Targeting DNA Damage Response-Mediated Resistance in Non-Small Cell Lung Cancer: From Mechanistic Insights to Drug Development


Simple Summary
Abstract
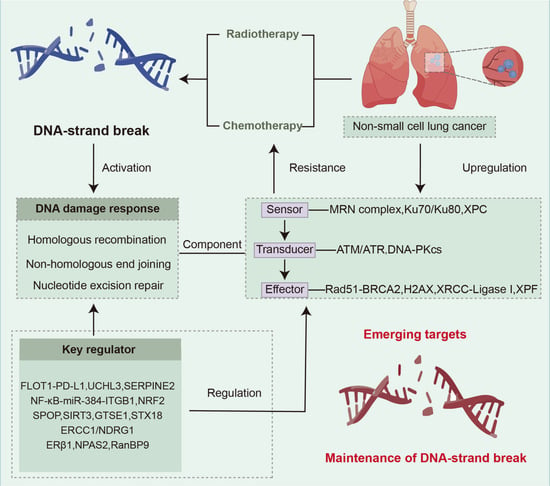
1. Introduction
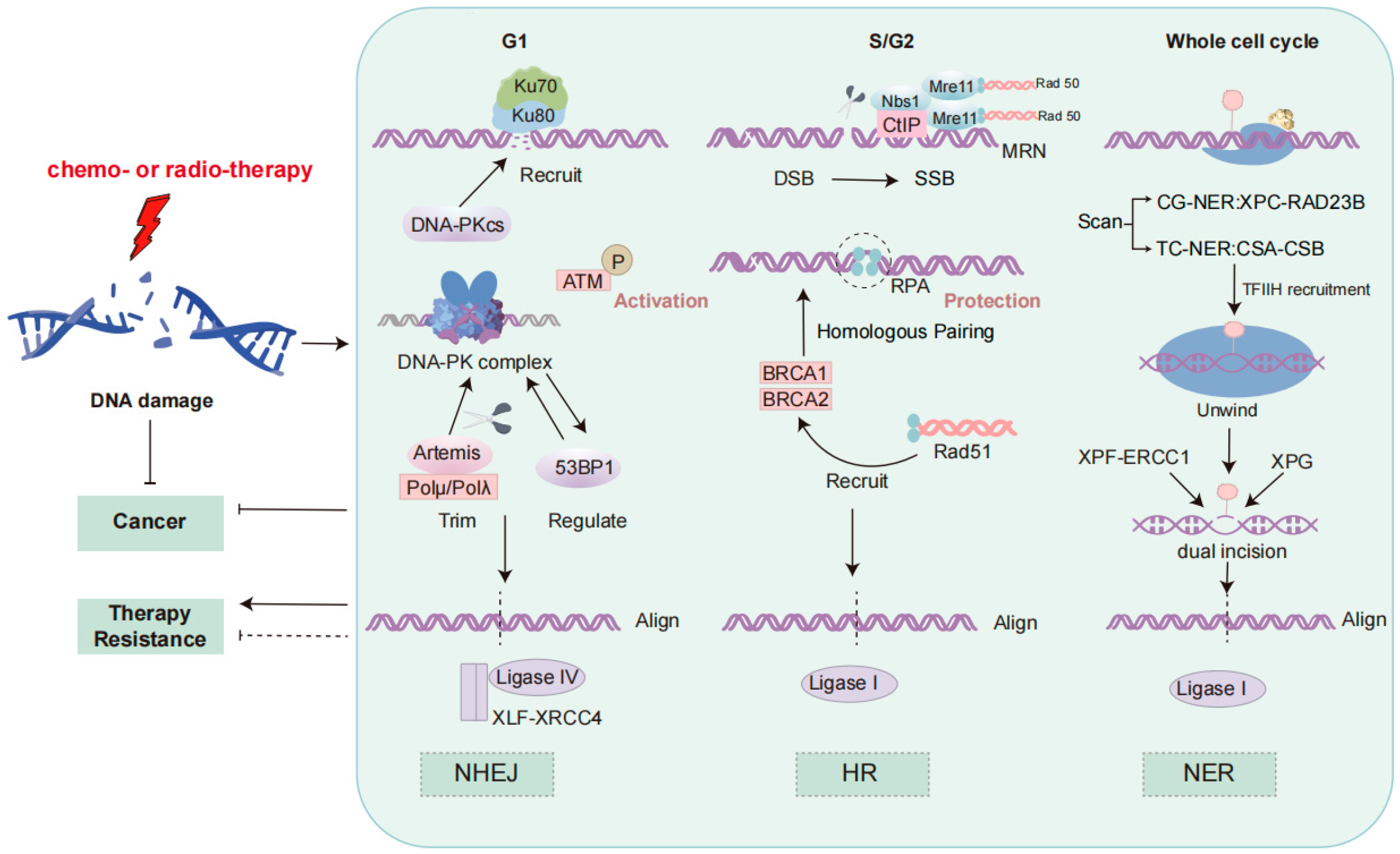
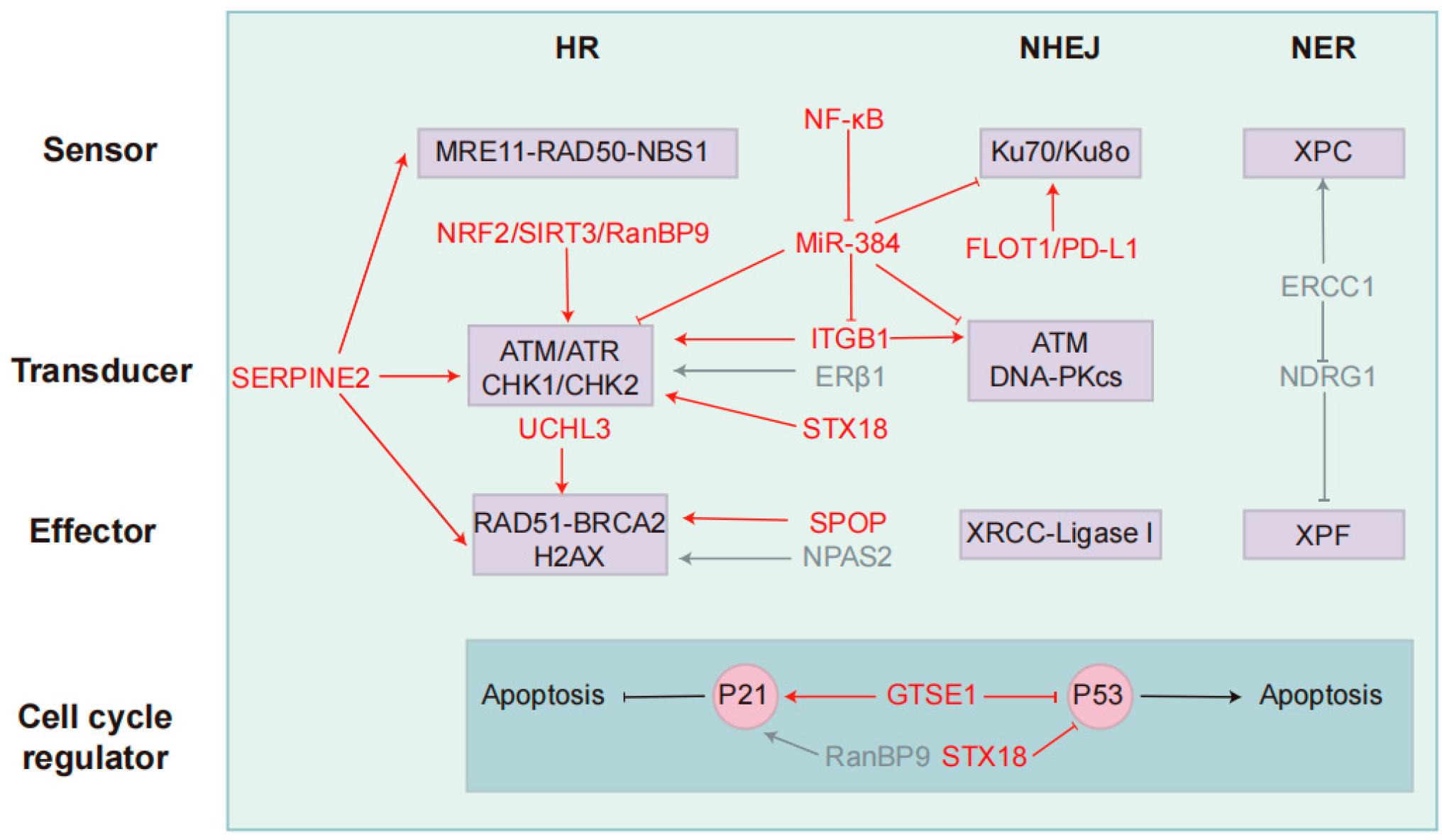
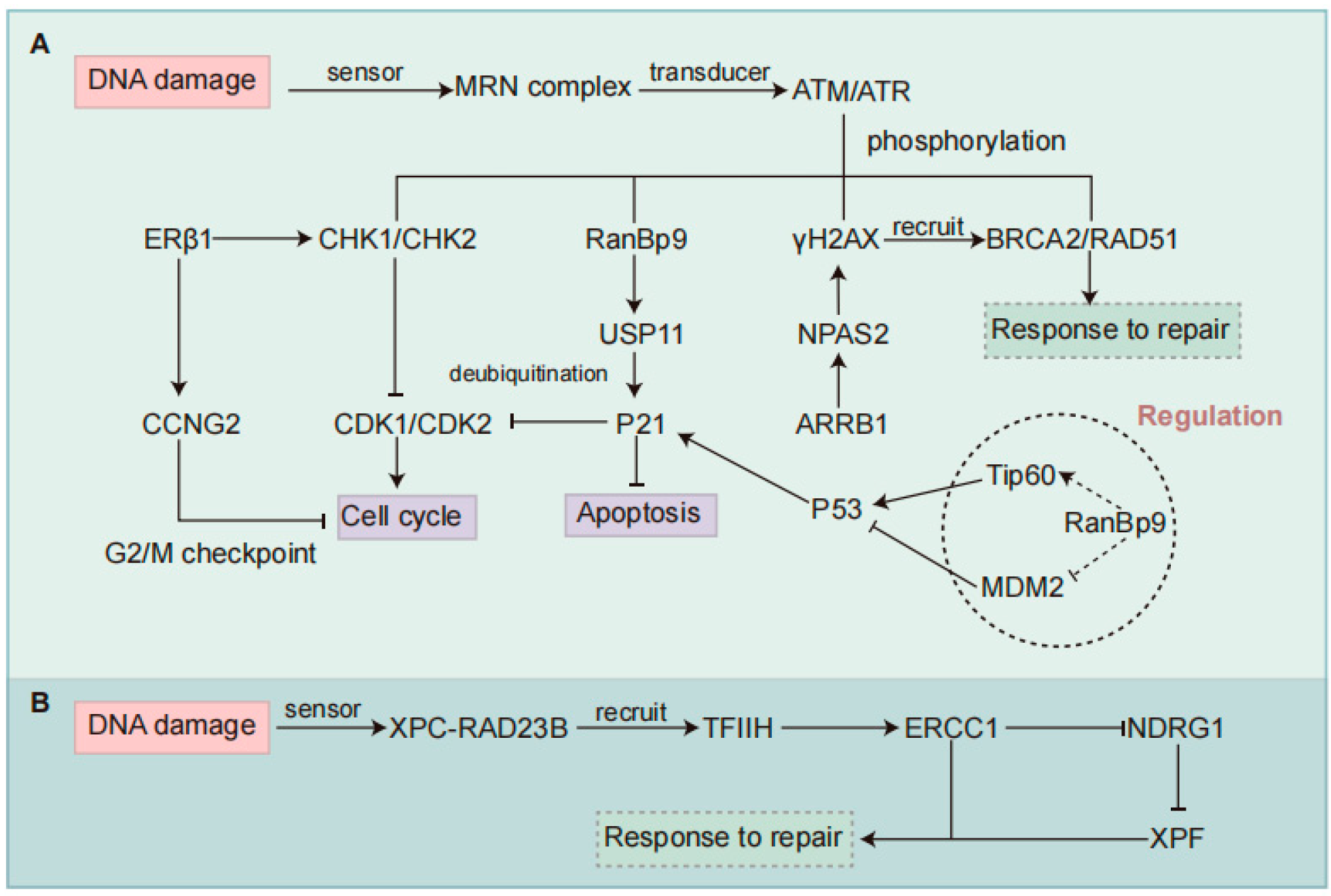
2. DDR Signaling Pathway
3. Radiotherapy
3.1. FLOT1-PD-L1
3.2. UCHL3
3.3. SERPINE2
3.4. NF-κB-miR-384-ITGB1
3.5. NRF2
3.6. SPOP
3.7. SIRT3
3.8. GTSE1
3.9. STX18
3.10. RanBP9
4. Chemotherapy
4.1. ERCC1/NDRG1
4.2. ERβ1
4.3. NPAS2
4.4. RanBP9
5. Summary and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, Y.; Liu, J.; Feng, L.; Yu, J.; Chen, D. Global burden of lung cancer in 2022 and projections to 2050: Incidence and mortality estimates from GLOBOCAN. Cancer Epidemiol. 2024, 93, 102693. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Wu, J.; Lin, Z. Non-Small Cell Lung Cancer Targeted Therapy: Drugs and Mechanisms of Drug Resistance. Int. J. Mol. Sci. 2022, 23, 15056. [Google Scholar] [CrossRef]
- Ricciuti, B.; Recondo, G.; Spurr, L.F.; Li, Y.Y.; Lamberti, G.; Venkatraman, D.; Umeton, R.; Cherniack, A.D.; Nishino, M.; Sholl, L.M.; et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 4135–4142. [Google Scholar] [CrossRef]
- Gu, W.; Zhuang, W.; Zhuang, M.; He, M.; Li, Z. DNA damage response and repair gene mutations are associated with tumor mutational burden and outcomes to platinum-based chemotherapy/immunotherapy in advanced NSCLC patients. Diagn. Pathol. 2023, 18, 119. [Google Scholar] [CrossRef]
- Dai, J.; Jiang, M.; He, K.; Wang, H.; Chen, P.; Guo, H.; Zhao, W.; Lu, H.; He, Y.; Zhou, C. DNA Damage Response and Repair Gene Alterations Increase Tumor Mutational Burden and Promote Poor Prognosis of Advanced Lung Cancer. Front. Oncol. 2021, 11, 708294. [Google Scholar] [CrossRef]
- Sun, S.; Wang, K.; Guo, D.; Zheng, H.; Liu, Y.; Shen, H.; Du, J. Identification of the key DNA damage response genes for predicting immunotherapy and chemotherapy efficacy in lung adenocarcinoma based on bulk, single-cell RNA sequencing, and spatial transcriptomics. Comput. Biol. Med. 2024, 171, 108078. [Google Scholar] [CrossRef]
- Besse, B.; Pons-Tostivint, E.; Park, K.; Hartl, S.; Forde, P.M.; Hochmair, M.J.; Awad, M.M.; Thomas, M.; Goss, G.; Wheatley-Price, P.; et al. Biomarker-directed targeted therapy plus durvalumab in advanced non-small-cell lung cancer: A phase 2 umbrella trial. Nat. Med. 2024, 30, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, T.; Gao, Y.F.; Zheng, W.; Wang, C.J.; Xiao, L.; Huang, M.S.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. Targeting DNA Damage Response in the Radio(Chemo)therapy of Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 839. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Roth, D.B.; Wilson, J.H. Relative rates of homologous and nonhomologous recombination in transfected DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 3355–3359. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Yan, Q.; Zhu, H.; Lan, L.; Yi, J.; Yang, J. Cleavage of Ku80 by caspase-2 promotes non-homologous end joining-mediated DNA repair. DNA Repair 2017, 60, 18–28. [Google Scholar] [CrossRef]
- Kara, A.; Özgür, A.; Nalbantoğlu, S.; Karadağ, A. DNA repair pathways and their roles in drug resistance for lung adenocarcinoma. Mol. Biol. Rep. 2021, 48, 3813–3825. [Google Scholar] [CrossRef]
- Kobayashi, J. Molecular mechanism of the recruitment of NBS1/hMRE11/hRAD50 complex to DNA double-strand breaks: NBS1 binds to gamma-H2AX through FHA/BRCT domain. J. Radiat. Res. 2004, 45, 473–478. [Google Scholar] [CrossRef]
