KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance

 , , ,
, , ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. KRAS Structure and Function
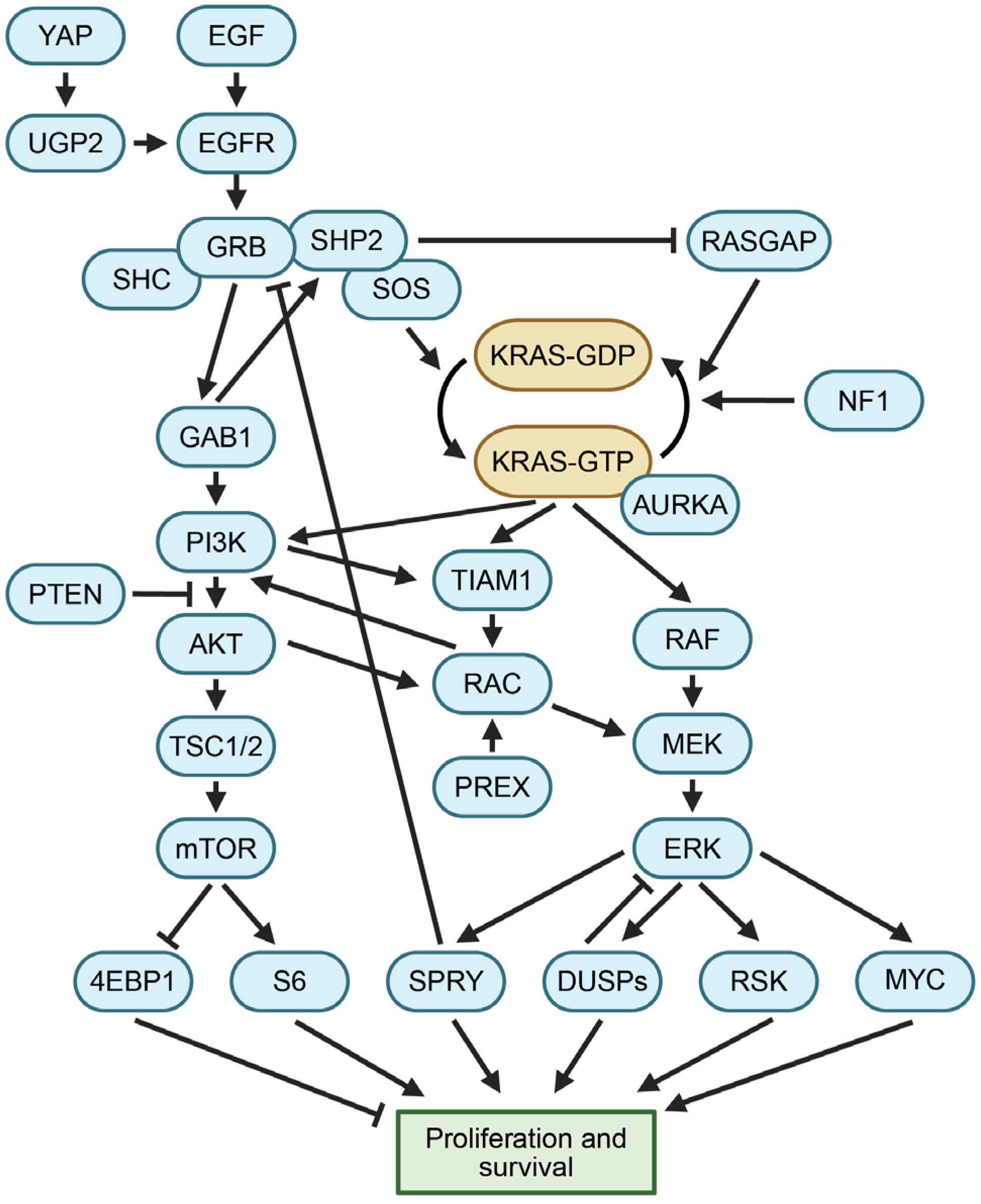
1.2. KRAS Activity Is Regulated at Multiple Levels
1.3. KRAS Regulates Growth through Multiple Mechanisms
1.4. Impact of KRAS on Metabolic Processes
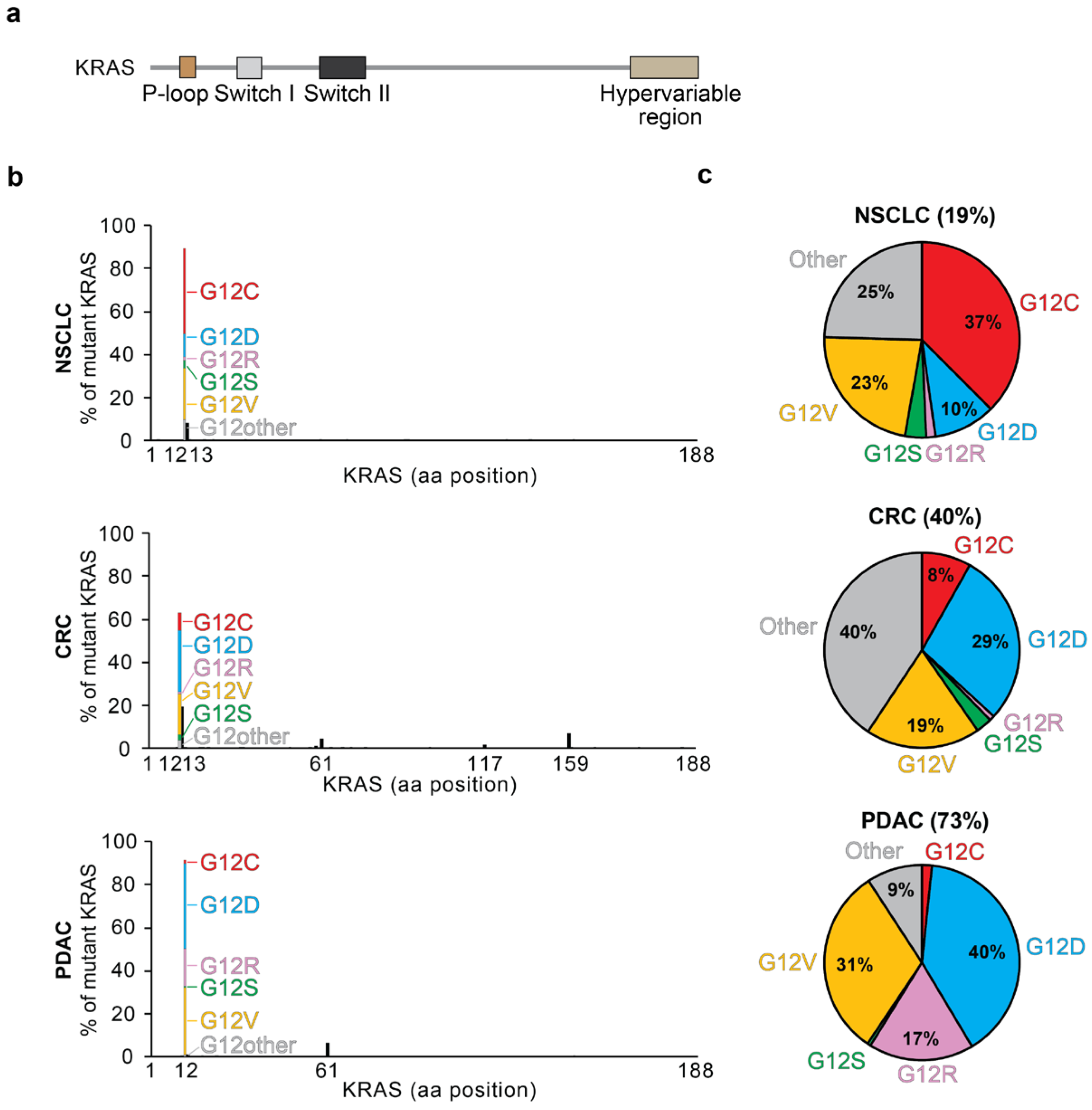
1.5. KRAS Mutations
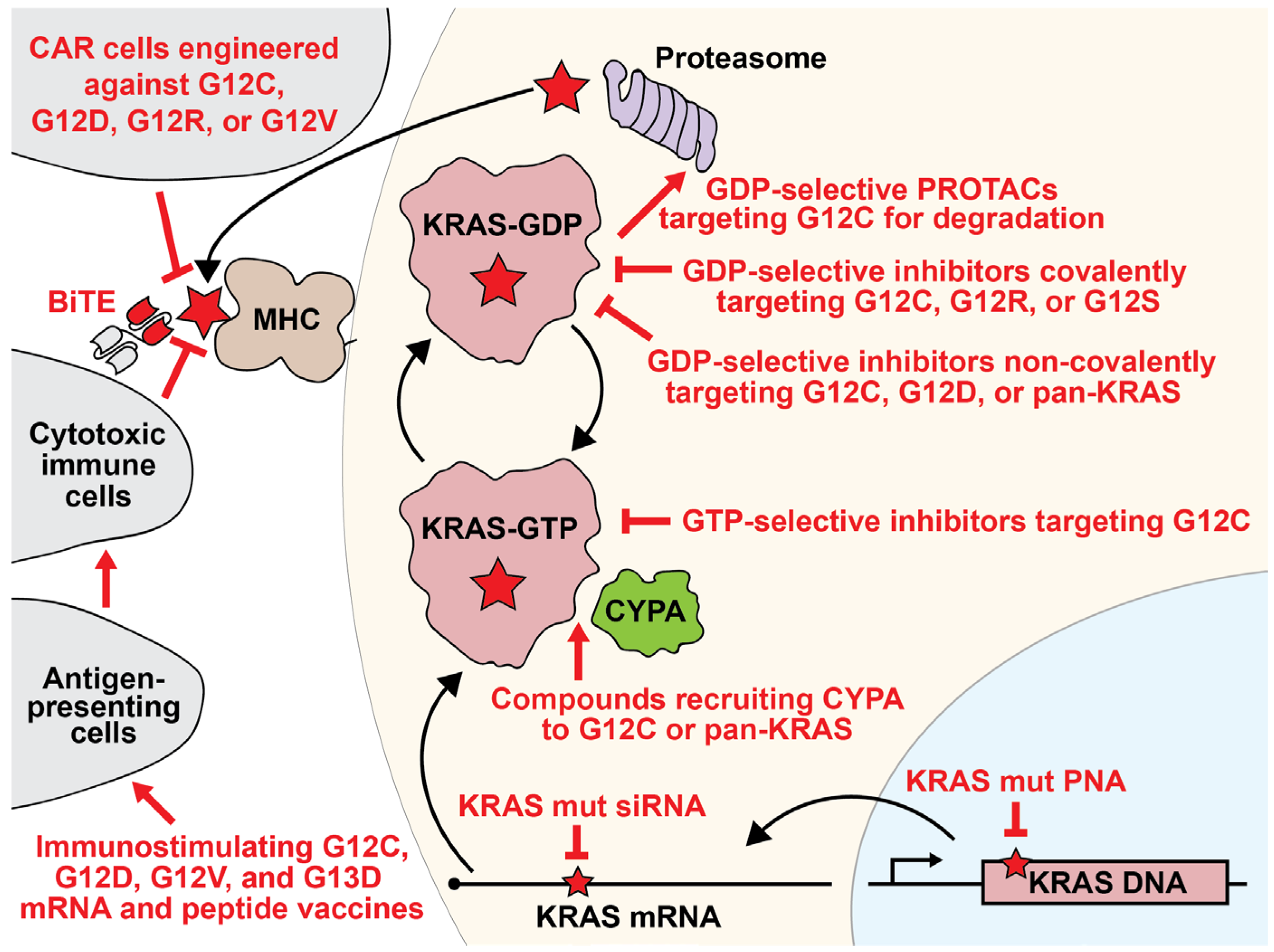
2. Targeted KRAS Inhibitors
2.1. KRAS as a Therapeutic Target
2.2. KRAS G12C-Selective Covalent Inhibitors
2.3. KRAS G12D Inhibitors
2.4. KRAS G12S and KRAS G12R Tool Compounds
2.5. Pan-KRAS Inhibitors
2.6. Cyclophilin A Recruitment
2.7. siRNAs
2.8. Peptide Nucleic Acids
2.9. Destabilizing and Degrading Agents
3. KRAS-Dependent Immunotherapies
3.1. Mutant KRAS Vaccines
3.2. Bispecific T-Cell Engagers
3.3. Adoptive Immunotherapies
4. Mutational Resistance
4.1. Resistance to KRAS Inhibitors
4.2. Drug Resistant Mutations to KRASi
4.3. Copy Number Alterations in KRAS
4.4. Resistance Caused by Other Genes
5. Non-Mutational Resistance
5.1. Rapid Adaptive Resistance
5.2. Reactivation of the RAS Pathway or Alternative Signaling
5.3. Epithelial-to-Mesenchymal Transition
5.4. Tumor Microenvironment
5.5. Drug Efflux Pumps
5.6. Combination Therapies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| 4EBP1 | Eukaryotic Translation Initiation Factor 4E Binding Protein 1, encoded by the gene EIF4EBP1. |
| ALK | ALK Receptor Tyrosine Kinase, also known as Anaplastic Lymphoma Kinase. |
| Amplification | An increase in the number of copies in a region of the genome. |
| APC | APC Regulator Of WNT Signaling Pathway, also known as Adenomatous Polyposis Coli. |
| ARAF | A-Raf Proto-Oncogene, Serine/Threonine Kinase. |
| ARF | One of the proteins encoded by the gene Cyclin Dependent Kinase Inhibitor 2A, or CDKN2A. Also known as p19-ARF. |
| AURKA | Aurora Kinase A. |
| BiTE | Bispecific T-cell engager. |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase. |
| CAB39 | Calcium-Binding Protein 39. |
| CAS9 | CRISPR-associated protein 9, used together with CRISPR to cleave DNA. |
| CCL2 | C-C Motif Chemokine Ligand 2. |
| CDK4 | Cyclin-Dependent Kinase 4. |
| CDK6 | Cyclin-Dependent Kinase 6. |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A, encodes p16-INK4A and ARF. |
| CDKN2B | Cyclin-Dependent Kinase Inhibitor 2B |
| COVID-19 | Coronavirus disease of 2019, caused by the virus SARS-CoV-2 |
| CRAF | Raf-1 Proto-Oncogene, Serine/Threonine Kinase, also known as RAF1. |
| CRC | Colorectal carcinoma or colorectal cancer. |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats, often used to knock out a gene of interest. |
| CRISPRi | Inhibitory form of CRISPR that uses dead Cas9 deficient at cutting DNA to recruit negative regulatory transcription factors to the promoter of a gene of interest. |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4, an immune checkpoint protein. |
| CTNNB1 | Catenin Beta 1, also known as β-catenin. |
| CYCA | Cyclophilin A. |
| Cyclins | A family of proteins that bind to cyclin-dependent kinases and regulate cell cycle. |
| DC | Dendritic cell. |
| DTP | Drug tolerant persister cells. |
| DUSPs | Dual-Specificity Phosphatase protein family. |
| E2F | E2F Transcription Factor. |
| EGF | Epidermal Growth Factor. |
| EGFR | Epidermal Growth Factor Receptor. |
| EMT | Epithelial-to-mesenchymal transition. |
| ERK1 | Mitogen-Activated Protein Kinase 3, also known as Extracellular Signal-Regulated Kinase 1, a protein encoded by the gene MAPK3. |
| ERK2 | Mitogen-Activated Protein Kinase 2, also known as Extracellular Signal-Regulated Kinase 2, a protein encoded by the gene MAPK1. |
| FDA | United States Food and Drug Administration. |
| FGFR | Fibroblast Growth Factor Receptor 1. |
| G12Ci | An inhibitor that is selective for KRAS with the 12th amino acid altered from glycine to cysteine. |
| GAB1 | GRB2-associated binding protein 1. |
