
Evolution of the Noncoding Features of Sea Snake Mitochondrial Genomes within Elapidae


Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Sequencing of mtDNAs
2.3. Annotation
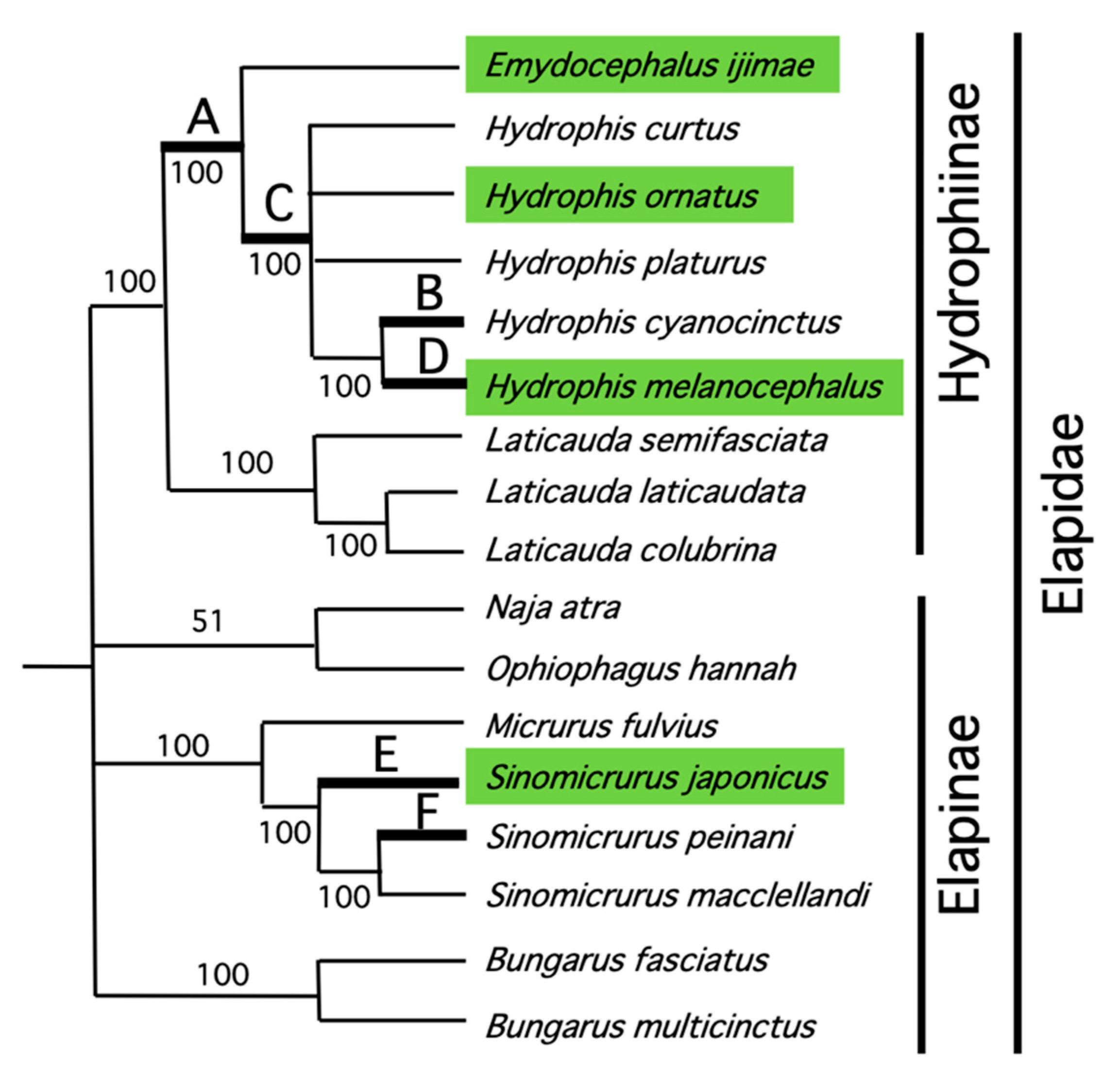
2.4. Phylogenetic Analysis
3. Results
3.1. Sequencing Snake Mitochondrial Genomes
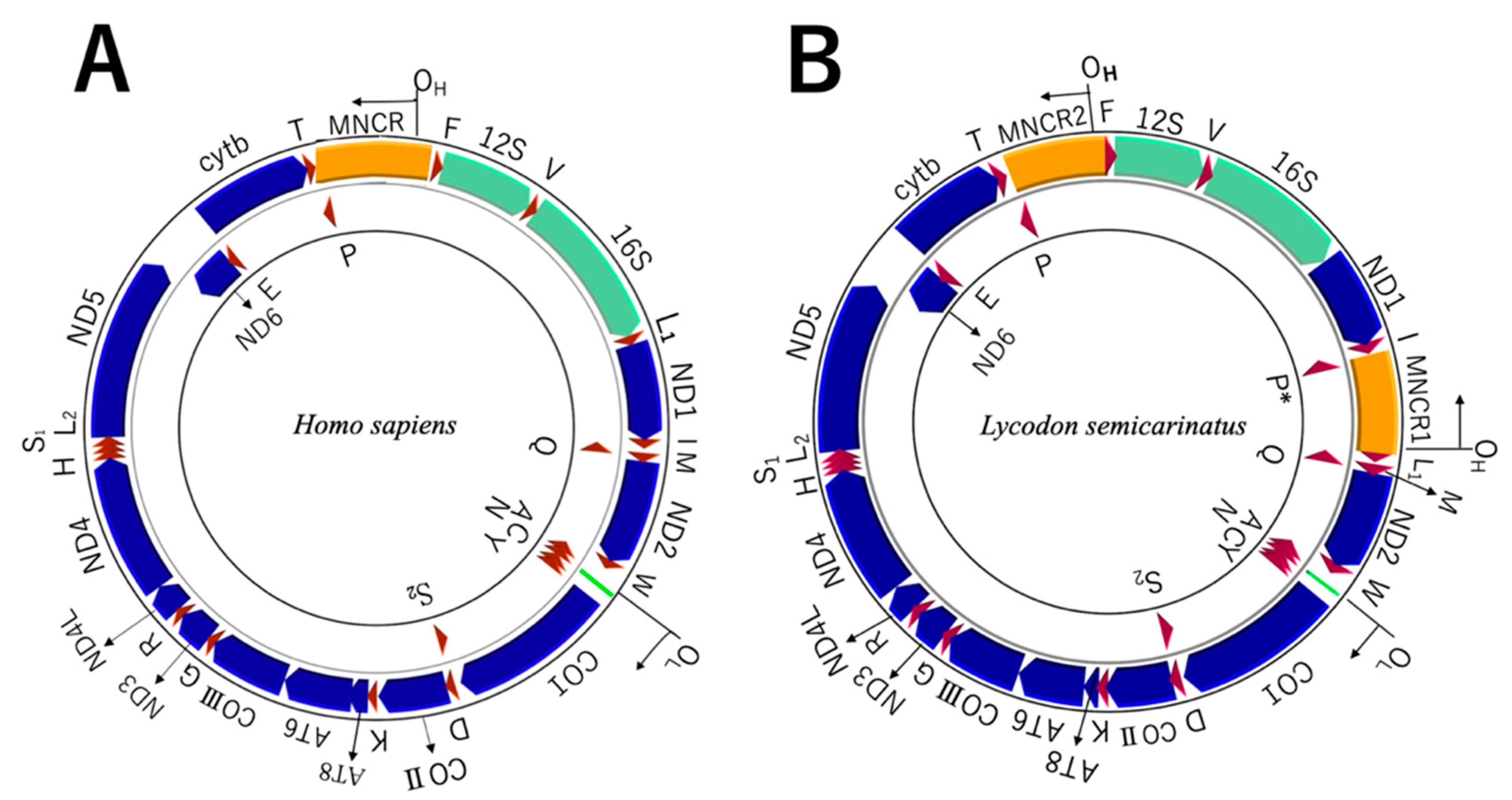
3.2. Encoded Genes
3.3. Non-Coding Regions in WANCY
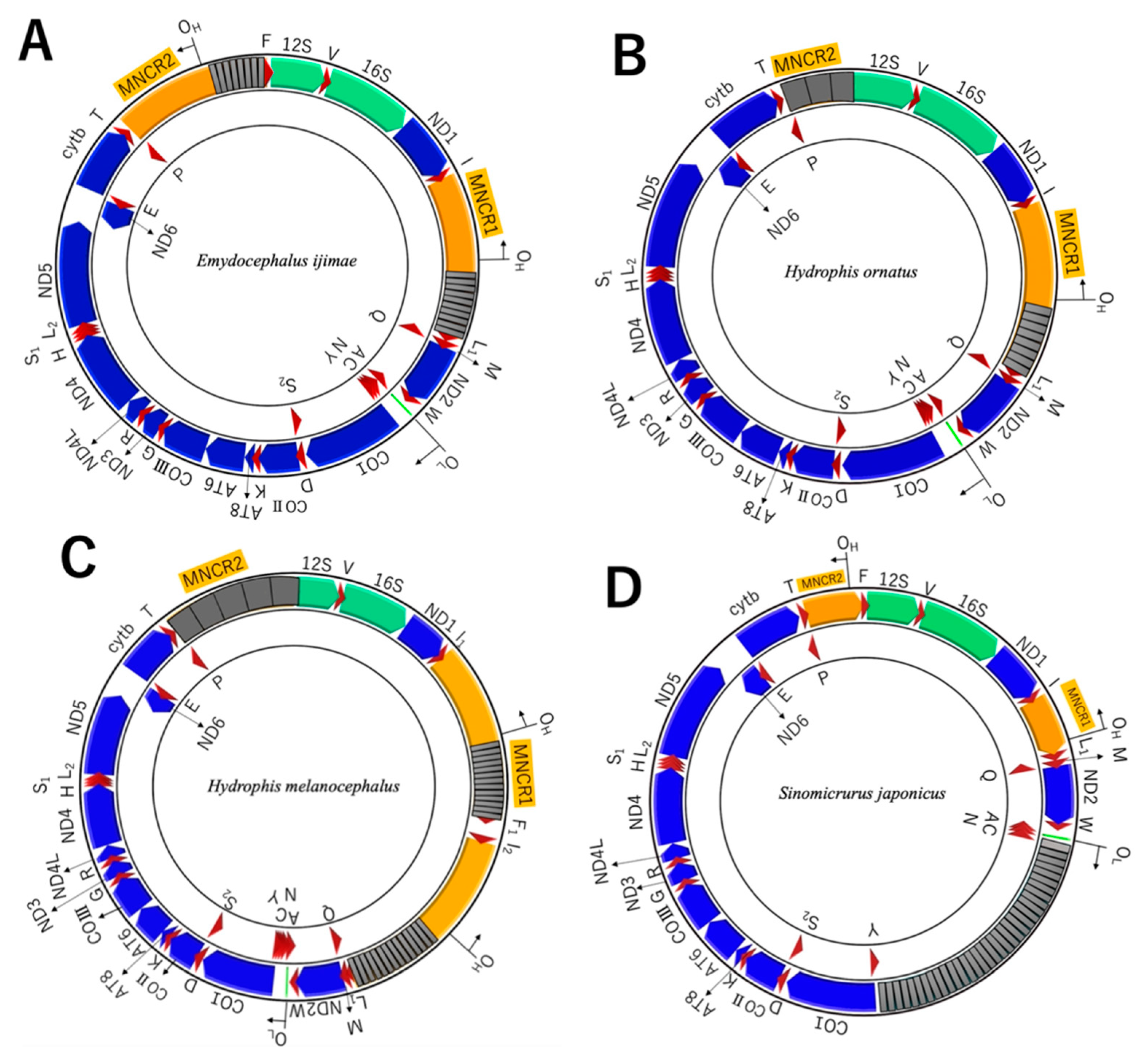
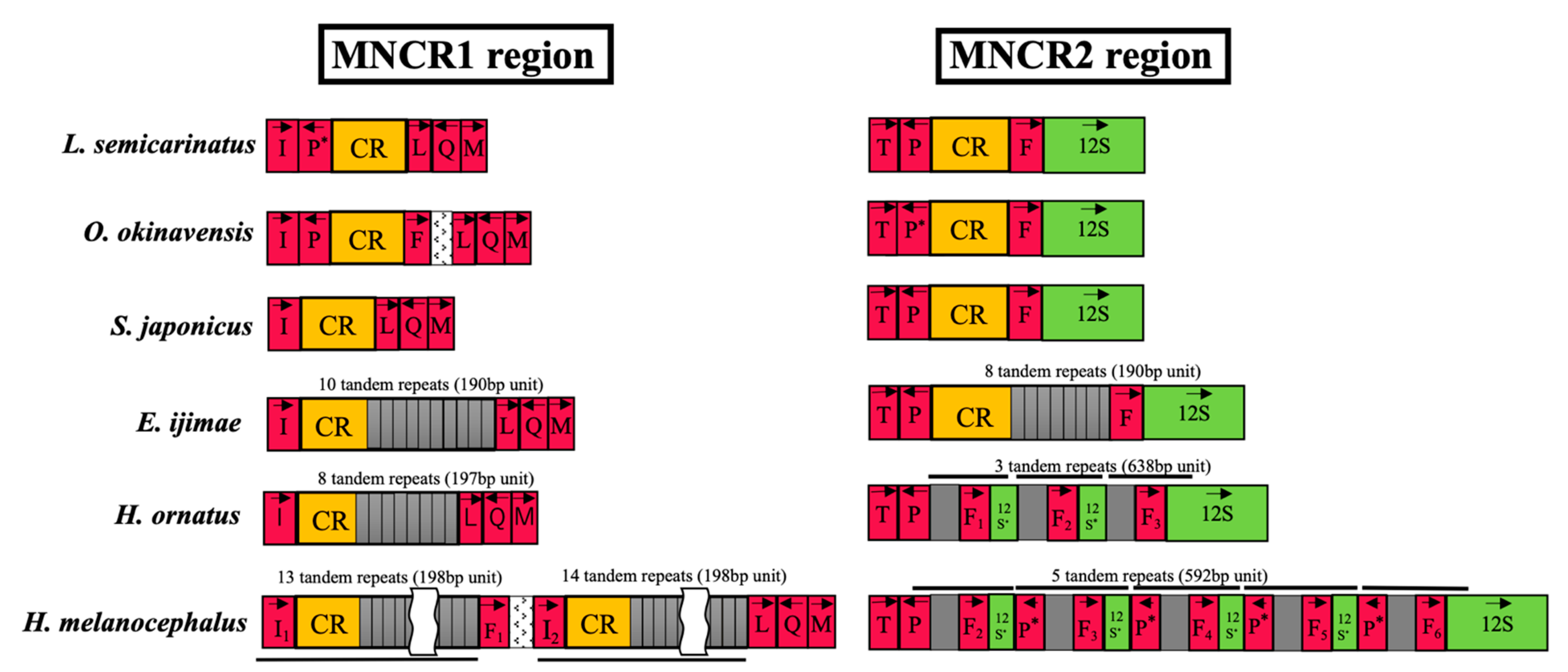
3.4. Non-Coding Regions in MNCRs
4. Discussion
4.1. Accuracy in Sequencing Mitochondrial Genomes
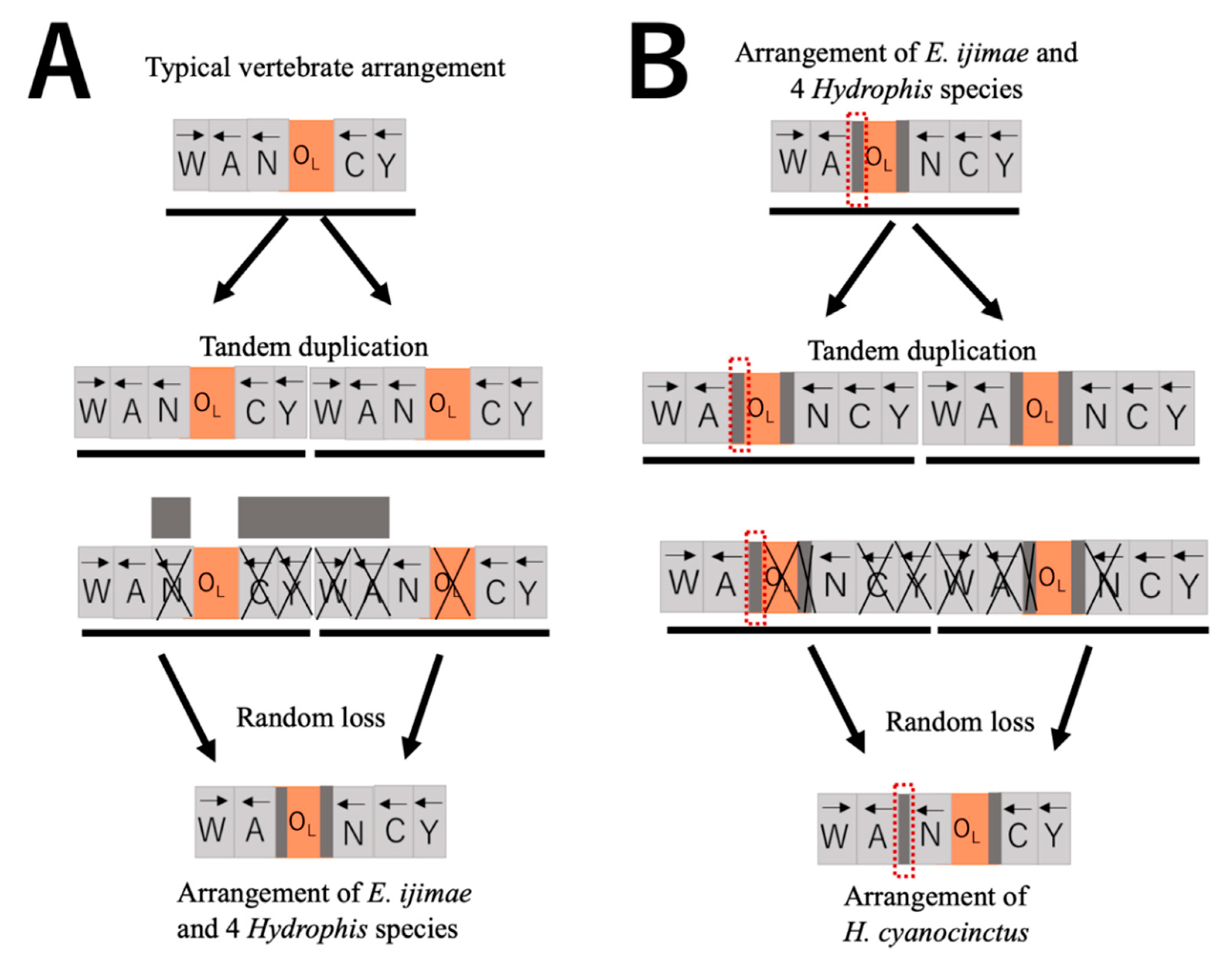
4.2. Rearrangement of L-Strand Replication Origin
4.3. Insertion of Long Repetitive Sequences
4.4. Loss of Duplicate Control Regions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Clayton, D.A. Transcription and replication of animal mitochondrial DNAs. Int. Rev. Cytol. 1992, 141, 217–232. [Google Scholar] [PubMed]
