Novel Tools for Diagnosis and Monitoring of AML

 , , ,
, , ,
Abstract
1. Introduction
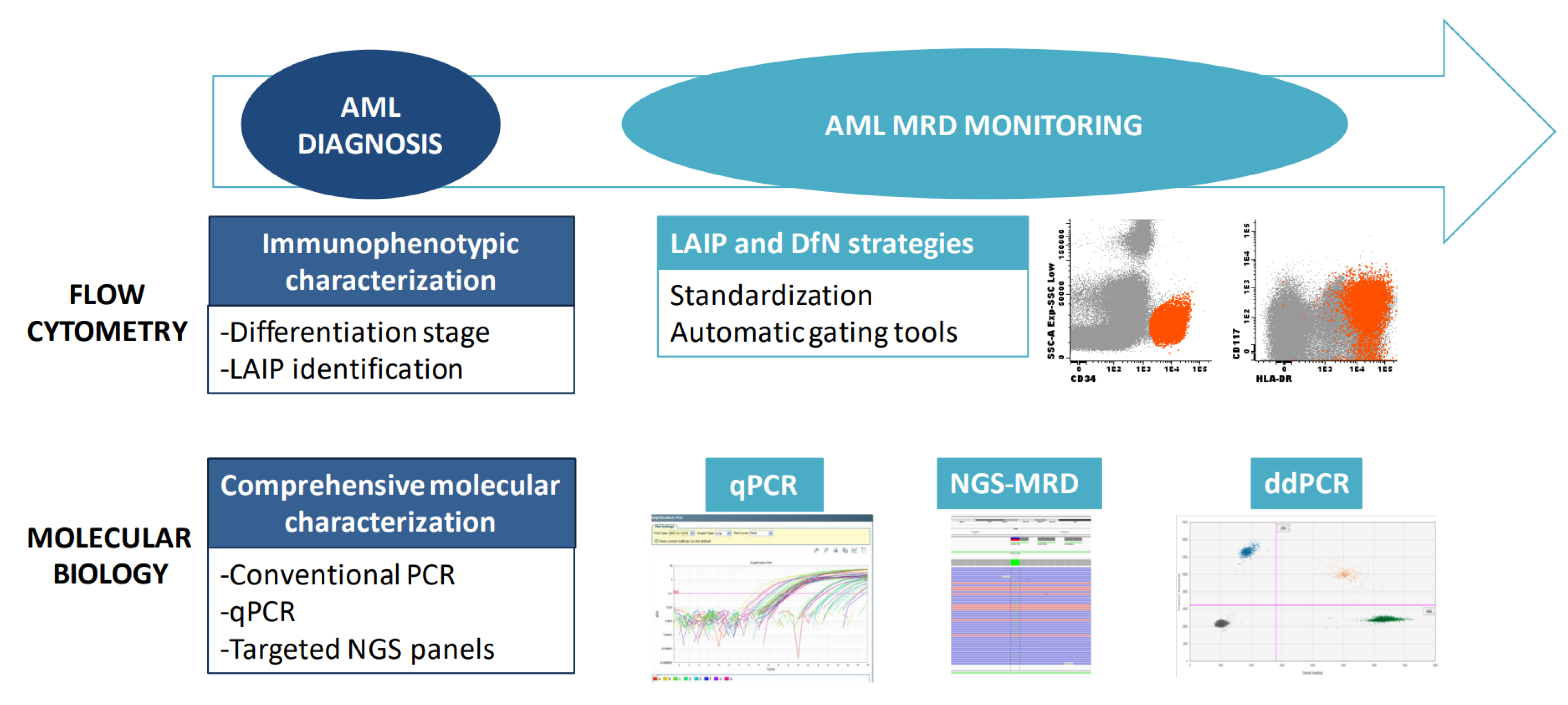
2. Diagnosis
2.1. Flow Cytometry for Lineage Assessment
2.2. Molecular Biology
3. Monitoring
3.1. Flow Cytometry for MRD Detection in AML
3.2. Molecular Monitoring
3.2.1. qPCR
3.2.2. Digital PCR
3.2.3. NGS
3.2.4. Selection of MRD Molecular Markers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A leukemia-associated CD34/CD123/CD25/CD99+ immunophenotype identifies FLT3-mutated clones in acute myeloid Leukemia. Clinical Cancer Research. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Kalina, T.; Flores-Montero, J.; van der Velden, V.H.J.; Martin-Ayuso, M.; Böttcher, S.; Ritgen, M.; Almeida, J.; Lhermitte, L.; Asnafi, V.; Mendonça, A.; et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012, 26, 1986–2010. [Google Scholar] [CrossRef]
- Lhermitte, L.; on behalf of Mthe EuroFlow Consortium; Mejstrikova, E.; van der Sluijs-Gelling, A.J.; Grigore, G.E.; Sedek, L.; Bras, A.E.; Gaipa, G.; da Costa, E.S.; Novakova, M.; et al. Automated database-guided expert-supervised orientation for immunophenotypic diagnosis and classification of acute leukemia. Leukemia 2018, 32, 874–881. [Google Scholar] [CrossRef]
- Lhermitte, L.; Barreau, S.; Morf, D.; Fernandez, P.; Grigore, G.; Barrena, S.; de Bie, M.; Flores-Montero, J.; Brüggemann, M.; Mejstrikova, E.; et al. Automated identification of leukocyte subsets improves standardization of database-guided expert-supervised diagnostic orientation in acute leukemia: A EuroFlow study. Mod. Pathol. 2021, 34, 59–69. [Google Scholar] [CrossRef]
- Martínez, D.S.; Tirado, N.; Mensurado, S.; Martínez-Moreno, A.; Romecín, P.; Agüera, F.G.; Correia, D.V.; Silva-Santos, B.; Menéndez, P. Generation and proof-of-concept for allogeneic CD123 CAR-Delta One T (DOT) cells in acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e005400. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.; Laird, P.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Eng. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Taube, F.; Georgi, J.A.; Kramer, M.; Stasik, S.; Middeke, J.M.; Röllig, C.; Krug, U.; Krämer, A.; Scholl, S.; Hochhaus, A.; et al. CEBPA mutations in 4708 patients with acute myeloid leukemia: Differential impact of bZIP and TAD mutations on outcome. Blood 2022, 139, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Wakita, S.; Sakaguchi, M.; Oh, I.; Kako, S.; Toya, T.; Najima, Y.; Doki, N.; Kanda, J.; Kuroda, J.; Mori, S.; et al. Prognostic impact of CEBPA bZIP domain mutation in acute myeloid leukemia. Blood Adv. 2022, 6, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Short, N.J.; Fu, C.; Berry, D.A.; Walter, R.B.; Freeman, S.D.; Hourigan, C.S.; Huang, X.; Gonzalez, G.N.; Hwang, H.; Qi, X.; et al. Association of hematologic response and assay sensitivity on the prognostic impact of measurable residual disease in acute myeloid leukemia: A systematic review and meta-analysis. Leukemia 2022, 36, 2817–2826. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Bataller, A.; Oñate, G.; Diaz-Beyá, M.; Guijarro, F.; Garrido, A.; Vives, S.; Tormo, M.; Arnan, M.; Salamero, O.; Sampol, A.; et al. Acute myeloid leukemia with NPM1 mutation and favorable European LeukemiaNet category: Outcome after preemptive intervention based on measurable residual disease. Br. J. Haematol. 2020, 191, 52–61. [Google Scholar] [CrossRef]
- Short, N.J.; Macaron, W.; Kadia, T.; Dinardo, C.; Issa, G.C.; Daver, N.; Wang, S.; Jorgensen, J.; Nguyen, D.; Bidikian, A.; et al. Clinical outcomes and impact of therapeutic intervention in patients with acute myeloid leukemia who experience measurable residual disease (MRD) recurrence following MRD -negative remission. Am. J. Hematol. 2022, 97, E408–E411. [Google Scholar] [CrossRef]
- Tiong, I.S.; Loo, S. Targeting Measurable Residual Disease (MRD) in Acute Myeloid Leukemia (AML): Moving beyond Prognostication. Int. J. Mol. Sci. 2023, 24, 4790. [Google Scholar] [CrossRef]
- Buckley, S.A.; Wood, B.L.; Othus, M.; Hourigan, C.S.; Ustun, C.; Linden, M.A.; DeFor, T.E.; Malagola, M.; Anthias, C.; Valkova, V.; et al. Minimal residual disease prior to allogeneic hematopoietic cell transplantation in acute myeloid leukemia: A meta-analysis. Haematologica 2017, 102, 865–873. [Google Scholar] [CrossRef]
- Hourigan, C.S.; Dillon, L.W.; Gui, G.; Logan, B.R.; Fei, M.; Ghannam, J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; et al. Impact of Conditioning Intensity of Allogeneic Transplantation for Acute Myeloid Leukemia with Genomic Evidence of Residual Disease. J. Clin. Oncol. 2020, 38, 1273–1283. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in acute myeloid leukemia: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, T.; Sauter, D.; Ringel, K.; Hoster, E.; Laubender, R.P.; Hubmann, M.; Bohlander, S.K.; Kakadia, P.M.; Schneider, S.; Dufour, A.; et al. Early assessment of minimal residual disease in AML by flow cytometry during aplasia identifies patients at increased risk of relapse. Leukemia 2015, 29, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Jorgensen, J.; Borthakur, G.; Jabbour, E.; Kadia, T.; Pierce, S.; Brandt, M.; Wang, S.; Konoplev, S.; Wang, X.; et al. Persistence of minimal residual disease assessed by multiparameter flow cytometry is highly prognostic in younger patients with acute myeloid leukemia. Cancer 2017, 123, 426–435. [Google Scholar] [CrossRef]