| GAP | GTPase-activating protein, promotes hydrolysis of GTP to GDP |
| GDP | Guanosine diphosphate. KRAS is inactive when bound to GDP. |
| GEF | Guanine nucleotide exchange factor, promotes exchange of GDP to GTP |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor, also known as colony-stimulating factor 2. |
| GRB2 | Growth Factor Receptor Bound Protein 2. |
| GTP | Guanosine triphosphate. KRAS is active when bound to GTP. |
| HDAC5 | Histone Deacetylase 5. |
| HLA | Human leukocyte antigens. |
| HRAS | HRas Proto-Oncogene, GTPase, also known as Harvey Rat Sarcoma Viral Oncoprotein. |
| ICMT | Isoprenylcysteine Carboxyl Methyltransferase. |
| IDH1 | Isocitrate Dehydrogenase (NADP(+)) 1. |
| IDH2 | Isocitrate Dehydrogenase (NADP(+)) 2. |
| IRE1α | Endoplasmic Reticulum To Nucleus Signaling 1, encoded by the gene ERN1. |
| KEAP1 | Kelch-Like ECH-Associated Protein 1. |
| KRAS | KRAS Proto-Oncogene, GTPase, also known as Kirsten Rat Sarcoma Viral Proto-Oncogene and also abbreviated at K-Ras. |
| KRASi | An entity that inhibits KRAS. |
| KSR | Kinase Suppressor Of Ras 1, encoded by the gene KSR1. |
| MDR1 | ATP-Binding Cassette Subfamily B Member 1, also known as Multidrug Resistance Protein 1, encoded by the gene ABCB1. |
| MEK | Mitogen-Activated Protein Kinase Kinase 1, encoded by the gene MAP2K1. |
| MET | MET Proto-Oncogene, Receptor Tyrosine Kinase. |
| MHC | Major histocompatibility complex. |
| MRAS | Muscle RAS Oncogene Homolog. |
| mTCR | Murine T-cell receptor. |
| mTOR | Mechanistic Target Of Rapamycin Kinase. |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor, also known as c-MYC and as Avian Myelocytomatosis Viral Oncogene Homolog. |
| NF1 | Neurofibromatosis type 1. |
| NRAS | NRAS Proto-Oncogene, GTPase, also known as Neuroblastoma RAS Viral (V-Ras) Oncogene Homolog. |
| NSCLC | Non-small cell lung cancer. |
| p53 | Tumor Protein P53, encoded by the gene TP53. |
| PD-1 | Programmed Cell Death Protein 1. |
| PDAC | Pancreatic ductal adenocarcinoma. |
| PD-L1 | CD274 Molecule, also known as Programmed Cell Death 1 Ligand 1, encoded by the gene CD274. |
| PTCH1 | Patched 1. |
| SHP2 | Protein Tyrosine Phosphatase Non-Receptor Type 11, encoded by the gene PTPN11. |
| PBL | Peripheral blood lymphocytes. |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha, also known as PtdIns-3-Kinase Subunit P110-Alpha, encodes the p110α subunit of PI3K. |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate. |
| PIP3 | Phosphatidylinositol-3,4,5-triphosphate. |
| PNA | Peptide nucleic acid, a synthetic molecule hybridizing DNA bases with a peptide polymer chain. |
| PREX1 | Phosphatidylinositol-3,4,5-Trisphosphate Dependent Rac Exchange Factor 1. |
| PROTAC | Proteolysis targeting chimeras containing a domain binding the protein of interest linked to a domain that recruits an E3 ubiquitin ligase. |
| PTEN | Phosphatase And Tensin Homolog, also known as Phosphatidylinositol 3,4,5-Trisphosphate 3-Phosphatase And Dual-Specificity Protein Phosphatase PTEN. |
| RAB | RAB1A, Member of RAS Oncogene Family. |
| RAN | RAN, Member of RAS Oncogene Family. |
| RAS-GAP | RAS P21 Protein Activator 1, also known as p120GAP, encoded by the gene RASA1. |
| RB1 | RB Transcriptional Corepressor 1, also known as Retinoblastoma 1. |
| RCE1 | Ras Converting CAAX Endopeptidase 1. |
| RET | Ret Proto-Oncogene, also known as Rearranged During Transfection. |
| RGS | Regulator Of G Protein Signaling. |
| RHO | Rhodopsin. |
| RICTOR | RPTOR Independent Companion Of MTOR Complex 2. |
| RSK1 | Ribosomal Protein S6 Kinase A1. |
| S6 | Ribosomal Protein S6. |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2, the virus that causes COVID-19. |
| SHOC2 | SHOC2 Leucine Rich Repeat Scaffold Protein. |
| siRNA | Small interfering ribonucleic acid, used to decrease expression of target messenger RNAs. |
| SMARCA4 | SWI/SNF Related, Matrix Associated, Actin-Dependent Regulator Of Chromatin, Subfamily A, Member 4. |