- Im, J.; Lawrence, J.; Seelig, D.; Nho, R.S. FoxM1-dependent RAD51 and BRCA2 signaling protects idiopathic pulmonary fibrosis fibroblasts from radiation-induced cell death. Cell Death Dis. 2018, 9, 584. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Rechkunova, N.I.; Krasikova, Y.S.; Lavrik, O.I. Nucleotide excision repair: DNA damage recognition and preincision complex assembly. Biochemistry 2011, 76, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Dokic, I.; Mairani, A.; Mein, S.; Brons, S.; Häring, P.; Haberer, T.; Jäkel, O.; Zimmermann, A.; Zenke, F.; et al. Overcoming hypoxia-induced tumor radioresistance in non-small cell lung cancer by targeting DNA-dependent protein kinase in combination with carbon ion irradiation. Radiat. Oncol. 2017, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshino, H.; Sato, K.; Kashiwakura, I.; Tsuruga, E. DAP3-mediated cell cycle regulation and its association with radioresistance in human lung adenocarcinoma cell lines. J. Radiat. Res. 2023, 64, 520–529. [Google Scholar] [CrossRef]
- Kim, H.; Jeong, I.H.; Choi, Y.K.; Lee, Y.K.; Moon, E.; Huh, Y.H.; Im, W.; Jin, J.O.; Kwak, M.; Lee, P.C. Suppression of Lung Cancer Malignancy by Micellized siRNA through Cell Cycle Arrest. Adv. Healthc. Mater. 2023, 12, e2202358. [Google Scholar] [CrossRef]
- Hao, J.; Song, Z.; Su, J.; Li, L.; Zou, L.; Zou, K. The PRX-1/TLR4 axis promotes hypoxia-induced radiotherapy resistance in non-small cell lung cancer by targeting the NF-κB/p65 pathway. Cell Signal 2023, 110, 110806. [Google Scholar] [CrossRef]
- Rasmi, R.R.; Sakthivel, K.M.; Guruvayoorappan, C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed. Pharmacother. 2020, 130, 110569. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, L.Y.; He, J.Z.; Miao, Z.M.; Li, Y.Y.; Zhang, Y.M.; Liu, Z.W.; Zhang, S.Z.; Chen, Y.; Zhou, G.C.; et al. Review: Mechanisms and perspective treatment of radioresistance in non-small cell lung cancer. Front. Immunol. 2023, 14, 1133899. [Google Scholar] [CrossRef]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef]
- Shu, Z.; Dwivedi, B.; Switchenko, J.M.; Yu, D.S.; Deng, X. PD-L1 deglycosylation promotes its nuclear translocation and accelerates DNA double-strand-break repair in cancer. Nat. Commun. 2024, 15, 6830. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Meng, L.; Meng, S.; Huang, L.; Luo, S.; Wu, X.; Gong, X. Flotillin-1 enhances radioresistance through reducing radiation-induced DNA damage and promoting immune escape via STING signaling pathway in non-small cell lung cancer. Cancer Biol. Ther. 2023, 24, 2203332. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Harrington, K.J.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.; Khilnani, A.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. 2018, 29, viii712. [Google Scholar] [CrossRef]
- Mtango, N.R.; Sutovsky, M.; Vandevoort, C.A.; Latham, K.E.; Sutovsky, P. Essential role of ubiquitin C-terminal hydrolases UCHL1 and UCHL3 in mammalian oocyte maturation. J. Cell Physiol. 2012, 227, 2022–2029. [Google Scholar] [CrossRef]
- Luo, K.; Li, L.; Li, Y.; Wu, C.; Yin, Y.; Chen, Y.; Deng, M.; Nowsheen, S.; Yuan, J.; Lou, Z. A phosphorylation-deubiquitination cascade regulates the BRCA2-RAD51 axis in homologous recombination. Genes. Dev. 2016, 30, 2581–2595. [Google Scholar] [CrossRef]
- Nishi, R.; Wijnhoven, P.W.G.; Kimura, Y.; Matsui, M.; Konietzny, R.; Wu, Q.; Nakamura, K.; Blundell, T.L.; Kessler, B.M. The deubiquitylating enzyme UCHL3 regulates Ku80 retention at sites of DNA damage. Sci. Rep. 2018, 8, 17891. [Google Scholar] [CrossRef]
- Xu, L.M.; Yuan, Y.J.; Yu, H.; Wang, S.; Wang, P. LINC00665 knockdown confers sensitivity in irradiated non-small cell lung cancer cells through the miR-582-5p/UCHL3/AhR axis. J. Transl. Med. 2022, 20, 350. [Google Scholar] [CrossRef]
- Liu, M.; Chen, H.; Chen, X.; Xiong, J.; Song, Z. Silencing UCHL3 enhances radio-sensitivity of non-small cell lung cancer cells by inhibiting DNA repair. Aging 2021, 13, 14277–14288. [Google Scholar] [CrossRef]
- Song, Z.; Tu, X.; Zhou, Q.; Huang, J.; Chen, Y.; Liu, J.; Lee, S.; Kim, W.; Nowsheen, S.; Luo, K.; et al. A novel UCHL(3) inhibitor, perifosine, enhances PARP inhibitor cytotoxicity through inhibition of homologous recombination-mediated DNA double strand break repair. Cell Death Dis. 2019, 10, 398. [Google Scholar] [CrossRef] [PubMed]
