Successful Multimodal Treatment of Intracranial Growing Teratoma Syndrome with Malignant Features

 , , , , , , and add
Show full author list
, , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
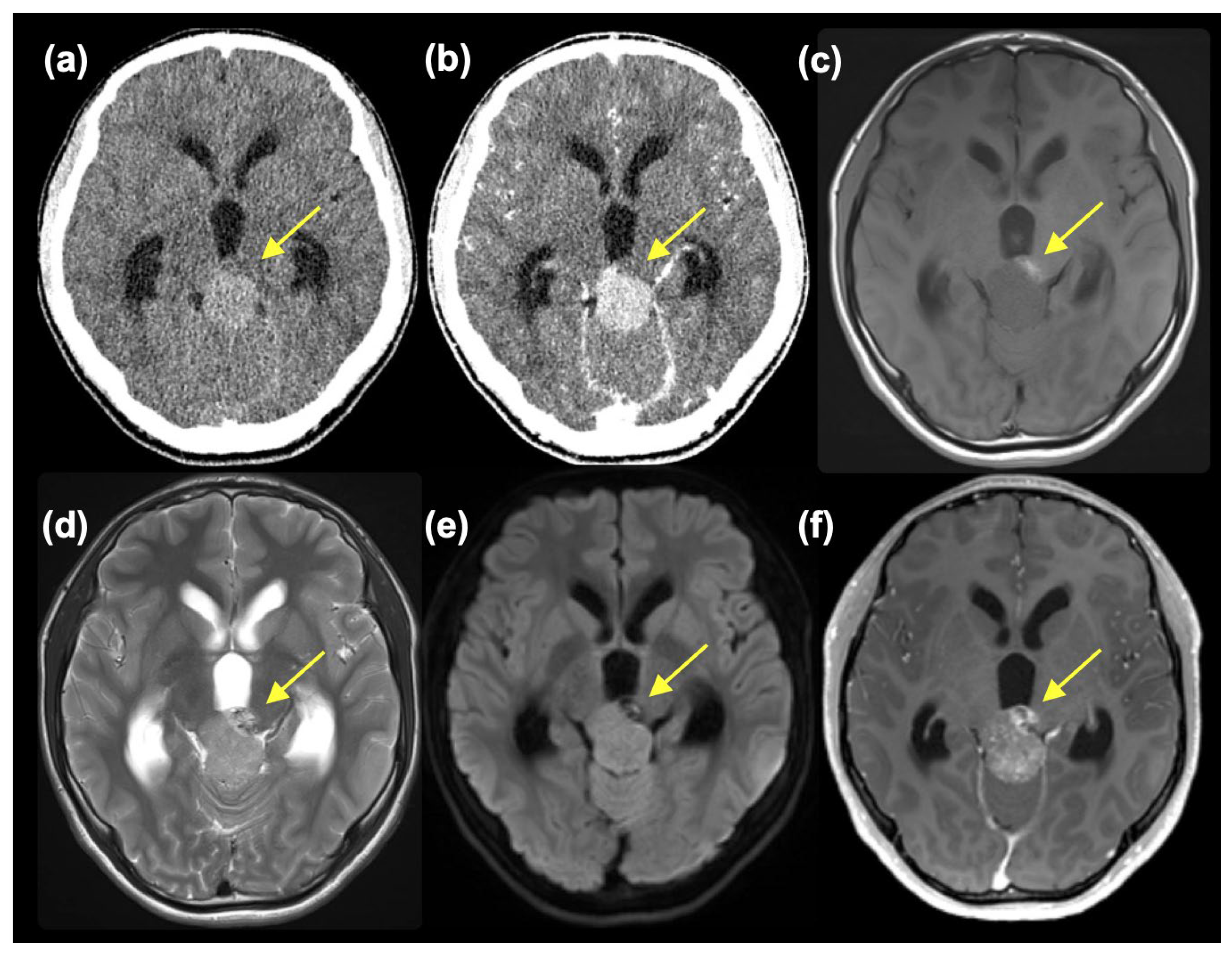
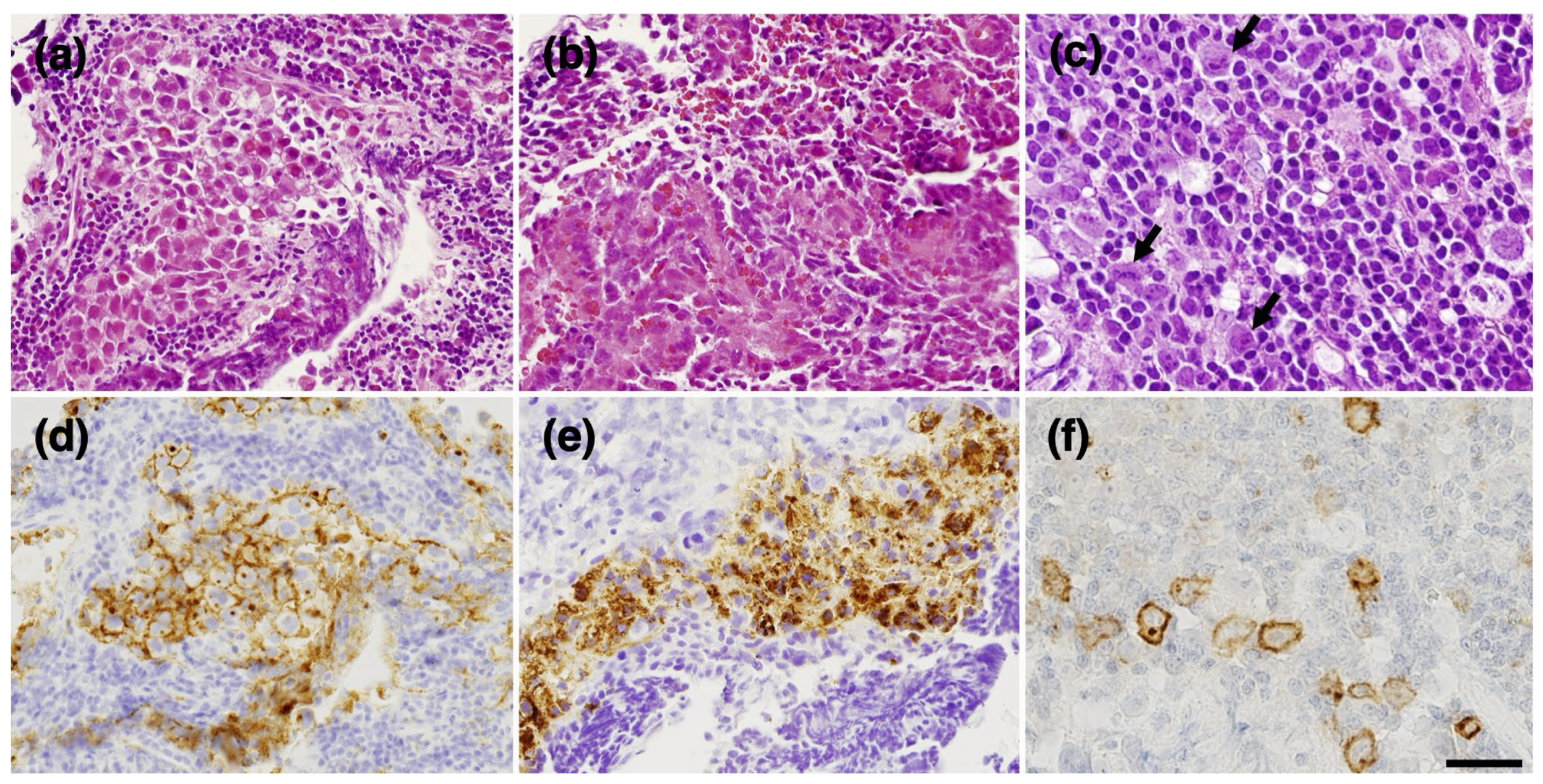
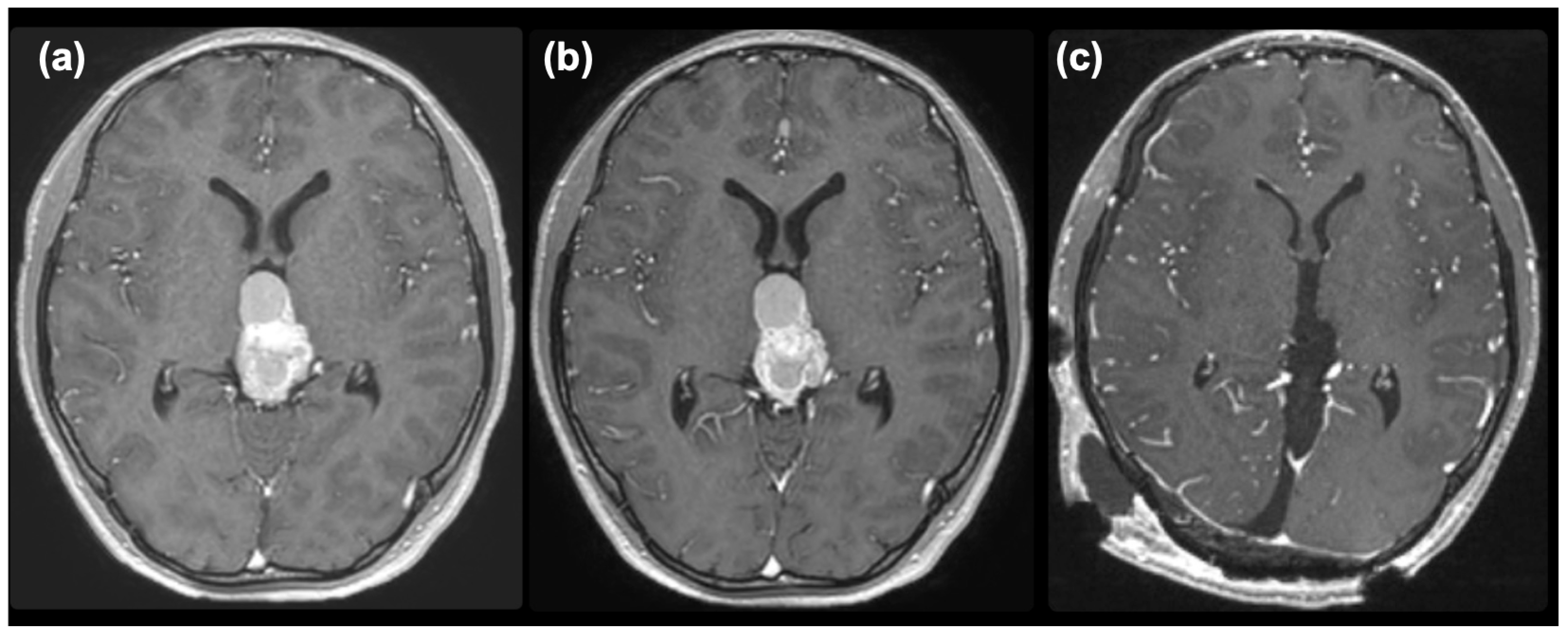
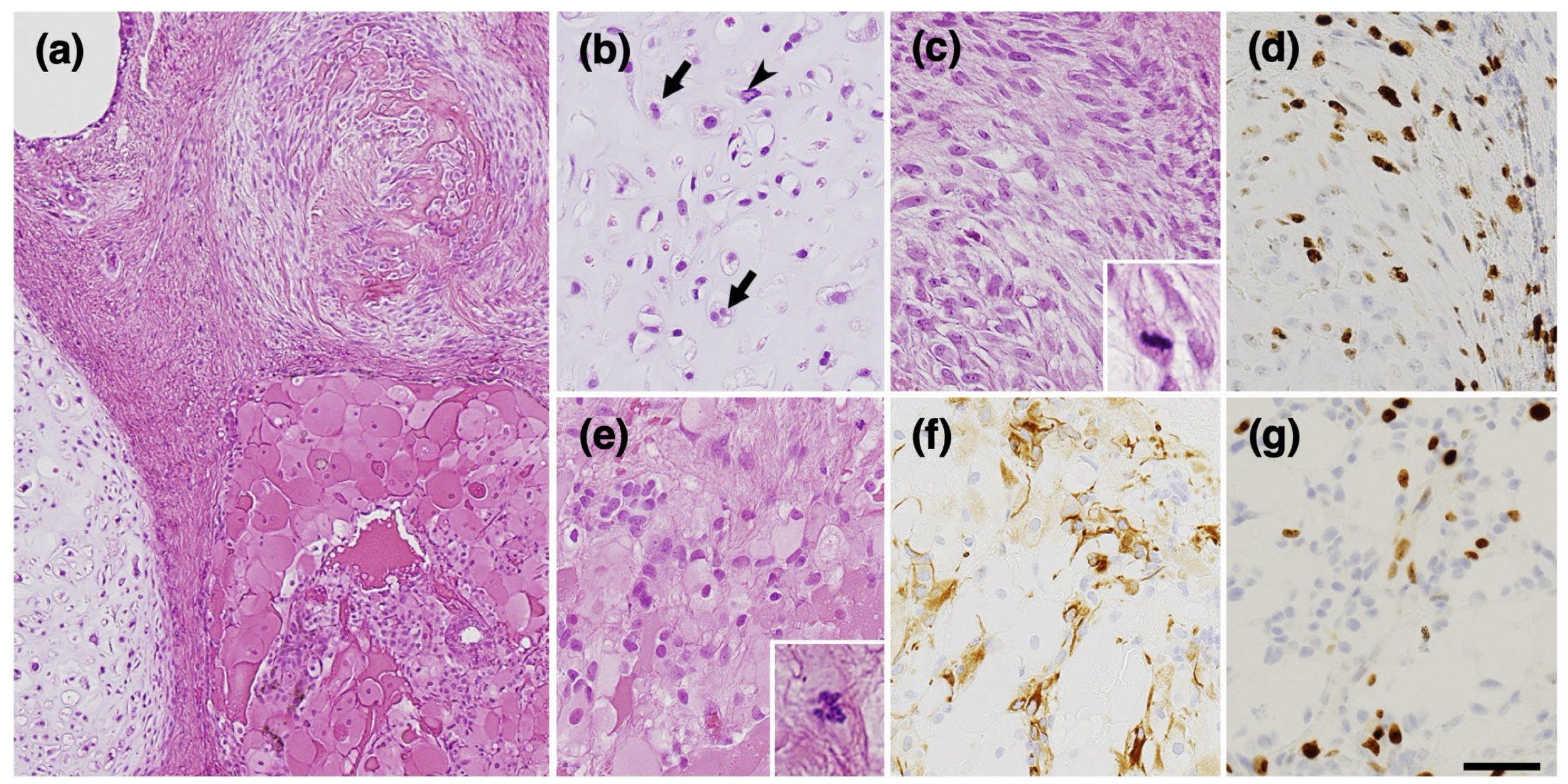
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Logothetis, C.J.; Samuels, M.L.; Trindade, A.; Johnson, D.E. The growing teratoma syndrome. Cancer 1982, 50, 1629–1635. [Google Scholar] [CrossRef]
- Matsutani, M.; Sano, K.; Takakura, K.; Fujimaki, T.; Nakamura, O.; Funata, N.; Seto, T. Primary intracranial germ cell tumors: A clinical analysis of 153 histologically verified cases. J. Neurosurg. 1997, 86, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takami, H.; Yanagisawa, T.; Kumabe, T.; Fujimaki, T.; Arakawa, Y.; Karasawa, K.; Terashima, K.; Yokoo, H.; Fukuoka, K.; et al. The Japan society for neuro-oncology guideline on the diagnosis and treatment of central nervous system germ cell tumors. Neuro-Oncology 2021, 24, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Tsumanuma, I.; Tanaka, R.; Fujii, Y. Occipital transtentorial approach and combined treatments