| SOS1 | SOS Ras/Rac Guanine Nucleotide Exchange Factor 1, also known as Son Of Sevenless Homolog 1. |
| SPRED1 | Sprouty-Related EVH1 Domain Containing 1. |
| SPRY1 | Sprouty RTK Signaling Antagonist 1. |
| Synthetic lethal/collateral dependency | Two genes whose alterations are lethal to cells only when they are in combination. |
| TCR | T-cell receptor. |
| TEAD | TEA Domain Transcription Factor 1. |
| TGFβ | Transforming Growth Factor Beta 1. |
| TME | Tumor microenvironment. |
| TSC1 | TSC Complex Subunit 1, also known as Tuberous Sclerosis 1 Protein. |
| TSC2 | TSC Complex Subunit 2, also known as Tuberous Sclerosis 2 Protein. |
| UGP2 | UDP-Glucose Pyrophosphorylase 2. |
| VEGF | Vascular Endothelial Growth Factor. |
| YAP | Yes1-Associated Transcriptional Regulator. |
References
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Milburn, M.V.; Tong, L.; deVos, A.M.; Brünger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.-H. Molecular Switch for Signal Transduction: Structural Differences Between Active and Inactive Forms of Protooncogenic Ras Proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted Therapies: Is the Undruggable Drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yuan, T.; Qian, M.; Yan, F.; Yang, L.; He, Q.; Yang, B.; Lu, J.; Zhu, H. Post-Translational Modification of KRAS: Potential Targets for Cancer Therapy. Acta Pharmacol. Sin. 2021, 42, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A Polybasic Domain or Palmitoylation Is Required in Addition to the CAAX Motif to Localize P21ras to the Plasma Membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Chardin, P.; Camonis, J.H.; Gale, N.W.; Van Aelst, L.; Schlessinger, J.; Wigler, M.H.; Bar-Sagi, D. Human Sos1: A Guanine Nucleotide Exchange Factor for Ras That Binds to GRB2. Science 1993, 260, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Hui, C.; Pawson, T. SH2-Containing Phosphotyrosine Phosphatase as a Target of Protein-Tyrosine Kinases. Science 1993, 259, 1607–1611. [Google Scholar] [CrossRef]
- Bonfini, L.; Karlovich, C.A.; Dasgupta, C.; Banerjee, U. The Son of Sevenless Gene Product: A Putative Activator of Ras. Science 1992, 255, 603–606. [Google Scholar] [CrossRef]
- Bollag, G.; McCormick, F. Differential Regulation of rasGAP and Neurofibromatosis Gene Product Activities. Nature 1991, 351, 576–579. [Google Scholar] [CrossRef]
- Li, C.; Vides, A.; Kim, D.; Xue, J.Y.; Zhao, Y.; Lito, P. The G Protein Signaling Regulator RGS3 Enhances the GTPase Activity of KRAS. Science 2021, 374, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-Canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Casteel, D.E.; Prakash, P.; Tan, L.; Van Der Hoeven, D.; Salim, A.A.; Kim, C.; Capon, R.J.; Lacey, E.; Cunha, S.R.; et al. AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol. Cell. Biol. 2016, 36, 3086–3099. [Google Scholar] [CrossRef] [PubMed]
- Durrant, D.E.; Morrison, D.K. Targeting the Raf Kinases in Human Cancer: The Raf Dimer Dilemma. Br. J. Cancer 2018, 118, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells with Wild-Type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of Receptor Tyrosine Kinase Signaling: Transient versus Sustained Extracellular Signal-Regulated Kinase Activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sanclemente, M.; Nieto, P.; Garcia-Alonso, S.; Fernández-García, F.; Esteban-Burgos, L.; Guerra, C.; Drosten, M.; Caleiras, E.; Martinez-Torrecuadrada, J.; Santamaría, D.; et al. RAF1 Kinase Activity Is Dispensable for KRAS/P53 Mutant Lung Tumor Progression. Cancer Cell 2021, 39, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; Zhu, H.; Silva, R.L.A.; Mills, J.R.; Teruya-Feldstein, J.; Lowe, S.W.; Tam, W.; Pelletier, J.; Wendel, H.-G. Tumorigenic Activity and Therapeutic Inhibition of Rheb GTPase. Genes Dev. 2008, 22, 2178–2188. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J. KRASG12R-Independent Macropinocytosis in Pancreatic Cancer. In Macropinocytosis; Commisso, C., Ed.; Subcellular Biochemistry; Springer International Publishing: Cham, Switzerland, 2022; Volume 98, pp. 205–221. ISBN 978-3-030-94003-4. [Google Scholar]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of Protein Is an Amino Acid Supply Route in Ras-Transformed Cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Camelo, F.; Le, A. The Intricate Metabolism of Pancreatic Cancers. In The Heterogeneity of Cancer Metabolism; Le, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2021; Volume 1311, pp. 77–88. ISBN 978-3-030-65767-3. [Google Scholar]
- Amendola, C.R.; Mahaffey, J.P.; Parker, S.J.; Ahearn, I.M.; Chen, W.-C.; Zhou, M.; Court, H.; Shi, J.; Mendoza, S.L.; Morten, M.J.; et al. KRAS4A Directly Regulates Hexokinase 1. Nature 2019, 576, 482–486. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-Based Functional Profiling of RASopathies and Oncogenic RAS Mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579. [Google Scholar] [CrossRef] [PubMed]
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.-J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-Specific Oncogenic Activity of KRASA146T. Cancer Discov. 2019, 9, 738–755. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The Origins and Genetic Interactions of KRAS Mutations Are Allele- and Tissue-Specific. Nat. Commun. 2021, 12, 1808. [Google Scholar] [CrossRef]
- Feig, L.A.; Cooper, G.M. Relationship among Guanine Nucleotide Exchange, GTP Hydrolysis, and Transforming Potential of Mutated Ras Proteins. Mol. Cell. Biol. 1988, 8, 2472–2478. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends. Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Patterson, M. The Trouble with Smoking. Nat. Rev. Genet. 2000, 1, 168. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Garland Science, Taylor & Francis Group, LLC: Abingdon, UK, 2014; ISBN 978-0-8153-4219-9. [Google Scholar]
- Hainaut, P. Patterns of P53 G->T Transversions in Lung Cancers Reflect the Primary Mutagenic Signature of DNA-Damage by Tobacco Smoke. Carcinogenesis 2001, 22, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Temko, D.; Tomlinson, I.P.M.; Severini, S.; Schuster-Böckler, B.; Graham, T.A. The Effects of Mutational Processes and Selection on Driver Mutations across Cancer Types. Nat. Commun. 2018, 9, 1857. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.H.; Adib, E.; Kwiatkowski, D.J. Distribution of KRAS G12C Somatic Mutations across Race, Sex, and Cancer Type. N. Engl. J. Med. 2021, 384, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Salmón, M.; Álvarez-Díaz, R.; Fustero-Torre, C.; Brehey, O.; Lechuga, C.G.; Sanclemente, M.; Fernández-García, F.; López-García, A.; Martín-Guijarro, M.C.; Rodríguez-Perales, S.; et al. Kras Oncogene Ablation Prevents Resistance in Advanced Lung Adenocarcinomas. J. Clin. Investig. 2023, 133, e164413. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Bergo, M.O.; Gavino, B.J.; Hong, C.; Beigneux, A.P.; McMahon, M.; Casey, P.J.; Young, S.G. Inactivation of Icmt Inhibits Transformation by Oncogenic K-Ras and B-Raf. J. Clin. Investig. 2004, 113, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Chai, T.F.; Manu, K.A.; Casey, P.J.; Wang, M. Isoprenylcysteine Carboxylmethyltransferase Is Required for the Impact of Mutant KRAS on TAZ Protein Level and Cancer Cell Self-Renewal. Oncogene 2020, 39, 5373–5389. [Google Scholar] [CrossRef]