- Monard, D. SERPINE2/Protease Nexin-1 in vivo multiple functions: Does the puzzle make sense? Semin. Cell Dev. Biol. 2017, 62, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, B.; Xing, A.Y.; Xu, K.S.; Li, G.X.; Yu, Z.H. Prognostic significance of SERPINE2 in gastric cancer and its biological function in SGC7901 cells. J. Cancer Res. Clin. Oncol. 2015, 141, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Biebl, A.; Neesse, A.; Wagner, M.; Iwamura, T.; Leder, G.; Adler, G.; Gress, T.M. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res. 2003, 63, 4945–4951. [Google Scholar]
- Pagliara, V.; Adornetto, A.; Mammì, M.; Masullo, M.; Sarnataro, D.; Pietropaolo, C.; Arcone, R. Protease Nexin-1 affects the migration and invasion of C6 glioma cells through the regulation of urokinase Plasminogen Activator and Matrix Metalloproteinase-9/2. Biochim. Biophys. Acta 2014, 1843, 2631–2644. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Q.; Zhu, L.; Xie, S.; Tu, L.; Yang, Y.; Wu, K.; Zhao, Y.; Wang, Y.; Xu, Y.; et al. SERPINE2/PN-1 regulates the DNA damage response and radioresistance by activating ATM in lung cancer. Cancer Lett. 2022, 524, 268–283. [Google Scholar] [CrossRef]
- Yang, Y.; Xin, X.; Fu, X.; Xu, D. Expression pattern of human SERPINE2 in a variety of human tumors. Oncol. Lett. 2018, 15, 4523–4530. [Google Scholar] [CrossRef]
- Kang, J.; Kim, W.; Kwon, T.; Youn, H.; Kim, J.S.; Youn, B. Plasminogen activator inhibitor-1 enhances radioresistance and aggressiveness of non-small cell lung cancer cells. Oncotarget 2016, 7, 23961–23974. [Google Scholar] [CrossRef]
- Smirnov, D.A.; Morley, M.; Shin, E.; Spielman, R.S.; Cheung, V.G. Genetic analysis of radiation-induced changes in human gene expression. Nature 2009, 459, 587–591. [Google Scholar] [CrossRef]
- Dokuni, R.; Nagano, T.; Jimbo, N.; Sato, H.; Kiriu, T.; Yasuda, Y.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; Maniwa, Y.; et al. High expression level of serpin peptidase inhibitor clade E member 2 is associated with poor prognosis in lung adenocarcinoma. Respir. Res. 2020, 21, 331. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, J.; Qiu, M.; Zhao, J.; Zou, F.; Meng, M.; Jiang, X.; Yuan, Z.; Mi, Z.; Wu, Z. MicroRNA-384 radiosensitizes human non-small cell lung cancer by impairing DNA damage response and repair signaling, which is inhibited by NF-κB. Cancer Biol. Med. 2024, 21, 1050–1066. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.A.; Addison, C.L. β1 integrin: An emerging player in the modulation of tumorigenesis and response to therapy. Cell Adh Migr. 2012, 6, 71–77. [Google Scholar] [CrossRef]
- Dingemans, A.M.; van den Boogaart, V.; Vosse, B.A.; van Suylen, R.J.; Griffioen, A.W.; Thijssen, V.L. Integrin expression profiling identifies integrin alpha5 and beta1 as prognostic factors in early stage non-small cell lung cancer. Mol. Cancer 2010, 9, 152. [Google Scholar] [CrossRef]
- Yin, H.L.; Wu, C.C.; Lin, C.H.; Chai, C.Y.; Hou, M.F.; Chang, S.J.; Tsai, H.P.; Hung, W.C.; Pan, M.R.; Luo, C.W. β1 Integrin as a Prognostic and Predictive Marker in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1432. [Google Scholar] [CrossRef]
- Eke, I.; Dickreuter, E.; Cordes, N. Enhanced radiosensitivity of head and neck squamous cell carcinoma cells by β1 integrin inhibition. Radiother. Oncol. 2012, 104, 235–242. [Google Scholar] [CrossRef]
- Hehlgans, S.; Eke, I.; Storch, K.; Haase, M.; Baretton, G.B.; Cordes, N. Caveolin-1 mediated radioresistance of 3D grown pancreatic cancer cells. Radiother. Oncol. 2009, 92, 362–370. [Google Scholar] [CrossRef]
