, UK, 2016. [Google Scholar]
- Terrell, T.R.; Bostick, R.M. APOE, APOE promoter, and Tau genotypes and risk for concussion in college athletes. Clin. J. Sport Med. 2008, 18, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hiskens, M.I.; Angoa-Pérez, M. Modeling sports-related mild traumatic brain injury in animals-A systematic review. J. Neurosci. Res. 2019, 97, 1194–1222. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Zhang, Z. Global hypomethylation defines a sub- population of reactive microglia/macrophages in experimental traumatic brain injury. Neurosci. Lett. 2007, 429, 1–6. [Google Scholar] [CrossRef]
- Wong, V.S.; Langley, B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci. Lett. 2016, 625, 26–33. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S. Long-term impact of mild traumatic brain injuries on multiple functional outcomes and epigenetics: A pilot study with college students. Appl. Sci. 2020, 10, 4131. [Google Scholar] [CrossRef]
- Hamdeh, S.A.; Ciuculete, D.M. Differential DNA Methylation of the Genes for Amyloid Precursor Protein, Tau, and Neurofilaments in Human Traumatic Brain Injury. J. Neurotrauma 2021, 38, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zusman, B.E.; Shaffer, J.R.; Li, Y.; Arockiaraj, A.I.; Liu, S.; Weeks, D.E.; Desai, S.M.; Kochanek, P.M.; Puccio, A.M.; et al. Decreased DNA Methylation of RGMA is Associated with Intracranial Hypertension After Severe Traumatic Brain Injury: An Exploratory Epigenome-Wide Association Study. Neurocrit. Care 2022, 37, 26–37. [Google Scholar] [CrossRef]
- Lundberg, J.; Karimi, M. Traumatic brain injury induces relocalization of DNA-methyltransferase 1. Neurosci. Lett. 2009, 457, 8–11. [Google Scholar] [CrossRef]
- Sagarkar, S.; Bhamburkar, T. Minimal traumatic brain injury causes persistent changes in DNA methylation at BDNF gene promoters in rat amygdala: A possible role in anxiety-like behaviors. Neurobiol. Dis. 2017, 106, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, M.S.; Winter, W.C. Sleep disturbances in athletic concussion. Brain Inj. 2015, 29, 221–227. [Google Scholar] [CrossRef]
- Aoun, R.; Rawal, H. Impact of traumatic brain injury on sleep: An overview. Nat. Sci. Sleep 2019, 11, 131–140. [Google Scholar] [CrossRef]
- Shein, N.A.; Grigoriadis, N. Histone deacetylase inhibitor ITF2357, is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 2009, 23, 4266–4275. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, X. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: Scriptaid protects against TBI via AKT. Neurotherapeutics 2013, 10, 124–142. [Google Scholar] [CrossRef]
- Dash, P.K.; Orsi, S.A. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience 2009, 163, 1–8. [Google Scholar] [CrossRef]
- Sada, N.; Fujita, Y. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell. Death Dis. 2020, 11, 655. [Google Scholar] [CrossRef]
- Meza-Sosa, K.F.; Valle-García, D. Role of microRNAs in central nervous system development and pathology. J. Neurosci. Res. 2012, 90, 1–12. [Google Scholar] [CrossRef]