- Buldini, B.; Rizzati, F.; Masetti, R.; Fagioli, F.; Menna, G.; Micalizzi, C.; Putti, M.C.; Rizzari, C.; Santoro, N.; Zecca, M.; et al. Prognostic significance of flow-cytometry evaluation of minimal residual disease in children with acute myeloid leukaemia treated according to the AIEOP-AML 2002/01 study protocol. Br. J. Haematol. 2017, 177, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; Van Putten, W.L.; Kelder, A.; Van Der Velden, V.H.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; De Greef, G.E.; Valk, P.J.; et al. High Prognostic Impact of Flow Cytometric Minimal Residual Disease Detection in Acute Myeloid Leukemia: Data From the HOVON/SAKK AML 42A Study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Jonas, B.A.; Pullarkat, V.; Recher, C.; Schuh, A.C.; Thirman, M.J.; Garcia, J.S.; DiNardo, C.D.; Vorobyev, V.; Fracchiolla, N.S.; et al. Measurable Residual Disease Response and Prognosis in Treatment-Naïve Acute Myeloid Leukemia with Venetoclax and Azacitidine. J. Clin. Oncol. 2022, 40, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Bergua, J.; Infante, J.; Esteve, J.; Guimaraes, J.E.; Sierra, J.; Sanz, M. Update on management and progress of novel therapeutics for R/R AML: An Iberian expert panel consensus. Ann. Hematol. 2019, 98, 2467–2483. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Kelder, A.; Cloos, J.; Schuurhuis, G.J. Immunophenotypic Detection of Measurable Residual (Stem Cell) Disease Using LAIP Approach in Acute Myeloid Leukemia. Curr. Protoc. Cytom. 2019, 91, e66. [Google Scholar] [CrossRef]
- Wood, B.L. Acute Myeloid Leukemia Minimal Residual Disease Detection: The Difference from Normal Approach. Curr. Protoc. Cytom. 2020, 93, e73. [Google Scholar] [CrossRef]
- Tettero, J.M.; Freeman, S.; Buecklein, V.; Venditti, A.; Maurillo, L.; Kern, W.; Walter, R.B.; Wood, B.L.; Roumier, C.; Philippé, J.; et al. Technical Aspects of Flow Cytometry-based Measurable Residual Disease Quantification in Acute Myeloid Leukemia: Experience of the European LeukemiaNet MRD Working Party. Hemasphere 2021, 6, e676. [Google Scholar] [CrossRef]
- Sui, J.; Chen, Q.; Zhang, Y.; Sheng, Y.; Wu, J.; Li, J.; Weng, X.; Chen, B. Identifying leukemia-associated immunophenotype-based individualized minimal residual disease in acute myeloid leukemia and its prognostic significance. Am. J. Hematol. 2019, 94, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Röhnert, M.A.; Kramer, M.; Schadt, J.; Ensel, P.; Thiede, C.; Krause, S.W.; Bücklein, V.; Hoffmann, J.; Jaramillo, S.; Schlenk, R.F.; et al. Reproducible measurable residual disease detection by multiparametric flow cytometry in acute myeloid leukemia. Leukemia 2022, 36, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, F.; Lechevalier, N.; Vial, J.P.; Béné, M.C. An R-Derived FlowSOM Process to Analyze Unsupervised Clustering of Normal and Malignant Human Bone Marrow Classical Flow Cytometry Data. Cytom. Part A 2019, 95, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Vial, J.P.; Lechevalier, N.; Lacombe, F.; Dumas, P.-Y.; Bidet, A.; Leguay, T.; Vergez, F.; Pigneux, A.; Béné, M.C. Unsupervised Flow Cytometry Analysis Allows for an Accurate Identification of Minimal Residual Disease Assessment in Acute Myeloid Leukemia. Cancers 2021, 13, 629. [Google Scholar] [CrossRef]