- Li, Y.; Sun, C.; Tan, Y.; Zhang, H.; Li, Y.; Zou, H. ITGB1 enhances the Radioresistance of human Non-small Cell Lung Cancer Cells by modulating the DNA damage response and YAP1-induced Epithelial-mesenchymal Transition. Int. J. Biol. Sci. 2021, 17, 635–650. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, X.; Zhang, Y.; Wang, Z. Curcumin suppresses the malignancy of non-small cell lung cancer by modulating the circ-PRKCA/miR-384/ITGB1 pathway. Biomed. Pharmacother. 2021, 138, 111439. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, D.; Liu, S.; Shao, M.; Liu, Y.; Li, A.; Lv, Y.; Huang, M.; Lou, D.; Fan, Q. Curcumin enhances radiosensitization of nasopharyngeal carcinoma by regulating circRNA network. Mol. Carcinog. 2020, 59, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Dong, M.; Li, J.; Sun, Y.; Gao, Y.; Wang, Y.; Du, L.; Liu, Y.; Ji, K.; He, N.; et al. NRF2 promotes radiation resistance by cooperating with TOPBP1 to activate the ATR-CHK1 signaling pathway. Theranostics 2024, 14, 681–698. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Kim, K.; Norris, A.J.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; Della Donna, L.; Ratikan, J.; Szelag, H.; Hlatky, L.; et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, H.; Sun, M.; Yang, J.; Zhang, W.; Han, S.; Xu, B. Speckle-type POZ protein, SPOP, is involved in the DNA damage response. Carcinogenesis 2014, 35, 1691–1697. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, D.; Cai, M.; Luo, Z.; Zhu, Y.; Gong, L.; Lei, Y.; Tan, X.; Zhu, Q.; Han, S. SPOP regulates the DNA damage response and lung adenocarcinoma cell response to radiation. Am. J. Cancer Res. 2019, 9, 1469–1483. [Google Scholar]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015, 4, e09207. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Y.; Huang, A.; Chen, Y.; Wang, J.; Liu, N.; Wang, X.; Gong, Y.; Wang, W.; Pan, J. Overexpression of SERPINA3 suppresses tumor progression by modulating SPOP/NF-κB in lung cancer. Int. J. Oncol. 2023, 63, 96. [Google Scholar] [CrossRef]
- Tian, S.; Xu, M.; Geng, X.; Fang, J.; Xu, H.; Xue, X.; Hu, H.; Zhang, Q.; Yu, D.; Guo, M.; et al. Network Medicine-Based Strategy Identifies Maprotiline as a Repurposable Drug by Inhibiting PD-L1 Expression via Targeting SPOP in Cancer. Adv. Sci. 2025, 12, e2410285. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Cheng, Y.; Huang, J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Iachettini, S.; Salvati, E.; Zizza, P.; Maresca, C.; D’Angelo, C.; Benarroch-Popivker, D.; Capolupo, A.; Del Gaudio, F.; Cosconati, S.; et al. SIRT6 interacts with TRF2 and promotes its degradation in response to DNA damage. Nucleic Acids Res. 2017, 45, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Li, Z.; Zhang, C.; Lu, X.; Tu, B.; Cao, Z.; Li, Y.; Chen, Y.; Jiang, L.; Wang, H.; et al. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci. Adv. 2019, 5, eaav1118. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, P.; Wang, H.; Li, L.; Li, Q. SIRT3 promotion reduces resistance to cisplatin in lung cancer by modulating the FOXO3/CDT1 axis. Cancer Med. 2021, 10, 1394–1404. [Google Scholar] [CrossRef]
- Huang, B.; Ding, J.; Guo, H.; Wang, H.; Xu, J.; Zheng, Q.; Zhou, L. SIRT3 Regulates the ROS-FPR1/HIF-1α Axis under Hypoxic Conditions to Influence Lung Cancer Progression. Cell Biochem. Biophys. 2023, 81, 813–821. [Google Scholar] [CrossRef]
- Cao, K.; Chen, Y.; Zhao, S.; Huang, Y.; Liu, T.; Liu, H.; Li, B.; Cui, J.; Cai, J.; Bai, C.; et al. Sirt3 Promoted DNA Damage Repair and Radioresistance Through ATM-Chk2 in Non-small Cell Lung Cancer Cells. J. Cancer 2021, 12, 5464–5472. [Google Scholar] [CrossRef]
- Ren, G.; Ma, Y.; Wang, X.; Zheng, Z.; Li, G. Aspirin blocks AMPK/SIRT3-mediated glycolysis to inhibit NSCLC cell proliferation. Eur. J. Pharmacol. 2022, 932, 175208. [Google Scholar] [CrossRef]
- Xu, T.; Ma, M.; Chi, Z.; Si, L.; Sheng, X.; Cui, C.; Dai, J.; Yu, S.; Yan, J.; Yu, H.; et al. High G2 and S-phase expressed 1 expression promotes acral melanoma progression and correlates with poor clinical prognosis. Cancer Sci. 2018, 109, 1787–1798. [Google Scholar] [CrossRef]
- Bublik, D.R.; Scolz, M.; Triolo, G.; Monte, M.; Schneider, C. Human GTSE-1 regulates p21(CIP1/WAF1) stability conferring resistance to paclitaxel treatment. J. Biol. Chem. 2010, 285, 5274–5281. [Google Scholar] [CrossRef]