- Doda, J.N.; Wright, C.T.; Clayton, D.A. Elongation of displacement-loop strands in human and mouse mitochondrial DNA is arrested near specific template sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 6116–6120. [Google Scholar] [CrossRef]
- Walberg, M.W.; Clayton, D.A. Sequence and properties of the human KB cell and mouse L cell D-loop regions of mitochondrial DNA. Nucleic Acids Res. 1981, 9, 5411–5421. [Google Scholar] [CrossRef]
- Saccone, C.; Attimonelli, M.; Sbisà, E. Structural elements highly preserved during the evolution of the D-loop-containing region in vertebrate mitochondrial DNA. J. Mol. Evol. 1987, 26, 205–211. [Google Scholar] [CrossRef]
- Sbisá, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian mitochondrial D-loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Brehm, A.; Harris, D.J.; Alves, C.; Jesus, J.; Thomarat, F.; Vicente, L. Structure and evolution of the mitochondrial DNA complete control region in the lizard Lacerta dugesii (Lacertidae, Sauria). J. Mol. Evol. 2003, 56, 46–53. [Google Scholar] [CrossRef]
- Jemt, E.; Persson, Ö.; Shi, Y.; Mehmedovic, M.; Uhler, J.P.; López, M.D.; Freyer, C.; Gustafsson, C.M.; Samuelsson, T.; Falkenberg, M. Regulation of DNA replication at the end of the mitochondrial D-loop involves the helicase TWINKLE and a conserved sequence element. Nucleic Acids Res. 2015, 43, 9265–9275. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, Y.; Ota, H.; Nishida, M.; Ozawa, T. The complete nucleotide sequence of snake (Dinodon semicarinatus) mitochondrial genome with two identical control regions. Genetics 1998, 150, 313–329. [Google Scholar] [CrossRef]