- Chiu, V.K.; Bivona, T.; Hach, A.; Sajous, J.B.; Silletti, J.; Wiener, H.; Johnson, R.L.; Cox, A.D.; Philips, M.R. Ras Signalling on the Endoplasmic Reticulum and the Golgi. Nat. Cell Biol. 2002, 4, 343–350. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G.; Kang, R. Oncogenic KRAS Blockade Therapy: Renewed Enthusiasm and Persistent Challenges. Mol. Cancer 2021, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging Ras Back in the Ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS G12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C -Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.-J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Xiao, H.; Shen, P.; Yang, Y.; Xue, J.; Yang, Y.; Shang, Y.; Zhang, L.; Li, X.; Zhang, Y.; et al. KRAS(G12D) Can Be Targeted by Potent Inhibitors via Formation of Salt Bridge. Cell Discov. 2022, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-Tumor Efficacy of a Potent and Selective Non-Covalent KRASG12D Inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.-K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Tria, S.M.; Burge, M.E.; Whitehall, V.L.J. The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers. Cancers 2023, 15, 2375. [Google Scholar] [CrossRef]
- Zhang, Z.; Guiley, K.Z.; Shokat, K.M. Chemical Acylation of an Acquired Serine Suppresses Oncogenic Signaling of K-Ras(G12S). Nat. Chem. Biol. 2022, 18, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Morstein, J.; Ecker, A.K.; Guiley, K.Z.; Shokat, K.M. Chemoselective Covalent Modification of K-Ras(G12R) with a Small Molecule Electrophile. J. Am. Chem. Soc. 2022, 144, 15916–15921. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Shokat, K.M. Disease-Causing Mutations in the G Protein Gαs Subvert the Roles of GDP and GTP. Cell 2018, 173, 1254–1264.e11. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS Inhibitor Disables Oncogenic Signalling and Tumour Growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef]
- Kim, S.; Wolfe, A.; Kim, S.E. Targeting Cancer’s Sweet Spot: UGP2 as a Therapeutic Vulnerability. Mol. Cell. Oncol. 2021, 8, 1990676. [Google Scholar] [CrossRef]
- Schulze, C.J.; Seamon, K.J.; Zhao, Y.; Yang, Y.C.; Cregg, J.; Kim, D.; Tomlinson, A.; Choy, T.J.; Wang, Z.; Sang, B.; et al. Chemical Remodeling of a Cellular Chaperone to Target the Active State of Mutant KRAS. Science 2023, 381, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.N.; Lee, S.-Y.; Lee, S.; Youn, H.; Im, H.-J. Lipid Nanoparticles for Delivery of RNA Therapeutics: Current Status and the Role of in Vivo Imaging. Theranostics 2022, 12, 7509–7531. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Azam, S.H.; Feng, A.Y.; Gutierrez-Ford, C.; Huggins, H.; Pallan, P.S.; Van Swearingen, A.E.D.; Egli, M.; Cox, A.D.; Der, C.J.; et al. Silencing of Oncogenic KRAS by Mutant-Selective Small Interfering RNA. ACS Pharmacol. Transl. Sci. 2021, 4, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Fellmann, C.; Lee, C.-S.; Ritchie, C.D.; Thapar, V.; Lee, L.C.; Hsu, D.J.; Grace, D.; Carver, J.O.; Zuber, J.; et al. Development of siRNA Payloads to Target KRAS-Mutant Cancer. Cancer Discov. 2014, 4, 1182–1197. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; De Stanchina, E.; Mazutis, L.; et al. Rapid Non-Uniform Adaptation to Conformation-Specific KRAS(G12C) Inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-Dependent Tumors with AZD4785, a High-Affinity Therapeutic Antisense Oligonucleotide Inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS Is a Druggable Target for Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [PubMed]
- Shai, A.; Galouk, E.; Miari, R.; Tareef, H.; Sammar, M.; Zeidan, M.; Rayan, A.; Falah, M. Inhibiting Mutant KRAS G12D Gene Expression Using Novel Peptide Nucleic Acid-based Antisense: A Potential New Drug Candidate for Pancreatic Cancer. Oncol. Lett. 2022, 23, 130. [Google Scholar] [CrossRef]
- Rothman, J.H.; Surriga, O.; de Stanchina, E.; Vasudeva, S.D.; Schwartz, G.K. Obstruction of BRAFV600E Transcription by Complementary PNA Oligomers as a Means to Inhibit BRAF-Mutant Melanoma Growth. Cancer Gene Ther. 2017, 24, 401–408. [Google Scholar] [CrossRef]