- Lin, F.; Xie, Y.J.; Zhang, X.K.; Huang, T.J.; Xu, H.F.; Mei, Y.; Liang, H.; Hu, H.; Lin, S.T.; Luo, F.F.; et al. GTSE1 is involved in breast cancer progression in p53 mutation-dependent manner. J. Exp. Clin. Cancer Res. 2019, 38, 152. [Google Scholar] [CrossRef]
- Subhash, V.V.; Tan, S.H.; Tan, W.L.; Yeo, M.S.; Xie, C.; Wong, F.Y.; Kiat, Z.Y.; Lim, R.; Yong, W.P. GTSE1 expression represses apoptotic signaling and confers cisplatin resistance in gastric cancer cells. BMC Cancer 2015, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, H.; Lian, Y.; Chen, L.; Gu, L.; Wang, J.; Huang, Y.; Deng, M.; Gao, Z.; Huang, Y. GTSE1 promotes cell migration and invasion by regulating EMT in hepatocellular carcinoma and is associated with poor prognosis. Sci. Rep. 2017, 7, 5129. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Du, L.; Zhang, P.; Ma, N.; Liang, Y.; Han, Y.; Qu, B. Knockdown GTSE1 enhances radiosensitivity in non-small-cell lung cancer through DNA damage repair pathway. J. Cell Mol. Med. 2020, 24, 5162–5167. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Yang, H.; Chen, X.; Wang, Y.; Zhang, S.; Wang, P.; Chen, C.; Wang, K.; Liu, Z.; Zheng, X. Discovery of pyrimidine-2,4-diamine analogues as efficiency anticancer drug by targeting GTSE1. Bioorg Chem. 2024, 151, 107700. [Google Scholar] [CrossRef]
- Hatsuzawa, K.; Hirose, H.; Tani, K.; Yamamoto, A.; Scheller, R.H.; Tagaya, M. Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis-Golgi vesicle trafficking. J. Biol. Chem. 2000, 275, 13713–13720. [Google Scholar] [CrossRef]
- Thumser-Henner, C.; Oeck, S.; Kalmbach, S.; Forster, J.; Kindl, F.; Sak, A.; Schramm, A.; Schuler, M. Syntaxin 18 regulates the DNA damage response and epithelial-to-mesenchymal transition to promote radiation resistance of lung cancer. Cell Death Dis. 2022, 13, 529. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Palmieri, D.; Scarpa, M.; Tessari, A.; Uka, R.; Amari, F.; Lee, C.; Richmond, T.; Foray, C.; Sheetz, T.; Braddom, A.; et al. Ran Binding Protein 9 (RanBP9) is a novel mediator of cellular DNA damage response in lung cancer cells. Oncotarget 2016, 7, 18371–18383. [Google Scholar] [CrossRef]
- Domingues, S.C.; Konietzko, U.; Henriques, A.G.; Rebelo, S.; Fardilha, M.; Nishitani, H.; Nitsch, R.M.; da Cruz, E.S.E.F.; da Cruz, E.S.O.A. RanBP9 modulates AICD localization and transcriptional activity via direct interaction with Tip60. J. Alzheimers Dis. 2014, 42, 1415–1433. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, C.; Price, B.D. Mechanistic links between ATM and histone methylation codes during DNA repair. Prog. Mol. Biol. Transl. Sci. 2012, 110, 263–288. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- McNeil, E.M.; Melton, D.W. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012, 40, 9990–10004. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, K.; Wang, X.; Chen, H.; Zhou, J.; Wu, X.; Liu, T.; Yang, Y.; Yang, X.; Cui, D.; et al. NDRG1 disruption alleviates cisplatin/sodium glycididazole-induced DNA damage response and apoptosis in ERCC1-defective lung cancer cells. Int. J. Biochem. Cell Biol. 2018, 100, 54–60. [Google Scholar] [CrossRef]
- Gómez-Casero, E.; Navarro, M.; Rodríguez-Puebla, M.L.; Larcher, F.; Paramio, J.M.; Conti, C.J.; Jorcano, J.L. Regulation of the differentiation-related gene Drg-1 during mouse skin carcinogenesis. Mol. Carcinog. 2001, 32, 100–109. [Google Scholar] [CrossRef]
- Qu, X.; Zhai, Y.; Wei, H.; Zhang, C.; Xing, G.; Yu, Y.; He, F. Characterization and expression of three novel differentiation-related genes belong to the human NDRG gene family. Mol. Cell Biochem. 2002, 229, 35–44. [Google Scholar] [CrossRef]
- Fan, W.; Huang, J.; Tian, F.; Hong, X.; Zhu, K.; Zhan, Y.; Li, X.; Wang, X.; Wang, X.; Cai, L.; et al. m(6)A-Modified SNRPA Controls Alternative Splicing of ERCC1 Exon 8 to Induce Cisplatin Resistance in Lung Adenocarcinoma. Adv. Sci. 2024, 11, e2404609. [Google Scholar] [CrossRef]