- Kumazawa, Y.; Ota, H.; Nishida, M.; Ozawa, T. Gene rearrangements in snake mitochondrial genomes: Highly concerted evolution of control-region-like sequences duplicated and inserted into a tRNA gene cluster. Mol. Biol. Evol. 1996, 13, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Kumazawa, Y. Complete mitochondrial DNA sequences of six snakes: Phylogenetic relationships and molecular evolution of genomic features. J. Mol. Evol. 2005, 61, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Wang, H.; Yan, J.; Pan, T.; Jiang, S.; Rao, D.; Zhang, B. Multiple independent structural dynamic events in the evolution of snake mitochondrial genomes. BMC Genom. 2018, 19, 354. [Google Scholar] [CrossRef]
- Eberhard, J.R.; Wright, T.F.; Bermingham, E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol. Biol. Evol. 2001, 18, 1330–1342. [Google Scholar] [CrossRef]
- Lee, J.S.; Miya, M.; Lee, Y.S.; Kim, C.G.; Park, E.H.; Aoki, Y.; Nishida, M. The complete DNA sequence of the mitochondrial genome of the self-fertilizing fish Rivulus marmoratus (Cyprinodontiformes, Rivulidae) and the first description of duplication of a control region in fish. Gene 2001, 280, 1–7. [Google Scholar] [CrossRef]
- Rest, J.S.; Ast, J.C.; Austin, C.C.; Waddell, P.J.; Tibbetts, E.A.; Hay, J.M.; Mindell, D.P. Molecular systematics of primary reptilian lineages and the tuatara mitochondrial genome. Mol. Phylogenet. Evol. 2003, 29, 289–297. [Google Scholar] [CrossRef]
- Kumazawa, Y.; Endo, H. Mitochondrial genome of the Komodo dragon: Efficient sequencing method with reptile-oriented primers and novel gene rearrangements. DNA Res. 2004, 11, 115–125. [Google Scholar] [CrossRef]
- Sano, N.; Kurabayashi, A.; Fujii, T.; Yonekawa, H.; Sumida, M. Complete nucleotide sequence of the mitochondrial genome of Schlegel’s tree frog Rhacophorus schlegelii (family Rhacophoridae): Duplicated control regions and gene rearrangements. Genes Genet. Syst. 2005, 80, 213–224. [Google Scholar] [CrossRef]
- Parham, J.F.; Feldman, C.R.; Boore, J.L. The complete mitochondrial genome of the enigmatic bigheaded turtle (Platysternon): Description of unusual genomic features and the reconciliation of phylogenetic hypotheses based on mitochondrial and nuclear DNA. BMC Evol. Biol. 2006, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.-L.; Nie, L.-W.; Pu, Y.-G. Complete mitochondrial genome of Chinese big-headed turtle, Platysternon megacephalum, with a novel gene organization in vertebrate mtDNA. Gene 2006, 380, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef]
- The Reptile Database. Available online: http://www.reptile-database.org (accessed on 24 July 2022).