- Hicks, S.D.; Ledy, J. Defining Biological Phenotypes of Mild Traumatic Brain Injury Using Saliva MicroRNA Profiles. J. Neurotrauma 2022, 39, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Jiao, J. MicroRNA-21 in the Pathogenesis of Traumatic Brain Injury. Neurochem. Res. 2018, 43, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, M. Neuroprotective effects of miR-27a against traumatic brain injury via suppressing FoxO3a-mediated neuronal autophagy. Biochem. Biophys. Res. Commun. 2017, 482, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Fonken, L.K. MicroRNAs: Roles in Regulating Neuroinflammation. Neuroscientist 2018, 24, 221–245. [Google Scholar] [CrossRef]
- Lv, J.; Zeng, Y. MicroRNA let-7c-5p improves neurological outcomes in a murine model of traumatic brain injury by suppressing neuroinflammation and regulating microglial activation. Brain. Res. 2018, 1685, 91–104. [Google Scholar] [CrossRef]
- Di Pietro, V.; Porto, E.; Ragusa, M.; Barbagallo, C.; Davies, D.; Forcione, M.; Logan, A.; Di Pietro, C.; Purrello, M.; Grey, M.; et al. Salivary MicroRNAs: Diagnostic Markers of Mild Traumatic Brain Injury in Contact-Sport. Front. Mol. Neurosci. 2018, 11, 290. [Google Scholar] [CrossRef]
- Svingos, A.M.; Asken, B.M.; Bauer, R.M.; DeKosky, S.T.; Hromas, G.A.; Jaffee, M.S.; Hayes, R.L.; Clugston, J.R. Exploratory study of sport-related concussion effects on peripheral micro-RNA expression. Brain Inj. 2019, 33, 1–7. [Google Scholar] [CrossRef]
- Shultz, S.R.; Taylor, C.J.; Aggio-Bruce, R.; O’Brien, W.T.; Sun, M.; Cioanca, A.V.; Neocleous, G.; Symons, G.F.; Brady, R.D.; Hardikar, A.A.; et al. Decrease in Plasma miR-27a and miR-221 After Concussion in Australian Football Players. Biomarker Insights 2022, 17, 11772719221081318. [Google Scholar] [CrossRef]
- Papa, L.; Slobounov, S.M.; Breiter, H.C.; Walter, A.; Bream, T.; Seidenberg, P.; Bailes, J.E.; Bravo, S.; Johnson, B.; Kaufman, D.; et al. Elevations in MicroRNA Biomarkers in Serum Are Associated with Measures of Concussion, Neurocognitive Function, and Subconcussive Trauma over a Single National Collegiate Athletic Association Division I Season in Collegiate Football Players. J. Neurotrauma 2019, 36, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Sandmo, S.B.; Matyasova, K. Changes in circulating microRNAs following head impacts in soccer. Brain Inj. 2022, 36, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Balakathiresan, N.; Bhomia, M. MicroRNA let-7i is a promising serum biomarker for blast-induced traumatic brain injury. J. Neurotrauma 2012, 29, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xu, J. MicroRNA-23a/b and microRNA-27a/b suppress Apaf-1 protein and alleviate hypoxia-induced neuronal apoptosis. Cell Death Dis. 2014, 5, e1132. [Google Scholar] [CrossRef]
- McDonald, R.P.; Horsburgh, K.J.; Graham, D.I.; Nicoll, J.A. Mitochondrial DNA deletions in acute brain injury. Neuroreport 1999, 10, 1875–1878. [Google Scholar] [CrossRef]
- Hicks, S.D.; Olympia, R.P.; Onks, C.; Kim, R.Y.; Zhen, K.J.; Fedorchak, G.; DeVita, S.; Rangnekar, A.; Heller, M.; Zwibel, H.; et al. Saliva microRNA Biomarkers of Cumulative Concussion. Int. J. Mol. Sci. 2020, 21, 7758. [Google Scholar] [CrossRef]



| Type of miRNA | Reference |
|---|---|
| ↑miR-1, ↑miR-126, ↑miR-133a, ↑miR-206, ↑miR-208, ↑miR-146a, ↑miR-221, ↑miR-222, ↑miR-499, mir-223, ↑miR-24, ↑miR-149, ↑miR-30a, ↑miR-125a-5p | [98,99,100,101,102,103,104,105,106,107,108] |
| ↓miR-26a, ↓miR-29b, ↓miR-15b | [100,105] |
| Type of miRNA | Results and Main Conclusion | Reference |
|---|---|---|
| miR-29b | Deterioration of tendon adhesion; | [147,148,149] |