- Lin, C.Y.; Hung, C.C.; Wang, C.C.N.; Lin, H.Y.; Huang, S.H.; Sheu, M.J. Demethoxycurcumin sensitizes the response of non-small cell lung cancer to cisplatin through downregulation of TP and ERCC1-related pathways. Phytomedicine 2019, 53, 28–36. [Google Scholar] [CrossRef]
- Park, K.C.; Paluncic, J.; Kovacevic, Z.; Richardson, D.R. Pharmacological targeting and the diverse functions of the metastasis suppressor, NDRG1, in cancer. Free Radic. Biol. Med. 2020, 157, 154–175. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Nikolos, F.; Thomas, C.; Rajapaksa, G.; Bado, I.; Gustafsson, J. ERβ regulates NSCLC phenotypes by controlling oncogenic RAS signaling. Mol. Cancer Res. 2014, 12, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Elebro, K.; Borgquist, S.; Rosendahl, A.H.; Markkula, A.; Simonsson, M.; Jirström, K.; Rose, C.; Ingvar, C.; Jernström, H. High Estrogen Receptor β Expression Is Prognostic among Adjuvant Chemotherapy-Treated Patients-Results from a Population-Based Breast Cancer Cohort. Clin. Cancer Res. 2017, 23, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Pinton, G.; Manente, A.G.; Daga, A.; Cilli, M.; Rinaldi, M.; Nilsson, S.; Moro, L. Agonist activation of estrogen receptor beta (ERβ) sensitizes malignant pleural mesothelioma cells to cisplatin cytotoxicity. Mol. Cancer 2014, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Ishii, A.; Washiya, K.; Konno, T.; Kon, H.; Yamaya, C.; Ono, I.; Ogawa, J. Combined overexpression of EGFR and estrogen receptor alpha correlates with a poor outcome in lung cancer. Anticancer Res. 2005, 25, 4693–4698. [Google Scholar]
- Rades, D.; Setter, C.; Dahl, O.; Schild, S.E.; Noack, F. The prognostic impact of tumor cell expression of estrogen receptor-α, progesterone receptor, and androgen receptor in patients irradiated for nonsmall cell lung cancer. Cancer 2012, 118, 157–163. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ding, X.; Shen, Z.; Liu, Z.; An, T.; Duan, J.; Zhong, J.; Wu, M.; Zhao, J.; et al. ERβ localization influenced outcomes of EGFR-TKI treatment in NSCLC patients with EGFR mutations. Sci. Rep. 2015, 5, 11392. [Google Scholar] [CrossRef]
- Nose, N.; Uramoto, H.; Iwata, T.; Hanagiri, T.; Yasumoto, K. Expression of estrogen receptor beta predicts a clinical response and longer progression-free survival after treatment with EGFR-TKI for adenocarcinoma of the lung. Lung Cancer 2011, 71, 350–355. [Google Scholar] [CrossRef]
- Skov, B.G.; Fischer, B.M.; Pappot, H. Oestrogen receptor beta over expression in males with non-small cell lung cancer is associated with better survival. Lung Cancer 2008, 59, 88–94. [Google Scholar] [CrossRef]
- Nikolos, F.; Thomas, C.; Bado, I.; Gustafsson, J. ERβ Sensitizes NSCLC to Chemotherapy by Regulating DNA Damage Response. Mol. Cancer Res. 2018, 16, 233–242. [Google Scholar] [CrossRef]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef]
- Liu, S.; Hu, C.; Li, M.; An, J.; Zhou, W.; Guo, J.; Xiao, Y. Estrogen receptor beta promotes lung cancer invasion via increasing CXCR4 expression. Cell Death Dis. 2022, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, W.; Wang, Y.; Jin, X.; Guo, H.; Wang, Y.; Tang, Y.; Yao, X. Explore the mechanism and substance basis of Mahuang FuziXixin Decoction for the treatment of lung cancer based on network pharmacology and molecular docking. Comput. Biol. Med. 2022, 151, 106293. [Google Scholar] [CrossRef] [PubMed]
- Reick, M.; Garcia, J.A.; Dudley, C.; McKnight, S.L. NPAS2: An analog of clock operative in the mammalian forebrain. Science 2001, 293, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Huang, W.; Zhou, Y.; Wang, Y.; Fu, K.; Zhuang, W. NPAS2 dampens chemo-sensitivity of lung adenocarcinoma cells by enhancing DNA damage repair. Cell Death Dis. 2024, 15, 101. [Google Scholar] [CrossRef]
- Wang, S.; Huang, C.; Zheng, Y.; Wu, X.; Zhong, Y. NPAS2, transcriptionally activated by ARRB1, promotes the malignant behaviours of lung adenocarcinoma cells and regulates the reprogramming of glucose metabolism. Clin. Exp. Pharmacol. Physiol. 2024, 51, e13860. [Google Scholar] [CrossRef]
