Myelodysplastic Neoplasms (MDS) with Ring Sideroblasts or SF3B1 Mutations: The Improved Clinical Utility of World Health Organization and International Consensus Classification 2022 Definitions, a Single-Centre Retrospective Chart Review

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
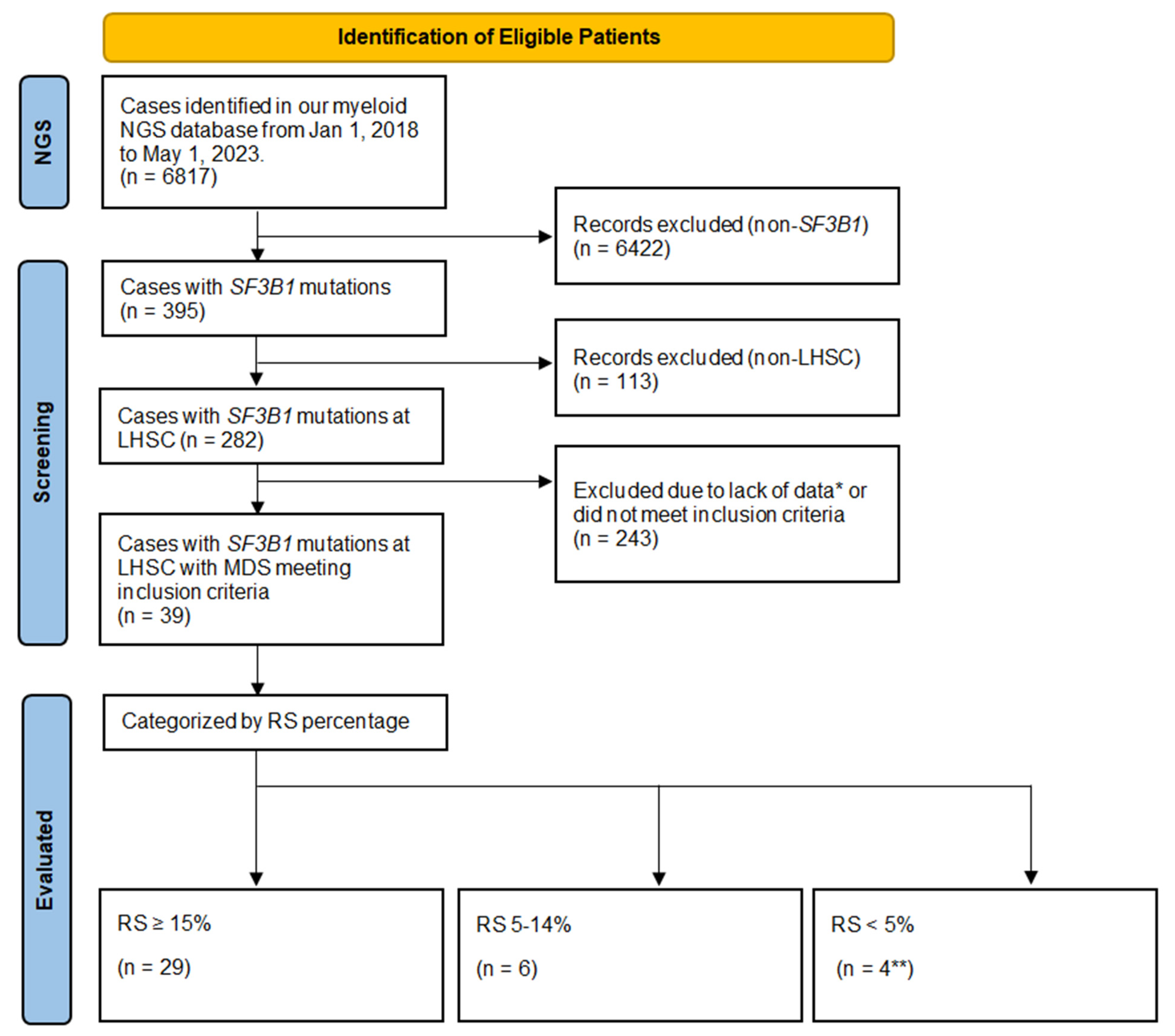
2.1. Patient Selection
2.2. NGS Assay
2.3. Bone Marrow Aspirate Morphologic and RS Assessments
2.4. Statistical Analysis
3. Results
3.1. Patient Cohort
3.2. Ring Sideroblasts
3.3. Patients with SF3B1 Mutation and No RSs
3.4. Patient Outcomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sekeres, M.A.; Taylor, J. Diagnosis and Treatment of Myelodysplastic Syndromes: A Review: A Review. JAMA 2022, 328, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vardiman, J.W. Myelodysplastic Syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J.; Lister, T.A.; Bloomfield, C.D. World Health Organization Classification of Neoplastic Diseases of the Hematopoietic and Lymphoid Tissues: Report of the Clinical Advisory Committee Meeting—Airlie House, Virginia, November 1997. J. Clin. Oncol. 1999, 17, 3835–3849. [Google Scholar] [CrossRef]
- World Health Organization Classification of Tumours. Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues; Jaffe, E.S., Harris, N.L., Stein, H., Vardiman, J.W., Eds.; IARC Press: Lyon, France, 2001. [Google Scholar]
- Komrokji, R.S.; Bennett, J.M. Evolving Classifications of the Myelodysplastic Syndromes. Curr. Opin. Hematol. 2007, 14, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Hanson, C.A.; Sulai, N.H.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Lasho, T.L.; Tefferi, A. Prognostic Irrelevance of Ring Sideroblast Percentage in World Health Organization–Defined Myelodysplastic Syndromes without Excess Blasts. Blood 2012, 119, 5674–5677. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 Mutation Identifies a Distinct Subset of Myelodysplastic Syndrome with Ring Sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-Mutant MDS as a Distinct Disease Subtype: A Proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Xiong, B.; Xue, M.; Yu, Y.; Wu, S.; Zuo, X. SF3B1 Mutation but Not Ring Sideroblasts Identifies a Specific Group of Myelodysplastic Syndrome–Refractory Cytopenia with Multilineage Dysplasia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 329–339.e3. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Myelodysplastic Syndromes. Mayo Clin. Proc. 2015, 90, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the Treatment of Anaemia in Patients with Lower-Risk Myelodysplastic Syndromes (PACE-MDS): A Multicentre, Open-Label Phase 2 Dose-Finding Study with Long-Term Extension Study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Della