for pineal parenchymal tumors. In Pineal Region Tumors: Diagnosis and Treatment Options; Kobayashi, T., Lunsford, L.D., Eds.; S.Karger AG: Basel, Switzerland, 2009; Volume 23. [Google Scholar]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Satomi, K.; Takami, H.; Fukushima, S.; Yamashita, S.; Matsushita, Y.; Nakazato, Y.; Suzuki, T.; Tanaka, S.; Mukasa, A.; Saito, N.; et al. 12p gain is predominantly observed in non-germinomatous germ cell tumors and identifies an unfavorable subgroup of central nervous system germ cell tumors. Neuro-Oncology 2021, 24, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, D.M.; Eleveld, T.F.; Sriram, S.; Dorssers, L.C.J.; Gillis, A.J.M.; Schmidtova, S.; Kalavska, K.; van de Werken, H.J.G.; Oing, C.; Honecker, F.; et al. Chromosome 3p25.3 gain is associated with cisplatin resistance and is an independent predictor of poor outcome in male malignant germ cell tumors. J. Clin. Oncol. 2022, 40, 3077–3087. [Google Scholar] [CrossRef]
- Takami, H.; Satomi, K.; Fukuoka, K.; Nakamura, T.; Tanaka, S.; Mukasa, A.; Saito, N.; Suzuki, T.; Yanagisawa, T.; Sugiyama, K.; et al. Distinct patterns of copy number alterations may predict poor outcome in central nervous system germ cell tumors. Sci. Rep. 2023, 13, 15760. [Google Scholar] [CrossRef]