- Zeng, C.; Xing, W.; Liu, Y. Identification of UGP2 as a Progression Marker That Promotes Cell Growth and Motility in Human Glioma. J. Cell. Biochem. 2019, 120, 12489–12499. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRAS G12C by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Janssen, K.; Claes, F.; Van De Velde, D.; Wehbi, V.L.; Houben, B.; Lampi, Y.; Nys, M.; Khodaparast, L.; Khodaparast, L.; Louros, N.; et al. Exploiting the Intrinsic Misfolding Propensity of the KRAS Oncoprotein. Proc. Natl. Acad. Sci. USA 2023, 120, e2214921120. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, P.; Bekaii-Saab, T.; McIntyre, K.; Rosemurgy, A.; Ross, S.B.; Richards, D.A.; Fisher, W.E.; Flynn, P.J.; Mattson, A.; Coeshott, C.; et al. A Phase 2 Randomized Placebo-Controlled Adjuvant Trial of GI-4000, a Recombinant Yeast Expressing Mutated RAS Proteins in Patients with Resected Pancreas Cancer. J. Pancreat. Cancer 2021, 7, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Chaft, J.E.; Litvak, A.; Arcila, M.E.; Patel, P.; D’Angelo, S.P.; Krug, L.M.; Rusch, V.; Mattson, A.; Coeshott, C.; Park, B.; et al. Phase II Study of the GI-4000 KRAS Vaccine After Curative Therapy in Patients with Stage I-III Lung Adenocarcinoma Harboring a KRAS G12C, G12D, or G12V Mutation. Clin. Lung Cancer 2014, 15, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhang, Q.; Sei, S.; Shoemaker, R.H.; Lubet, R.A.; Wang, Y.; You, M. Immunoprevention of KRAS-Driven Lung Adenocarcinoma by a Multipeptide Vaccine. Oncotarget 2017, 8, 82689–82699. [Google Scholar] [CrossRef]
- Gritstone Bio. A Study of a Personalized Cancer Vaccine Targeting Shared Neoantigens; ClinicalTrials.Gov ID NCT03953235; Gritstone Bio: Emeryville, CA, USA, 2023. [Google Scholar]
- Lv, X.; Lu, X.; Cao, J.; Luo, Q.; Ding, Y.; Peng, F.; Pataer, A.; Lu, D.; Han, D.; Malmberg, E.; et al. Modulation of the Proteostasis Network Promotes Tumor Resistance to Oncogenic KRAS Inhibitors. Science 2023, 381, eabn4180. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Hui, A.-M. The Paradigm Shift in Treatment from Covid-19 to Oncology with mRNA Vaccines. Cancer Treat. Rev. 2022, 107, 102405. [Google Scholar] [CrossRef]
- Xie, W.; Chen, B.; Wong, J. Evolution of the Market for mRNA Technology. Nat. Rev. Drug Discov. 2021, 20, 735–736. [Google Scholar] [CrossRef]
- Zhu, W.M.; Middleton, M.R. Combination Therapies for the Optimisation of Bispecific T-Cell Engagers in Cancer Treatment. Immunother. Adv. 2023, 3, ltad013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shokat, K.M. Bifunctional Small-Molecule Ligands of K-Ras Induce Its Association with Immunophilin Proteins. Angew. Chem. Int. Ed. 2019, 58, 16314–16319. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Maso, L.; Araki, K.Y.; Koide, A.; Hayman, J.; Akkapeddi, P.; Bang, I.; Neel, B.G.; Koide, S. Creating MHC-Restricted Neoantigens with Covalent Inhibitors That Can Be Targeted by Immune Therapy. Cancer Discov. 2023, 13, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.; Restifo, N.P.; Yang, J.C. Identification of T-Cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Blanchard, T.; Cesare, J.; Ford, M.J.; Richman, L.P.; Xu, C.; Baroja, M.L.; McCuaig, S.; Costeas, C.; Gabunia, K.; et al. Biochemical and Functional Characterization of Mutant KRAS Epitopes Validates This Oncoprotein for Immunological Targeting. Nat. Commun. 2021, 12, 4365. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Administering Peripheral Blood Lymphocytes Transduced with a Murine T-Cell Receptor Recognizing the G12D Variant of Mutated RAS in HLA-A*11:01 Patients 2023; National Cancer Institute: Bethesda, MD, USA, 2023. [Google Scholar]
- National Cancer Institute. Administering Peripheral Blood Lymphocytes Transduced with a Murine T-Cell Receptor Recognizing the G12V Variant of Mutated RAS in HLA-A*11:01 Patients 2024; National Cancer Institute: Bethesda, MD, USA, 2024. [Google Scholar]
- University of Pennsylvania. DC Vaccine in Pancreatic Cancer; ClinicalTrials.Gov ID NCT03592888; University of Pennsylvania: Philadelphia, PA, USA, 2023. [Google Scholar]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRAS G12C Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse Alterations Associated with Resistance to KRAS(G12C) Inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Preston, B.D.; Albertson, T.M.; Herr, A.J. DNA Replication Fidelity and Cancer. Semin. Cancer Biol. 2010, 20, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef]