Porta, M.G.; Santini, V.; Zeidan, A.M.; Komrokji, R.S.; Shortt, J.; Valcarcel, D.; Jonasova, A.; Dimicoli-Salazar, S.; Tiong, I.S.; et al. Efficacy and Safety of Luspatercept versus Epoetin Alfa in Erythropoiesis-Stimulating Agent-Naive, Transfusion-Dependent, Lower-Risk Myelodysplastic Syndromes (COMMANDS): Interim Analysis of a Phase 3, Open-Label, Randomised Controlled Trial. Lancet 2023, 402, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International Scoring System for Evaluating Prognosis in Myelodysplastic Syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Bhai, P.; Hsia, C.C.; Schenkel, L.C.; Hedley, B.D.; Levy, M.A.; Kerkhof, J.; Santos, S.; Stuart, A.; Lin, H.; Broadbent, R.; et al. Clinical Utility of Implementing a Frontline NGS-Based DNA and RNA Fusion Panel Test for Patients with Suspected Myeloid Malignancies. Mol. Diagn. Ther. 2022, 26, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.A.; Santos, S.; Kerkhof, J.; Stuart, A.; Aref-Eshghi, E.; Guo, F.; Hedley, B.; Wong, H.; Rauh, M.; Feilotter, H.; et al. Implementation of an NGS-based Sequencing and Gene Fusion Panel for Clinical Screening of Patients with Suspected Hematologic Malignancies. Eur. J. Haematol. 2019, 103, 178–189. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1. [Google Scholar] [CrossRef] [PubMed]
- Kawata, E.; Lazo-Langner, A.; Xenocostas, A.; Hsia, C.C.; Howson-Jan, K.; Deotare, U.; Saini, L.; Yang, P.; Broadbent, R.; Levy, M.; et al. Clinical value of next-generation sequencing compared to cytogenetics in patients with suspected myelodysplastic syndrome. Br. J. Haematol. 2021, 192, 729–736. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the Classification of the Myelodysplastic Syndromes. Br. J. Haematol. 1982, 51, 189–199. [Google Scholar] [CrossRef]
- Steensma, D.P. The Changing Classification of Myelodysplastic Syndromes: What’s in a Name? Hematology Am. Soc. Hematol. Educ. Program 2009, 2009, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Silverman, L.R. Modulation of the Clone: Altering the Course of Myelodysplastic Syndrome. Blood Bone Marrow Trans-Plant. Rev. 2006, 16, 5–8. [Google Scholar]
- Greenberg, P.L.; Sun, Z.; Miller, K.B.; Bennett, J.M.; Tallman, M.S.; Dewald, G.; Paietta, E.; van der Jagt, R.; Houston, J.; Thomas, M.L.; et al. Treatment of Myelodysplastic Syndrome Patients with Erythropoietin with or without Granulocyte Colony-Stimulating Factor: Results of a Prospective Randomized Phase 3 Trial by the Eastern Cooperative Oncology Group (E1996). Blood 2009, 114, 2393–2400. [Google Scholar] [CrossRef]
- Jädersten, M.; Malcovati, L.; Dybedal, I.; Giovanni Della Porta, M.; Invernizzi, R.; Montgomery, S.M.; Pascutto, C.; Porwit, A.; Cazzola, M.; Hellström-Lindberg, E. Erythropoietin and Granulocyte-Colony Stimulating Factor Treatment Associated with Improved Survival in Myelodysplastic Syndrome. J. Clin. Oncol. 2008, 26, 3607–3613. [Google Scholar] [CrossRef]