- Slowinski, J.B.; Knight, A.; Rooney, A.P. Inferring species trees from gene trees: A phylogenetic analysis of the Elapidae (Serpentes) based on the amino acid sequences of venom proteins. Mol. Phylogenet. Evol. 1997, 8, 349–362. [Google Scholar] [CrossRef]
- Castoe, T.A.; Smith, E.N.; Brown, R.M.; Parkinson, C.L. Higher-level phylogeny of Asian and American coralsnakes, their placement within the Elapidae (Squamata), and the systematic affinities of the enigmatic Asian coralsnake Hemibungarus calligaster (Wiegmann, 1834). Zool. J. Linn. Soc. 2007, 151, 809–831. [Google Scholar] [CrossRef][Green Version]
- Sanders, K.L.; Lee, M.S.Y.; Leys, R.; Foster, R.; Scott Keogh, J. Molecular phylogeny and divergence dates for Australasian elapids and sea snakes (hydrophiinae): Evidence from seven genes for rapid evolutionary radiations. J. Evol. Biol. 2008, 21, 682–695. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. Solexa QA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Kumazawa, Y.; Nishida, M. Sequence evolution of mitochondrial tRNA genes and deep-branch animal phylogenetics. J. Mol. Evol. 1993, 37, 380–398. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post–analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; Mclnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.W.; Clayton, D.A. In vitro replication of human mitochondrial DNA: Accurate initiation at the origin of light-strand synthesis. Cell 1985, 42, 951–958. [Google Scholar] [CrossRef]
- Franco-Sierra, N.D.; Díaz-Nieto, J.F. Rapid mitochondrial genome sequencing based on Oxford Nanopore Sequencing and a proxy for vertebrate species identification. Ecol. Evol. 2020, 10, 3544–3560. [Google Scholar] [CrossRef] [PubMed]
- Filipovíc, I.; Hereward, J.P.; Rašić, G.; Devine, G.J.; Furlong, M.J.; Etebari, K. The complete mitochondrial genome sequence of Oryctes rhinoceros (Coleoptera: Scarabaeidae) based on long-read nanopore sequencing. PeerJ 2021, 9, e10552. [Google Scholar] [CrossRef]
- Kinkar, L.; Gasser, R.B.; Webster, B.L.; Rollinson, D.; Littlewood, D.T.J.; Chang, B.C.H.; Stroehlein, A.J.; Korhonen, P.K.; Young, N.D. Nanopore sequencing resolves elusive long tandem-repeat regions in mitochondrial genomes. Int. J. Mol. Sci. 2021, 22, 1811. [Google Scholar] [CrossRef]
- Sahoo, B.; Das, G.; Sahoo, L.; Mahapatra, K.D.; Meher, P.K.; Udit, U.K.; Sundaray, J.K.; Das, P. The complete mitochondrial genome of Labeo catla (Hamilton, 1822) using long read sequencing. Mitochondrial DNA Part B 2021, 6, 402–403. [Google Scholar] [CrossRef]
- Kim, I.-H.; Park, J.; Suk, H.Y.; Bae, H.-G.; Min, M.-S.; Tsai, T.-S.; Park, D. Phylogenetic relationships of three representative sea krait species (genus Laticauda; elapidae; serpentes) based on 13 mitochondrial genes. Mitochondrial DNA Part A 2018, 29, 772–777. [Google Scholar] [CrossRef]
- Kim, J.-G.; Park, J.; Yi, C.-H.; Kim, M.-S.; Cho, I.-Y.; Kim, J.-M.; Kim, I.-H. The complete mitochondrial genome of a yellow-bellied sea snake (Hydrophis platurus) (Squamata, Elapidae). Mitochondrial DNA Part B 2020, 5, 891–892. [Google Scholar] [CrossRef]