| miR-28, miR-17-92 | mediate oxidative stress and induce tenocyte apoptosis; | |
| miR-135a | regulates TSPC senescence by targeting ROCK1; | |
| miR-608, miR-499 | related to pathogenesis of tendinopathy; regulate MYB and CUGBP2; | |
| miR-210 | regulates angiogenesis; | |
| miR-378, miR-133, miR-206, miR-140, let-7a, let-7e, miR-338, miR-381, miR-743 | related to mechanical stimuli | |
| miR-421-5p | Regulates the local expression of MMP2, MMP9, and drives angiogenesis by increasing VEGF | [150] |
| miR-30a-5p, miR-140-3p, miR-210-3p, miR-222-3p, miR-324-3p, miR-425-5p | Deregulated in patients in chronic tendinopathy; | [151,152] |
| miR-29c | progressive repression depending on the severity of the tendon pathology | [152] |
| miR-324 | Inhibits the expressions of MMP-2 and MMP-9 and might promote tendon disorganization | |
| miR-140-3p | Negatively regulates nuclear factor-κB (NF-κB) inflammatory signalling. | [153] |
| hsa-miR-145-5p, hsa-miR-99a-5p, hsa-miR-100-5p, hsa-miR-150-5p, hsa-miR-193b-3p, hsa-miR-103a-3p, hsa-miR-31-5p, hsa-miR-195-5p, hsa-miR-497-5p, hsa-miR-15a-5p, hsa-miR-16-5p, hsa-let-7b-5p | Downregulate miRNA associated with 216 genes in rotator cuff tendon injury | [154] |
| hsa-miR-297 | Upregulates miRNA associated with 216 genes in rotator cuff tendon injury | |
| miR-148a-3p | Promotes thrombospondin-4 expression and enhances angiogenesis during tendinopathy; | [155] |
| miR-124 | suppresses collagen formation of human tendon | |
| miR-124-3p | Directly binds and suppresses the expression of EGR1 and suppresses the synthesis of collagen during tenogenic differentiation | [149] |
| miR-30b-5p | Involved in cartilage degradation in rats and regulates chondrocyte apoptosis and migration by targeting Hoxa1 | [156] |
| mir-500, miR-548j | Regulates the processes associated with matrix remodelling | [157] |
| miR-214-5p | Regulates osteoblastic cell viability and apoptosis | [156] |
| miR-140–3p, miR-140-5p, miR-181a-5p, miR-181d-5p, miR-451 | Highly expressed miRNAs in standard healing fractures on day 14 | [158] |
| miR-21-5p, miR-23a-3p, miR-24-3p, miR-25-3p, miR-27a-3p, miR-29, miR-31, miR-100-5p, miR-122a-5p, miR-124-3p, miR-125b-5p, miR-148a-3p, miR-223-3p, miR-3679-3p, miR-4274 | Associated with the development of osteoporosis and bone fracture risk | [159,160,161] |
| Type of miRNA | Functional Activity | Reference |
|---|---|---|
| miR-1, miR-21, miR-28-5p, miR-34 family, miR-100, miR-133a, miR-133b, miR-205, miR-221, miR-222, miR-337-3p, miR-378 | Tendon homeostasis | [147,171] |
| miR-34 family, miR-199 family, miR-205-5p, miR-499 | Proliferation | [171,172] |
| miR-21-5p, miR-21a-3p, miR-29b, miR-34 family | Tendon adhesion | [147,171] |
| miR-21-5p, miR-34 family, miR-125a-5p, miR-145-5p, miR-151a-3p, miR-199a-5p, miR-382-5p, miR-498 | Tendon ECM | [147,171] |
| miR-17-92, miR-28, miR-34 family, miR-181 family | Apoptosis | [150,171,172] |
| miR-199 family | Cell survival | [172] |
| Biological Material Analysed | Examined Group | Type of miRNA | Results and Main Conclusion | Ref. |
|---|---|---|---|---|
| saliva | A total of 6 rugby professional and semi-professional athletes and 6 controls | miR-27b-3p miR-142-3p miR-let-7i-5p miR-107 miR-135b-5p | Expression was significantly upregulated in concussed athletes; univariate ROC curve analysis showed that the differentially expressed miRNAs could be considered good classifiers of concussion. | [263] |