- He, Y.; Wang, G.; Wang, Q.; Zhao, Z.; Gan, L.; Yang, S.; Wang, Y.; Guo, S.; An, J.; Zhang, J.; et al. Genetic variants in NPAS2 gene and clinical outcomes of resectable non-small-cell lung cancer. Future Oncol. 2021, 17, 795–805. [Google Scholar] [CrossRef]
- Wang, D.; Li, Z.; Messing, E.M.; Wu, G. Activation of Ras/Erk pathway by a novel MET-interacting protein RanBPM. J. Biol. Chem. 2002, 277, 36216–36222. [Google Scholar] [CrossRef]
- Wang, D.; Li, Z.; Schoen, S.R.; Messing, E.M.; Wu, G. A novel MET-interacting protein shares high sequence similarity with RanBPM, but fails to stimulate MET-induced Ras/Erk signaling. Biochem. Biophys. Res. Commun. 2004, 313, 320–326. [Google Scholar] [CrossRef]
- Braun, B.; Pfirrmann, T.; Menssen, R.; Hofmann, K.; Scheel, H.; Wolf, D.H. Gid9, a second RING finger protein contributes to the ubiquitin ligase activity of the Gid complex required for catabolite degradation. FEBS Lett. 2011, 585, 3856–3861. [Google Scholar] [CrossRef]
- Deng, T.; Xie, L.; Xiaofang, C.; Zhang, Z.; Xiao, Y.; Peng, Y.; Yin, L.; Fu, Y.; Li, X. ATM-Mediated translocation of RanBPM regulates DNA damage response by stabilizing p21 in non-small cell lung cancer cells. Cell Oncol. 2024, 47, 245–258. [Google Scholar] [CrossRef]
- Tessari, A.; Parbhoo, K.; Pawlikowski, M.; Fassan, M.; Rulli, E.; Foray, C.; Fabbri, A.; Embrione, V.; Ganzinelli, M.; Capece, M.; et al. RANBP9 affects cancer cells response to genotoxic stress and its overexpression is associated with worse response to platinum in NSCLC patients. Oncogene 2018, 37, 6463–6476. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- DeWeirdt, P.C.; Sangree, A.K.; Hanna, R.E.; Sanson, K.R.; Hegde, M.; Strand, C.; Persky, N.S.; Doench, J.G. Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat. Commun. 2020, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- Borea, R.; Saldanha, E.F.; Maheswaran, S.; Nicolo, E.; Singhal, S.; Pontolillo, L.; de Miguel Perez, D.; Venetis, K.; Dipasquale, A.; Ghazali, N.; et al. Cancer in a drop: Advances in liquid biopsy in 2024. Crit. Rev. Oncol. Hematol. 2025, 213, 104776. [Google Scholar] [CrossRef]
- Smirnov, A.V.; Yunusova, A.M. Novel CRISPR/Cas9-Based Approaches for Quantitative Study of DSB Repair Mechanics. Biochemistry 2025, 90, 437–456. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target/Pathway | Function/Mechanism | Radio-Resistance Role | Therapeutic Strategies | Major Challenges | References |
|---|---|---|---|---|---|
| FLOT1-PD-L1 | FLOT1 regulates PD-L1 expression; PD-L1-Ku interaction enhances DNA repair | Promotes NHEJ repair; suppresses CD8+ T-cell infiltration | Anti-PD-L1 antibodies + RT; FLOT1 inhibitors + STING agonists | Immune-related adverse events; standardization of nuclear PD-L1 assessment | [29,30,31,32,33,34,35] |
| UCHL3 | Deubiquitinase stabilizing Rad51/BRCA2; upregulates PD-L1 via AHR | Enhances HR repair; drives immune evasion | UCHL3 inhibitors (e.g., Perifosine) + PARP inhibitors | Off-target effects; cross-regulation with the AHR–PD-L1 axis | [36,37,38,39,40,41] |
| SERPINE2 | Binds ATM/MRE11 to activate Rad51; induces G2/M arrest | Facilitates HR repair; promotes metastasis | SERPINE2 inhibitors; serum SERPINE2 as biomarker | Lack of high-affinity inhibitors; association with invasive phenotypes | [42,43,44,45,46,47,48,49,50] |
| NF-κB-miR-384-ITGB1 | miR-384 targets ATM/Ku70/Ku80; ITGB1 activates YAP1/ATM-CHK2 | Suppresses HR/NHEJ; promotes EMT | miR-384 restoration (Curcumin); ITGB1/circRNA targeting | Delivery efficiency of miRNAs; complex role of ITGB1 in the EMT | [51,52,53,54,55,56,57,58,59,60,61] |
| NRF2 | Activates ATR/CHK1 and RPA32/TOPBP1; boosts antioxidant/HR pathways | Enhances ROS scavenging and HR repair | NRF2 inhibitor (e.g., ML385) + radiotherapy; NAC to reverse resistance | Protective role of NRF2 in normal tissue; dose-dependent effects | [62,63,64,65] |