- Piñero, P.; Morillas, M.; Gutierrez, N.; Barragán, E.; Such, E.; Breña, J.; García-Hernández, M.C.; Gil, C.; Botella, C.; González-Navajas, J.M.; et al. Identification of Leukemia-Associated Immunophenotypes by Databaseguided Flow Cytometry Provides a Highly Sensitive and Reproducible Strategy for the Study of Measurable Residual Disease in Acute Myeloblastic Leukemia. Cancers 2022, 14, 4010. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34+CD38− leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef]
- Li, S.-Q.; Xu, L.-P.; Wang, Y.; Zhang, X.-H.; Chen, H.; Chen, Y.-H.; Wang, F.-R.; Han, W.; Sun, Y.-Q.; Yan, C.-H.; et al. An LSC-based MRD assay to complement the traditional MFC method for prediction of AML relapse: A prospective study. Blood 2022, 140, 516–520. [Google Scholar] [CrossRef]
- Soh, K.T.; Conway, A.; Liu, X.; Wallace, P.K. Development of a 27-color panel for the detection of measurable residual disease in patients diagnosed with acute myeloid leukemia. Cytom. Part A 2022, 101, 970–983. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer Program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E.; et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef]
- Corbacioglu, A.; Scholl, C.; Schlenk, R.F.; Eiwen, K.; Du, J.; Bullinger, L.; Fröhling, S.; Reimer, P.; Rummel, M.; Derigs, H.-G.; et al. Prognostic Impact of Minimal Residual Disease inCBFB-MYH11–Positive Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 3724–3729. [Google Scholar] [CrossRef]
- Beillard, E.; Pallisgaard, N.; van der Velden, V.H.J.; Bi, W.; Dee, R.; van der Schoot, E.; Delabesse, E.; Macintyre, E.; Gottardi, E.; Saglio, G.; et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)—A Europe against cancer program. Leukemia 2003, 17, 2474–2486. [Google Scholar] [CrossRef]
- Gorello, P.; Cazzaniga, G.; Alberti, F.; Dell’Oro, M.G.; Gottardi, E.; Specchia, G.; Roti, G.; Rosati, R.; Martelli, M.F.; Diverio, D.; et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 2006, 20, 1103–1108. [Google Scholar] [CrossRef]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Dillon, L.W.; Hayati, S.; Roloff, G.; Tunc, I.; Pirooznia, M.; Mitrofanova, A.; Hourigan, C.S. Targeted RNA-sequencing for the quantification of measurable residual disease in acute myeloid leukemia. Haematologica 2019, 104, 297–304. [Google Scholar] [CrossRef]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M.; et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef]
- Schnittger, S.; Kern, W.; Tschulik, C.; Weiss, T.; Dicker, F.; Falini, B.; Haferlach, C.; Haferlach, T. Minimal residual disease levels assessed by NPM1 mutation–specific RQ-PCR provide important prognostic information in AML. Blood 2009, 114, 2220–2231. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Freeman, S.D. Evaluating measurable residual disease in acute myeloid leukemia. Blood Adv. 2018, 2, 1356–1366. [Google Scholar] [CrossRef]