- Diaz Jr, L.A.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The Molecular Evolution of Acquired Resistance to Targeted EGFR Blockade in Colorectal Cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Mezzadra, R.; Sinopoli, J.; Bian, Y.; Marasco, M.; Kaplun, E.; Gao, Y.; Zhao, H.; Paula, A.D.C.; Zhu, Y.; et al. Molecular Characterization of Acquired Resistance to KRASG12C-EGFR Inhibition in Colorectal Cancer. Cancer Discov. 2022, 13, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS–MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Secq, V.; Villeret, J.; Fina, F.; Carmassi, M.; Carcopino, X.; Garcia, S.; Metellus, I.; Boubli, L.; Iovanna, J.; Charpin, C. Triple Negative Breast Carcinoma EGFR Amplification Is Not Associated with EGFR, Kras or ALK Mutations. Br. J. Cancer 2014, 110, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. Amplifikation von MYCN in Neuroblastomen: Paradigma für die klinische Anwendung einer Onkogenveränderung. Klin. Padiatr. 1990, 202, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Der, C.J.; Cox, A.D. The Role of Wild Type RAS Isoforms in Cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef]
- Timar, J.; Kashofer, K. Molecular Epidemiology and Diagnostics of KRAS Mutations in Human Cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Sum, E.Y.M.; Lechuga, C.G.; Simón-Carrasco, L.; Jacob, H.K.C.; García-Medina, R.; Huang, S.; Beijersbergen, R.L.; Bernards, R.; Barbacid, M. Loss of P53 Induces Cell Proliferation via Ras-Independent Activation of the Raf/Mek/Erk Signaling Pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 15155–15160. [Google Scholar] [CrossRef]
- Paniagua, G.; Jacob, H.K.C.; Brehey, O.; García-Alonso, S.; Lechuga, C.G.; Pons, T.; Musteanu, M.; Guerra, C.; Drosten, M.; Barbacid, M. KSR Induces RAS-independent MAPK Pathway Activation and Modulates the Efficacy of KRAS Inhibitors. Mol. Oncol. 2022, 16, 3066–3081. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Orozco, P.; Gutmann, D.H. RAS and beyond: The Many Faces of the Neurofibromatosis Type 1 Protein. Dis. Models Mech. 2022, 15, dmm049362. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903.e15. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS G12C Inhibition Produces a Driver-Limited State Revealing Collateral Dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef] [PubMed]
- Bhang, H.C.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying Clonal Dynamics in Response to Cancer Therapy Using High-Complexity Barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Dobson, S.M.; García-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs. Cancer Discov. 2020, 10, 568–587. [Google Scholar] [CrossRef]
- Ning, W.; Marti, T.M.; Dorn, P.; Peng, R.-W. Non-Genetic Adaptive Resistance to KRASG12C Inhibition: EMT Is Not the Only Culprit. Front. Oncol. 2022, 12, 1004669. [Google Scholar] [CrossRef]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Portell, A.; Ivanova, E.V.; Lizotte, P.H.; Mahadevan, N.R.; Greene, J.R.; Vajdi, A.; Gurjao, C.; Teceno, T.; Taus, L.J.; et al. Dynamic Single-Cell RNA Sequencing Identifies Immunotherapy Persister Cells Following PD-1 Blockade. J. Clin. Investig. 2021, 131, e135038. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK Intercepts Heterogeneous Mechanisms of Acquired Resistance to Anti-EGFR Therapies in Colorectal Cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Yang, S. Overcoming Resistance to Drugs Targeting KRAS Mutation. Innovation 2020, 1, 100035. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative Feedback Regulation of the ERK1/2 MAPK Pathway. Cell Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef]