- Hsieh, M.-Y.; Chen, H.-H.; Lee, C.-Y.; Hung, G.-Y.; Chang, T.-Y.; Chen, S.-H.; Lai, J.-Y.; Jaing, T.-H.; Cheng, C.-N.; Chen, J.-S.; et al. A case series and literature review on 98 pediatric patients of germ cell tumor developing growing teratoma syndrome. Cancer Med. 2023, 12, 13256–13269. [Google Scholar] [CrossRef]
- Michaiel, G.; Strother, D.; Gottardo, N.; Bartels, U.; Coltin, H.; Hukin, J.; Wilson, B.; Zelcer, S.; Hansford, J.R.; Hassall, T.; et al. Intracranial growing teratoma syndrome (igts): An international case series and review of the literature. J. Neuro-Oncol. 2020, 147, 721–730. [Google Scholar] [CrossRef]
- Tsuyuguchi, S.; Sugiyama, K.; Kinoshita, Y.; Kolakshyapati, M.; Takayasu, T.; Usui, S.; Takano, M.; Yonezawa, U.; Taguchi, A.; Amatya, V.J.; et al. Primary and recurrent growing teratoma syndrome in central nervous system nongerminomatous germ cell tumors: Case series and review of the literature. World Neurosurg. 2020, 134, e360–e371. [Google Scholar] [CrossRef]
- Tullius, B.P.; Shankar, S.P.; Cole, S.; Triano, V.; Aradhya, S.; Huang, E.C.; Sanchez, T.; Pawar, A. Novel heterozygous mutation in the pten gene associated with ovarian germ cell tumor complicated by growing teratoma syndrome and overgrowth in a two-year-old female. Pediatr. Blood Cancer 2019, 66, e27788. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, J.; Shibata, H.; Kabata, M.; Kato, M.; Fukuda, K.; Tanaka, A.; Ohta, S.; Ukai, T.; Mitsunaga, K.; Yamada, Y.; et al. Dmrt1-mediated reprogramming drives development of cancer resembling human germ cell tumors with features of totipotency. Nat. Commun. 2021, 12, 5041. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.D.; Murphy, M.W.; Kim, S.; Cook, M.S.; Capel, B.; Zhu, R.; Matin, A.; Sarver, A.L.; Parker, K.L.; Griswold, M.D.; et al. The dm domain protein dmrt1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc. Natl. Acad. Sci. USA 2009, 106, 22323–22328. [Google Scholar] [CrossRef]
- Looijenga, L.H.J.; Van der Kwast, T.H.; Grignon, D.; Egevad, L.; Kristiansen, G.; Kao, C.-S.; Idrees, M.T. Report from the international society of urological pathology (isup) consultation conference on molecular pathology of urogenital cancers: Iv: Current and future utilization of molecular-genetic tests for testicular germ cell tumors. Am. J. Surg. Pathol. 2020, 44, e66–e79. [Google Scholar] [CrossRef]
- Atkin, N.B.; Baker, M. Specific chromosome change, i(12p), in testicular tumours? Lancet 1982, 320, 1349. [Google Scholar] [CrossRef] [PubMed]
- Kraggerud, S.M.; Skotheim, R.I.; Szymanska, J.; Eknæs, M.; Fosså, S.D.; Stenwig, A.E.; Peltomäki, P.; Lothe, R.A. Genome profiles of familial/bilateral and sporadic testicular germ cell tumors. Genes Chromosomes Cancer 2002, 34, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Dorssers, L.C.J.; Gillis, A.J.M.; Stoop, H.; van Marion, R.; Nieboer, M.M.; van Riet, J.; van de Werken, H.J.G.; Oosterhuis, J.W.; de Ridder, J.; Looijenga, L.H.J. Molecular heterogeneity and early metastatic clone selection in testicular germ cell cancer development. Br. J. Cancer 2019, 120, 444–452. [Google Scholar] [CrossRef]
- Pongratanakul, P.; Bremmer, F.; Pauls, S.; Poschmann, G.; Kresbach, C.; Parmaksiz, F.; Skowron, M.A.; Fuß, J.; Stephan, A.; Paffenholz, P.; et al. Assessing the risk to develop a growing teratoma syndrome based on molecular and epigenetic subtyping as well as novel secreted biomarkers. Cancer Lett. 2024, 585, 216673. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satake, D.; Natsumeda, M.; Satomi, K.; Tada, M.; Sato, T.; Okubo, N.; Kawabe, K.; Takahashi, H.; Tsukamoto, Y.; Okada, M.; et al. Successful Multimodal Treatment of Intracranial Growing Teratoma Syndrome with Malignant Features. Curr. Oncol. 2024, 31, 1831-1838. https://doi.org/10.3390/curroncol31040138