- Galimberti, S.; Balducci, S.; Guerrini, F.; Del Re, M.; Cacciola, R. Digital Droplet PCR in Hematologic Malignancies: A New Useful Molecular Tool. Diagnostics 2022, 12, 1305. [Google Scholar] [CrossRef] [PubMed]
- Coccaro, N.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Digital PCR: A Reliable Tool for Analyzing and Monitoring Hematologic Malignancies. Int. J. Mol. Sci. 2020, 21, 3141. [Google Scholar] [CrossRef]
- Bacher, U.; Dicker, F.; Haferlach, C.; Alpermann, T.; Rose, D.; Kern, W.; Haferlach, T.; Schnittger, S. Quantification of rare NPM1 mutation subtypes by digital PCR. Br. J. Haematol. 2014, 167, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Brambati, C.; Galbiati, S.; Xue, E.; Toffalori, C.; Crucitti, L.; Greco, R.; Sala, E.; Crippa, A.; Chiesa, L.; Soriani, N.; et al. Droplet digital polymerase chain reaction for DNMT3A and IDH1/2 mutations to improve early detection of acute myeloid leukemia relapse after allogeneic hematopoietic stem cell transplantation. Haematologica 2016, 101, e157–e161. [Google Scholar] [CrossRef]
- Crowgey, E.L.; Mahajan, N.; Wong, W.H.; Gopalakrishnapillai, A.; Barwe, S.P.; Kolb, E.A.; Druley, T.E. Error-corrected sequencing strategies enable comprehensive detection of leukemic mutations relevant for diagnosis and minimal residual disease monitoring. BMC Med. Genom. 2020, 13, 32. [Google Scholar] [CrossRef]
- Patkar, N.; Kakirde, C.; Shaikh, A.F.; Salve, R.; Bhanshe, P.; Chatterjee, G.; Rajpal, S.; Joshi, S.; Chaudhary, S.; Kodgule, R.; et al. Clinical impact of panel-based error-corrected next generation sequencing versus flow cytometry to detect measurable residual disease (MRD) in acute myeloid leukemia (AML). Leukemia 2021, 35, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Vonk, C.M.; Al Hinai, A.S.A.; Hanekamp, D.; Valk, P.J.M. Molecular Minimal Residual Disease Detection in Acute Myeloid Leukemia. Cancers 2021, 13, 5431. [Google Scholar] [CrossRef]
- McGowan, P.F.; Hyter, S.D.; Cui, W.; Plummer, R.M.; Godwin, A.K.; Zhang, D. Comparison of flow cytometry and next-generation sequencing in minimal residual disease monitoring of acute myeloid leukemia: One institute’s practical clinical experience. Int. J. Lab. Hematol. 2022, 44, 118–126. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Steensma, D.P.; Graubert, T.A.; Ebert, B.L. Clonal hematopoiesis and measurable residual disease assessment in acute myeloid leukemia. Blood 2020, 135, 1729–1738. [Google Scholar] [CrossRef]