| SPOP | Regulates HR repair via Rad51 interaction; stabilizes DDR components | Promotes HR repair; suppresses apoptosis | Maprotiline (SPOP modulator) + immunotherapy | Dual effect on NF-κB pathway; genomic instability risk | [66,67,68,69,70] |
| SIRT3 | NAD-dependent deacetylase; activates ATM-CHK2 signaling in HR repair | Enhances HR repair; induces G2/M arrest | Aspirin activates the AMPK/SIRT3/HK-II axis; SIRT3 inhibitors (e.g., AGK7) | Dual role in metabolism and DNA repair; systemic effects of SIRT3 modulation | [71,72,73,74,75,76,77] |
| GTSE1 | Stabilizes p21; suppresses p53 via MDM2-mediated degradation | Facilitates HR repair; inhibits DDR checkpoint activation | Y18 (GTSE1 inhibitor) + DNA-damaging agents | Efficacy differs by p53 status; long-term toxicity unknown | [78,79,80,81,82,83,84] |
| STX18 | Activates ATR/CHK1 signaling; reduces p53 stability | Enhances HR repair; promotes EMT and cell migration | siRNA-mediated STX18 silencing + radiotherapy; ER-Golgi transport inhibition (e.g., Brefeldin A analogs) | Ubiquitin-dependent regulation may disrupt secretory pathways; potential link to EMT | [85,86,87] |
| RanBP9 | ATM-dependent DDR activator; synergizes with KAT5 for ATM activation | Enhances HR repair; delays senescence | RanBP9 inhibition; combined ATM/KAT5 targeting | Genomic stability concerns in normal tissue; lack of specific inhibitors | [88,89,90] |
| Target/Pathway | Function/Mechanism | Resistance Role | Therapeutic Strategies | Major Challenges | References |
|---|---|---|---|---|---|
| ERCC1/NDRG1 | The ERCC1-XPF complex mediates NER/DSB repair; ERCC1 suppresses NDRG1 | Promotes cisplatin resistance via hypoxia tolerance and apoptosis inhibition | siRNA targeting the ERCC1-E8(+) isoform; DMC inhibiting PI3K/Akt/Snail; iron chelators (e.g., thiosemicarbazones) to upregulate NDRG1 | Hematologic toxicity; splicing plasticity contributing to resistance; dual role (tumor suppressor/promoter); microenvironment dependency | [93,94,95,96,97,98,99] |
| ERβ1 | Upregulates CHK1/CHK2; induces G2-M arrest via cyclin G2 | Enhances chemotherapy sensitivity (cisplatin/doxorubicin) in p53-deficient cells | SERMs; traditional formula MFXD targeting the ERβ/EGFR/HIF1α axis | Gender and subcellular localization affecting efficacy; pro-metastatic risk via circ-TMX4/CXCR4 | [100,101,102,103,104,105,106,107,108,109,110,111,112] |
| NPAS2 | Stabilizes H2AX mRNA to activate HR repair; regulates circadian rhythm | Drives cisplatin resistance via HR repair and glycolysis | CRISPR knockout or small-molecule inhibitors; combination with PARPi leveraging the “BRCAness” phenotype | Circadian disruption from systemic inhibition; glycolytic reprogramming may impair efficacy | [113,114,115,116] |
| RanBP9 | Scaffolds the ATM-p21-USP11 axis; stabilizes p21 via deubiquitination | Supports the DDR and chemoresistance by enhancing DNA repair | Disrupting RanBP9-USP11 interaction; combination with ATR or PARP inhibitors | Genomic stability concerns in normal tissue; lack of specific inhibitors | [117,118,119,120,121,122,123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, X.; Zhou, Y.; Deng, Y. Targeting DNA Damage Response-Mediated Resistance in Non-Small Cell Lung Cancer: From Mechanistic Insights to Drug Development. Curr. Oncol. 2025, 32, 367. https://doi.org/10.3390/curroncol32070367
Gong X, Zhou Y, Deng Y. Targeting DNA Damage Response-Mediated Resistance in Non-Small Cell Lung Cancer: From Mechanistic Insights to Drug Development. Current Oncology. 2025; 32(7):367. https://doi.org/10.3390/curroncol32070367
Chicago/Turabian StyleGong, Xue, Yongzhao Zhou, and Yi Deng. 2025. "Targeting DNA Damage Response-Mediated Resistance in Non-Small Cell Lung Cancer: From Mechanistic Insights to Drug Development" Current Oncology 32, no. 7: 367. https://doi.org/10.3390/curroncol32070367
APA StyleGong, X., Zhou, Y., & Deng, Y. (2025). Targeting DNA Damage Response-Mediated Resistance in Non-Small Cell Lung Cancer: From Mechanistic Insights to Drug Development. Current Oncology, 32(7), 367. https://doi.org/10.3390/curroncol32070367