| serum | A total of 27 collegiate athletes after sports-related concussion (~41% ♂, ~75% ♀ white, age 18.8 ± 0.8 years) | miR-153-3p miR-223-3p miR-26a-5p miR-423-3p miR-let-7a-5p | Significant increase in expression following SRC for miR153-3p (59% of the participants increased post-SRC), miR223-3p (70% increased), miR-let-7a-5p (65% increased); no statistically significant associations between changes in miRNA expression and clinical test scores, acute symptom severity, or clinical recovery time. | [264] |
| plasma | A total of 28 amateur Australian rules football players after sports-related concussions (20 ♂ and 8 ♀) and the control group, 99 Australian rules; football players (62 ♂, 37 ♀) | miR-19b-1-5p miR-20b-5p miR-21-5p miR-27a-3p miR-28-5p miR-103a-3p miR-106a-5p miR-125-5p miR-142-3p miR-194-5p miR-221-3p miR-223-3p miR-301b miR-338-5p miR-643 miR-769-5p miR-1260a miR-1290 | miR-27a and miR-221 were decreased in the sub-acute stages after SRC; plasma levels of these miRNAs were inversely correlated with SRC symptom severity. | [265] |
| Serum | A total of 53♂ (30 non-athlete control subjects and 23 collegiate student football athletes) | miR-20a miR-505* miR-362-3p miR-30d miR-92a miR-486 miR-195 miR-9-3p miR-151-5p | In athletes with declining neurocognitive functioning over the season, concentrations of miRNAs increased. Significant negative correlations with miR-505*, miR-30d, miR-92; miRNAs correlating with balance problems: miR-505*, miR-30d, miR-151-5p; correlating with poor reaction times: miR-20a, miR-505*, miR-30d, miR-92, and miR-151-5p. | [266] |
| Serum | Professional soccer players (44 after accidental head impact and 68 after repetitive headers), controls—from a bank of serum (young healthy individuals) | miR-1-3p miR-7-5p miR-16-p miR-17-5p miR-18a-5p miR-20a-5p miR-24-3p miR-27a-3p miR-93-5p miR-106b-5p miR-107 miR-130b-3p miR-122-5p mi-143-3p miR-150-5p miR-204-5p miR-206 miR-499a-5p miR-885-5p let-7c-5p | Dysregulation of expression depends on the type of injury (accidental head impacts or repetitive headers) and time (1 or 12 h after injury) | [267] |
| saliva | A total of 310 individuals: with no history of concussion (n = 230), single concussion (n = 56), recurrent concussion (n = 24) | 20 miRNAs | miR-28-3p and miR-339-3p demonstrated relationships with the number of prior concussions | [258] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarnowski, M.; Tomasiak, P.; Tkacz, M.; Zgutka, K.; Piotrowska, K. Epigenetic Alterations in Sports-Related Injuries. Genes 2022, 13, 1471. https://doi.org/10.3390/genes13081471
Tarnowski M, Tomasiak P, Tkacz M, Zgutka K, Piotrowska K. Epigenetic Alterations in Sports-Related Injuries. Genes. 2022; 13(8):1471. https://doi.org/10.3390/genes13081471
Chicago/Turabian StyleTarnowski, Maciej, Patrycja Tomasiak, Marta Tkacz, Katarzyna Zgutka, and Katarzyna Piotrowska. 2022. "Epigenetic Alterations in Sports-Related Injuries" Genes 13, no. 8: 1471. https://doi.org/10.3390/genes13081471
APA StyleTarnowski, M., Tomasiak, P., Tkacz, M., Zgutka, K., & Piotrowska, K. (2022). Epigenetic Alterations in Sports-Related Injuries. Genes, 13(8), 1471. https://doi.org/10.3390/genes13081471