Satake D, Natsumeda M, Satomi K, Tada M, Sato T, Okubo N, Kawabe K, Takahashi H, Tsukamoto Y, Okada M, et al. Successful Multimodal Treatment of Intracranial Growing Teratoma Syndrome with Malignant Features. Current Oncology. 2024; 31(4):1831-1838. https://doi.org/10.3390/curroncol31040138
Chicago/Turabian StyleSatake, Daiken, Manabu Natsumeda, Kaishi Satomi, Mari Tada, Taro Sato, Noritaka Okubo, Keita Kawabe, Haruhiko Takahashi, Yoshihiro Tsukamoto, Masayasu Okada, and et al. 2024. "Successful Multimodal Treatment of Intracranial Growing Teratoma Syndrome with Malignant Features" Current Oncology 31, no. 4: 1831-1838. https://doi.org/10.3390/curroncol31040138
APA StyleSatake, D., Natsumeda, M., Satomi, K., Tada, M., Sato, T., Okubo, N., Kawabe, K., Takahashi, H., Tsukamoto, Y., Okada, M., Sano, M., Iwabuchi, H., Shibata, N., Imamura, M., Imai, C., Takami, H., Ichimura, K., Nishikawa, R., Umezu, H., ... Oishi, M. (2024). Successful Multimodal Treatment of Intracranial Growing Teratoma Syndrome with Malignant Features. Current Oncology, 31(4), 1831-1838. https://doi.org/10.3390/curroncol31040138