- Hellström-Lindberg, E.; Gulbrandsen, N.; Lindberg, G.; Ahlgren, T.; Dahl, I.M.S.; Dybedal, I.; Grimfors, G.; Hesse-Sundin, E.; Hjorth, M.; Kanter-Lewensohn, L.; et al. A Validated Decision Model for Treating the Anaemia of Myelodysplastic Syndromes with Erythropoietin + Granulocyte Colony-stimulating Factor: Significant Effects on Quality of Life. Br. J. Haematol. 2003, 120, 1037–1046. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Xie, Z.; Bejar, R.; Borate, U.; Boultwood, J.; Brunner, A.M.; Buckstein, R.; Carraway, H.E.; Churpek, J.E.; Daver, N.G.; et al. Current Landscape of Translational and Clinical Research in Myelodysplastic Syndromes/Neoplasms (MDS): Proceedings from the 1st International Workshop on MDS (iwMDS) Of the International Consortium for MDS (icMDS). Blood Rev. 2023, 60, 101072. [Google Scholar] [CrossRef]


{kind=link}
| Total Patients | Male | Female | |
|---|---|---|---|
| Patient Characteristics | |||
| Total Patients; number (%) | 39 (100) | 20 (51) | 19 (49) |
| Age; median (range) years | 77 (57–92) | 78 (57–90) | 76 (64–92) |
| Laboratory Findings * | |||
| Haemoglobin; median (range) g/L | 96 (65–151) | 95 (65–151) | 96 (71–112) |
| MCV; median (range) fL | 105 (89–124) | 108 (89–124) | 104 (93–117) |
| Leukocytes; median (range) × 109/L | 6 (2–12) | 6 (2–12) | 6 (4–12) |
| Platelets; median (range) × 109/L | 220 (24–651) | 203 (24–651) | 253 (54–371) |
| Ferritin; median (range) mcg/L | 554 (59–4519) | 562 (59–4519) | 544 (172–2894) |
| Serum EPO; median (range) IU/L | 43 (9–1070) | 34 (9–1070) | 47 (18–615) |
| Management and Outcomes | |||
| Transfusions **; number (%) | 18 (46) | 8 (40) | 10 (53) |
| Transfusion-dependent ***; number (%) | 15 (38) | 7 (35) | 8 (42) |
| ESA; number (%) | 17 (44) | 8 (40) | 9 (47) |
| EMA; number (%) | 5 (13) | 1 (5) | 4 (21) |
| Other treatments; number (%) | 2 (5) | 2 (10) | 0 |
| Duration of follow-up †; median (range) months | 35 (3–180) | 34 (3–137) | 37 (4–180) |
| Total Patients N = 39 | Male N = 20 | Female N = 19 | |
|---|---|---|---|
| IPSS Risk Classification | |||
| Low; n (%) | 32 (82%) | 17 (85%) | 15 (79%) |
| Intermediate-1; n (%) | 7 (18%) | 3 (15%) | 4 (21%) |
| R-IPSS Risk Classification | |||
| Very Low; n (%) | 10 (26%) | 6 (30%) | 4 (21%) |
| Low; n (%) | 29 (74%) | 14 (70%) | 15 (79%) |
| M-IPSS Classification | |||
| Very Low, n (%); Score median (range) | 15 (39%); −1.64 (−2.69 to −1.52) | 7 (35%); −1.66 (−2.69 to −0.85) | 8 (42%); −1.63 (−1.93 to −1.57) |
| Low, n (%); Score median (range) | 24 (62%); −1.13 (−1.46 to −0.59) | 13 (65%); −1.17 (−1.46 to −0.59) | 11 (58%); −1.09 (−1.46 to −0.81) |
| Patient Serial ID | Age (yrs) | Sex | MDS Subtype (WHO 2016) | RS Count (%) | SF3B1 Mutation VAF (%) | Other Gene Mutations | Cytogenetics |