- Heuser, M.; Heida, B.; Büttner, K.; Wienecke, C.P.; Teich, K.; Funke, C.; Brandes, M.; Klement, P.; Liebich, A.; Wichmann, M.; et al. Posttransplantation MRD monitoring in patients with AML by next-generation sequencing using DTA and non-DTA mutations. Blood Adv. 2021, 5, 2294–2304. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Hutter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; et al. Indeterminate and oncogenic potential: CHIP vs CHOP mutations in AML with NPM1 alteration. Leukemia 2022, 36, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Höllein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Diagnostic Markers | |
|---|---|
| Lineage assigning antigens | |
| Myeloperoxidase | Myeloid lineage |
| CD11c, CD14, CD64, lysozyme | Myeloid lineage with monocytic differentiation |
| CD19 | B-lineage. Requires also at least one (CD19 strong) or two (CD19 weak) from CD22, CD10 and CD79a |
| CD3 (surface or cytoplasmic) | T-lineage |
| Myeloid differentiation-associated antigens | |
| CD13, CD33, CD11b, CD15, CD64 | Myeloid |
| CD14, CD36, CD64, CD4, CD11c | Monocytic |
| CD41, CD42b, CD61, CD36 | Megakaryocytic |
| CD235a, CD71 strong, CD105, CD36 | Erythroid |
| CD203c, CD123 | Basophil |
| CD123, CD4, HLA-DR strong, CD303, CD304 | Dendritic |
| CD117 strong | Mastocytic |
| MRD markers | |
| Basic markers | |
| CD34, CD117, HLA-DR, CD45, CD13, CD33 | Myeloid precursor identification |
| CD7, CD56 | Lymphoid antigen aberrancies |
| Other useful markers | |
| CD64, CD14, CD11b, CD4 | Monocytic |
| CD19, CD2, CD5 | Lymphoid antigen aberrancies |
| CD38, CD123, CD133 | Leukemia stem cell identification |
| ICC 2022 | WHO 2022 | ||
|---|---|---|---|
| Category | Blasts Required for Diagnosis | Category | Blasts Required for Diagnosis |
| AML with Recurrent Genetic Abnormalities | AML with Defining Genetic Abnormalities | ||
| Acute promyelocytic leukemia (APL) with t(15;17)(q24.1;q21.2)/PML::RARA | ≥10% | Acute promyelocytic leukemia (APL) with PML::RARA fusion | No threshold |
| APL with other RARA rearrangements | ≥10% | ||
| AML with t(8;21)(q22;q22.1)/RUNX1::RUNX1T1 | ≥10% | AML with RUNX1::RUNX1T1 fusion | No threshold |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB::MYH11 | ≥10% | AML with CBFB::MYH11 fusion | No threshold |
| AML with t(9;11)(p21.3;q23.3)/MLLT3::KMT2A | ≥10% | AML with KMT2A rearrangement | No threshold |
| AML with other KMT2A rearrangements | ≥10% | ||
| AML with t(6;9)(p22.3;q34.1)/DEK::NUP214 | ≥10% | AML with DEK::NUP214 fusion | No threshold |
| AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)/GATA2; MECOM(EVI1) | ≥10% | AML with MECOM rearrangement | No threshold |
| AML with other MECOM rearrangements | ≥10% | ||
| AML with other rare recurring translocations | ≥10% | AML with NUP98 rearrangement | No threshold |
| AML with RBM15::MRTFA fusion | No threshold | ||
| AML with t(9;22)(q34.1;q11.2)/BCR::ABL1 | ≥20% | AML with BCR::ABL1 fusion | ≥20% |
| AML with mutated NPM1 | ≥10% | AML with NPM1 mutation | No threshold |
| AML with in-frame bZIP CEBPA mutations | ≥10% | AML with CEBPA mutation | ≥20% |