- Hagenbeek, T.J.; Zbieg, J.R.; Hafner, M.; Mroue, R.; Lacap, J.A.; Sodir, N.M.; Noland, C.L.; Afghani, S.; Kishore, A.; Bhat, K.P.; et al. An Allosteric Pan-TEAD Inhibitor Blocks Oncogenic YAP/TAZ Signaling and Overcomes KRAS G12C Inhibitor Resistance. Nat. Cancer 2023, 4, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Kimura, R.; Hirade, K.; Yanase, S.; Nishioka, Y.; Kasuga, N.; Yamaguchi, R.; Ebi, H. Scribble Mis-Localization Induces Adaptive Resistance to KRAS G12C Inhibitors through Feedback Activation of MAPK Signaling Mediated by YAP-Induced MRAS. Nat. Cancer 2023, 4, 829–843. [Google Scholar] [CrossRef]
- Tammaccaro, S.L.; Prigent, P.; Le Bail, J.-C.; Dos-Santos, O.; Dassencourt, L.; Eskandar, M.; Buzy, A.; Venier, O.; Guillemot, J.-C.; Veeranagouda, Y.; et al. TEAD Inhibitors Sensitize KRASG12C Inhibitors via Dual Cell Cycle Arrest in KRASG12C-Mutant NSCLC. Pharmaceuticals 2023, 16, 553. [Google Scholar] [CrossRef]
- Solanki, H.S.; Welsh, E.A.; Fang, B.; Izumi, V.; Darville, L.; Stone, B.; Franzese, R.; Chavan, S.; Kinose, F.; Imbody, D.; et al. Cell Type–Specific Adaptive Signaling Responses to KRASG12C Inhibition. Clin. Cancer Res. 2021, 27, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Massagué, J. Epithelial-Mesenchymal Transitions. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, K.K.; McAndrews, K.M.; LeBleu, V.S.; Yang, S.; Lyu, H.; Li, B.; Sockwell, A.M.; Kirtley, M.L.; Morse, S.J.; Moreno Diaz, B.A.; et al. KRASG12D Inhibition Reprograms the Microenvironment of Early and Advanced Pancreatic Cancer to Promote FAS-Mediated Killing by CD8+ T Cells. Cancer Cell 2023, 41, 1606–1620.e8. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 Inhibition Diminishes KRASG12C Cycling and Promotes Tumor Microenvironment Remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef] [PubMed]
- Kurimchak, A.M.; Herrera-Montávez, C.; Montserrat-Sangrà, S.; Araiza-Olivera, D.; Hu, J.; Neumann-Domer, R.; Kuruvilla, M.; Bellacosa, A.; Testa, J.R.; Jin, J.; et al. The Drug Efflux Pump MDR1 Promotes Intrinsic and Acquired Resistance to PROTACs in Cancer Cells. Sci. Signal. 2022, 15, eabn2707. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-D.; Zhang, M.; Cai, C.-Y.; Teng, Q.-X.; Wang, J.-Q.; Fu, Y.-G.; Cui, Q.; Patel, K.; Wang, D.-T.; Chen, Z.-S. Overexpression of ABCB1 Associated with the Resistance to the KRAS-G12C Specific Inhibitor ARS-1620 in Cancer Cells. Front. Pharmacol. 2022, 13, 843829. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Malek, S. The Promise and Peril of KRAS G12C Inhibitors. Cancer Cell 2021, 39, 1059–1061. [Google Scholar] [CrossRef]
- O’Sullivan, É.; Keogh, A.; Henderson, B.; Finn, S.P.; Gray, S.G.; Gately, K. Treatment Strategies for KRAS-Mutated Non-Small-Cell Lung Cancer. Cancers 2023, 15, 1635. [Google Scholar] [CrossRef]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRASG12C Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.-H.I. KRAS Inhibitors– Yes but What next? Direct Targeting of KRAS–Vaccines, Adoptive T Cell Therapy and Beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ash, L.J.; Busia-Bourdain, O.; Okpattah, D.; Kamel, A.; Liberchuk, A.; Wolfe, A.L. KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance. Curr. Oncol. 2024, 31, 2024-2046. https://doi.org/10.3390/curroncol31040150
Ash LJ, Busia-Bourdain O, Okpattah D, Kamel A, Liberchuk A, Wolfe AL. KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance. Current Oncology. 2024; 31(4):2024-2046. https://doi.org/10.3390/curroncol31040150
Chicago/Turabian StyleAsh, Leonard J., Ottavia Busia-Bourdain, Daniel Okpattah, Avrosina Kamel, Ariel Liberchuk, and Andrew L. Wolfe. 2024. "KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance" Current Oncology 31, no. 4: 2024-2046. https://doi.org/10.3390/curroncol31040150
APA StyleAsh, L. J., Busia-Bourdain, O., Okpattah, D., Kamel, A., Liberchuk, A., & Wolfe, A. L. (2024). KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance. Current Oncology, 31(4), 2024-2046. https://doi.org/10.3390/curroncol31040150