|---|---|---|---|---|---|---|---|
| 1 | 76 | F | RS-SLD | 15 | SF3B1:c.2098A>G,p.(Lys700Glu) (23.4%) | None | Normal |
| 2 | 78 | M | RS-SLD | 15+ | SF3B1:c.1997A>C,p.(Lys666Thr) (38%) | None | Normal |
| 3 | 76 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (42.2%) | TET2 | Del 13q |
| 4 | 64 | F | SLD | 0 | SF3B1:c.2098A>G,p.(Lys700Glu) (38.9%) | TET2, TP53 | Normal |
| 5 | 64 | F | SLD | 9 | SF3B1:c.2098A>G,p.(Lys700Glu) (40.8%) | None | Normal |
| 6 | 82 | F | MLD | 10 | SF3B1:c.1873C>T,p.(Arg625Cys) (33.8%) | None | Normal |
| 7 | 83 | M | RS-MLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (39.8%) | DNMT3A | Normal |
| 8 | 65 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (45%) | IDH2, DNMT3A | Normal |
| 9 | 84 | F | RS-MLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (40%) | TET2, DNMT3A | Normal |
| 10 | 64 | M | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (25.6%) | None | Normal |
| 11 | 88 | F | SLD | 5–14% | SF3B1:c.2098A>G,p.(Lys700Glu) (43.2%) | TET2, DNMT3A, SH2B3 | Del 20q |
| 12 | 73 | M | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (35.9%) | TET2 | Inv 13q |
| 13 | 74 | M | RS-MLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (36.1%) | KIT | Normal |
| 14 | 80 | F | RS-SLD | 15+ | SF3B1:c.1873C>T,p.(Arg625Cys) (26.8%) | None | Normal |
| 15 | 85 | M | RS-SLD | 15+ | SF3B1:c.1997A>G,p.(Lys666Arg) (36.6%) | TET2 | Normal |
| 16 | 65 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (28.5%) | None | Normal |
| 17 | 63 | M | RS-SLD | 15+ | SF3B1:c.1986C>G,p.(His662Gln) (29.5%) | None | Y minus |
| 18 | 90 | M | MLD | 8 | SF3B1:c.2098A>G,p.(Lys700Glu) (39.2%) | None | Y minus |
| 19 | 78 | M | RS-SLD | 15+ | SF3B1:c.1997A>G,p.(Lys666Arg) (33.3%) | None | Normal |
| 20 | 67 | M | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (44.6%) | None | Normal |
| 21 | 57 | M | SLD | 12 | SF3B1:c.1998G>T,p.(Lys666Asn) (33.5%) | None | Normal |
| 22 | 80 | F | RS-MLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (37.9%) | TET2 | Normal |
| 23 | 81 | M | MLD | 0 | SF3B1:c.1986C>G,p.(His662Gln) (7.4%) | TET2, KIT | Normal |
| 24 | 70 | F | RS-SLD | 15+ | SF3B1:c.1984C>T,p.(His662Tyr) (45.6%) | TET2 | Normal |
| 25 | 92 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (42.7%) | TET2 | Normal |
| 26 | 79 | M | SLD | 0 | SF3B1:c.1998G>C,p.(Lys666Asn) (21.8%) SF3B1:c.2098A>G, p.(Lys700Glu) (20.1%) | None | Normal |
| 27 | 86 | M | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (28.7%) | DNMT3A | Normal |
| 28 | 88 | M | SLD | 1 | SF3B1:c.1997A>C,p.(Lys666Thr) (39.6%) | None | Normal |
| 29 | 70 | F | RS-SLD | 15+ | SF3B1:c.1986C>A,p.(His662Gln) (37.4%) | None | Normal |
| 30 | 81 | M | RS-SLD | 65 | SF3B1:c.2098A>G,p.(Lys700Glu) (46%) | DNMT3A, TET2 | Normal |