| AML and MDS/AML with mutated TP53 | 10–19% (MDS/AML) and ≥20% (AML) | ||
| AML and MDS/AML with myelodysplasia-related gene mutations Defined by mutations in ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, or ZRSR2 | 10–19% (MDS/AML) and ≥20% (AML) | AML, myelodysplasia-related Defined by one or more cytogenetics of molecular alterations:
| ≥20% |
| AML with myelodysplasia-related cytogenetic abnormalities Defined by a complex karyotype (≥3 unrelated clonal chromosomal abnormalities in the absence of other class-defining recurring genetic abnormalities), del(5q)/t(5q)/add(5q), −7/del(7q), +8, del(12p)/t(12p)/add(12p), i(17q),−17/add(17p) or del(17p), del(20q), and/or idic(X)(q13) clonal abnormalities | 10–19% (MDS/AML) and ≥20% (AML) | ||
| AML with other defined genetic alterations | No threshold | ||
| AML without recurrent genetic abnormalities | AML without defining genetic abnormalities | ||
| AML not otherwise specified (NOS) | 10–19% (MDS/AML) and ≥20% (AML) | AML defined by differentiation | ≥20% |
| Gene | Diagnostic | Prognostic | Therapeutic | MRD Marker |
|---|---|---|---|---|
| FLT3 | ||||
| IDH1 | ||||
| IDH2 | ||||
| NPM1 | ||||
| CEBPA | * | |||
| DDX41 | * | |||
| TP53 | * | |||
| ASXL1 | ||||
| BCOR | ||||
| EZH2 | ||||
| RUNX1 | * | |||
| SF3B1 | ||||
| SRSF2 | ||||
| STAG2 | ||||
| U2AF1 | ||||
| ZRSR2 | ||||
| ANKRD26 | * | |||
| BCORL1 | ||||
| BRAF | ||||
| CBL | ||||
| CSF3R | ||||
| DNMT3A | ||||
| ETV6 | * | |||
| GATA2 | * | |||
| JAK2 | ||||
| KIT | ||||
| KRAS | ||||
| NRAS | ||||
| NF1 | ||||
| PHF6 | ||||
| PPM1D | ||||
| PTPN11 | ||||
| RAD21 | ||||
| SETBP1 | ||||
| TET2 | ||||
| WT1 | ||||
| Fusion Gene | Diagnostic | Prognostic | Therapeutic | MRD Marker |
| PML::RARA | ||||
| CBFB::MYH11 | ||||
| RUNX1:RUNX1T1 | ||||
| KMT2Ar | ||||
| BCR::ABL1 | ||||
| DEK::NUP214 | ||||
| MECOMr | ||||
| NUP98r | ||||
| KAT6A::CREBBP | ||||
| RBM15::MRTFA | ||||
| X::RARA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guijarro, F.; Garrote, M.; Villamor, N.; Colomer, D.; Esteve, J.; López-Guerra, M. Novel Tools for Diagnosis and Monitoring of AML. Curr. Oncol. 2023, 30, 5201-5213. https://doi.org/10.3390/curroncol30060395
Guijarro F, Garrote M, Villamor N, Colomer D, Esteve J, López-Guerra M. Novel Tools for Diagnosis and Monitoring of AML. Current Oncology. 2023; 30(6):5201-5213. https://doi.org/10.3390/curroncol30060395
Chicago/Turabian StyleGuijarro, Francesca, Marta Garrote, Neus Villamor, Dolors Colomer, Jordi Esteve, and Mónica López-Guerra. 2023. "Novel Tools for Diagnosis and Monitoring of AML" Current Oncology 30, no. 6: 5201-5213. https://doi.org/10.3390/curroncol30060395
APA StyleGuijarro, F., Garrote, M., Villamor, N., Colomer, D., Esteve, J., & López-Guerra, M. (2023). Novel Tools for Diagnosis and Monitoring of AML. Current Oncology, 30(6), 5201-5213. https://doi.org/10.3390/curroncol30060395