- Yi, C.-H.; Park, J.; Sasai, T.; Kim, H.S.; Kim, J.-G.; Kim, M.-S.; Cho, I.-Y.; Kim, I.-H. Complete mitochondrial genome of the Ijima’s Sea Snake (Emydocephalus ijimae) (Squamata, Elapidae). Mitochondrial DNA Part B 2019, 4, 2658–2659. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.-H.; Park, J.; Sasai, T.; Kim, H.S.; Kim, J.-G.; Kim, M.-S.; Cho, I.-Y.; Kim, J.-M.; Kim, I.-H. Complete mitochondrial genome of the Hydrophis melanocephalus slender-necked sea snake (Squamata, Elapidae). Mitochondrial DNA Part B 2020, 5, 1406–1407. [Google Scholar] [CrossRef]
- Qiu, Q.; Chen, Z.; Zhang, Z.; Du, Y.; Lin, C.; Ji, X. Next-generation sequencing yields the complete mitogenome of the Shaw’s sea–snake (Squamata: Elapidae). Mitochondrial DNA Part B 2019, 4, 3628–3629. [Google Scholar] [CrossRef]
- Qiu, Q.; Liu, Q.; Zhao, Y.; Du, Y.; Lin, C.; Ji, X. Next-generation sequencing yields the complete mitogenome of the blue-banded sea-snake (Squamata: Elapidae). Mitochondrial DNA Part B 2019, 4, 2328–2329. [Google Scholar] [CrossRef] [PubMed]
- Zaher, H.; Murphy, R.W.; Arredondo, J.C.; Graboski, R.; Machado-Filho, P.R.; Mahlow, K.; Montingelli, G.G.; Quadros, A.B.; Orlov, N.L.; Wilkinson, M.; et al. Large-scale molecular phylogeny, morphology, divergence-time estimation, and the fossil record of advanced caenophidian snakes (Squamata: Serpentes). PLoS ONE 2019, 14, e0216148. [Google Scholar]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Jiang, Z.J.; Castoe, T.A.; Austin, C.C.; Burbrink, F.T.; Herron, M.D.; McGuire, J.A.; Parkinson, C.L.; Pollock, D.D. Comparative mitochondrial genomics of snakes: Extraordinary substitution rate dynamics and functionality of the duplicate control region. BMC Evol. Biol. 2007, 7, 123. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scientific Name | Classification | Museum Voucher No. | mtDNA Accession No. | mtDNA Length (bp) | MNCR1 Length (bp) | MNCR2 Length (bp) |
|---|---|---|---|---|---|---|
| Emydocephalus ijimae | Hydrophiinae; Hydrophiini | KUZ28042 | LC648431 | 20,598 | 2783 | 2586 |
| Hydrophis ornatus | Hydrophiinae; Hydrophiini | KUZ21782 | LC648430 | 19,682 | 2733 | 1692 |
| Hydrophis melanocephalus | Hydrophiinae; Hydrophiini | NUM-A0368 | LC648429 | 26,316 | 8330 | 2727 |
| Sinomicrurus japonicus | Elapinae; Calliophini | SDNCU-A4882 | LC648432 | 21,581 | 1028 | 1008 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiaokaiti, X.; Hashiguchi, Y.; Ota, H.; Kumazawa, Y. Evolution of the Noncoding Features of Sea Snake Mitochondrial Genomes within Elapidae. Genes 2022, 13, 1470. https://doi.org/10.3390/genes13081470
Xiaokaiti X, Hashiguchi Y, Ota H, Kumazawa Y. Evolution of the Noncoding Features of Sea Snake Mitochondrial Genomes within Elapidae. Genes. 2022; 13(8):1470. https://doi.org/10.3390/genes13081470
Chicago/Turabian StyleXiaokaiti, Xiakena, Yasuyuki Hashiguchi, Hidetoshi Ota, and Yoshinori Kumazawa. 2022. "Evolution of the Noncoding Features of Sea Snake Mitochondrial Genomes within Elapidae" Genes 13, no. 8: 1470. https://doi.org/10.3390/genes13081470
APA StyleXiaokaiti, X., Hashiguchi, Y., Ota, H., & Kumazawa, Y. (2022). Evolution of the Noncoding Features of Sea Snake Mitochondrial Genomes within Elapidae. Genes, 13(8), 1470. https://doi.org/10.3390/genes13081470