| 31 | 76 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (38.5%) | None | Normal |
| 32 | 78 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (40.8%) | None | Normal |
| 33 | 87 | F | SLD | 5–14% | SF3B1:c.2098A>G,p.(Lys700Glu) (32.2%) | None | Normal |
| 34 | 75 | M | RS-SLD | 15+ | SF3B1:c.1997A>G,p.(Lys666Arg) (43.6%) | ZRSR2, TET2 | Normal |
| 35 | 73 | M | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (31.7%) | DNMT3A | Normal |
| 36 | 73 | F | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (37.1%) | None | Del 20q |
| 37 | 81 | M | RS-SLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (25.1%) | DNMT3A | Normal |
| 38 | 77 | F | RS-MLD | 15+ | SF3B1:c.1986C>A,p.(His662Gln) (23.4%) | TET2 | Normal |
| 39 | 75 | M | RS-MLD | 15+ | SF3B1:c.2098A>G,p.(Lys700Glu) (39.7%) | TET2 | Normal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortuza, S.; Chin-Yee, B.; James, T.E.; Chin-Yee, I.H.; Hedley, B.D.; Ho, J.M.; Saini, L.; Lazo-Langner, A.; Schenkel, L.; Bhai, P.; et al. Myelodysplastic Neoplasms (MDS) with Ring Sideroblasts or SF3B1 Mutations: The Improved Clinical Utility of World Health Organization and International Consensus Classification 2022 Definitions, a Single-Centre Retrospective Chart Review. Curr. Oncol. 2024, 31, 1762-1773. https://doi.org/10.3390/curroncol31040134
Mortuza S, Chin-Yee B, James TE, Chin-Yee IH, Hedley BD, Ho JM, Saini L, Lazo-Langner A, Schenkel L, Bhai P, et al. Myelodysplastic Neoplasms (MDS) with Ring Sideroblasts or SF3B1 Mutations: The Improved Clinical Utility of World Health Organization and International Consensus Classification 2022 Definitions, a Single-Centre Retrospective Chart Review. Current Oncology. 2024; 31(4):1762-1773. https://doi.org/10.3390/curroncol31040134
Chicago/Turabian StyleMortuza, Shamim, Benjamin Chin-Yee, Tyler E. James, Ian H. Chin-Yee, Benjamin D. Hedley, Jenny M. Ho, Lalit Saini, Alejandro Lazo-Langner, Laila Schenkel, Pratibha Bhai, and et al. 2024. "Myelodysplastic Neoplasms (MDS) with Ring Sideroblasts or SF3B1 Mutations: The Improved Clinical Utility of World Health Organization and International Consensus Classification 2022 Definitions, a Single-Centre Retrospective Chart Review" Current Oncology 31, no. 4: 1762-1773. https://doi.org/10.3390/curroncol31040134
APA StyleMortuza, S., Chin-Yee, B., James, T. E., Chin-Yee, I. H., Hedley, B. D., Ho, J. M., Saini, L., Lazo-Langner, A., Schenkel, L., Bhai, P., Sadikovic, B., Keow, J., Sangle, N., & Hsia, C. C. (2024). Myelodysplastic Neoplasms (MDS) with Ring Sideroblasts or SF3B1 Mutations: The Improved Clinical Utility of World Health Organization and International Consensus Classification 2022 Definitions, a Single-Centre Retrospective Chart Review. Current Oncology, 31(4), 1762-1773. https://doi.org/10.3390/curroncol31040134