Pediatric Brain Tumours: Lessons from the Immune Microenvironment



Abstract
1. Introduction
2. Immune Surveillance and Trafficking in the Developing Central Nervous System
3. Immunoediting and Immunomodulation in Pediatric Brain Tumours
3.1. Medulloblastoma
3.1.1. Tumour-Infiltrating Lymphocytes (TILs)
Natural Killer (NK) Cells
T and B Lymphocytes
3.1.2. Tumour-Associated Macrophages
3.2. High-Grade Pediatric Gliomas
3.2.1. Tumour-Infiltrating Lymphocytes
3.2.2. Tumour-Associated Macrophages
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, D.A.; Richardson, L.C.; Henley, S.J.; Wilson, R.J.; Dowling, N.F.; Weir, H.K.; Tai, E.W.; Buchanan Lunsford, N. Pediatric Cancer Mortality and Survival in the United States, 2001–2016. Cancer 2020, 126, 4379–4389. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer Heterogeneity: Implications for Targeted Therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.M.; Shatz, C.J. Immune Signalling in Neural Development, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2004, 5, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Marc, T. Chapter 110—Brain Development and the Immune System: An Introduction to Inflammatory and Infectious Diseases of the Child’s Brain. In Handbook of Clinical Neurology; Dulac, O., Lassonde, M., Sarnat, H.B., Eds.; Pediatric Neurology Part II; Elsevier: Amsterdam, The Netherlands, 2013; Volume 112, pp. 1087–1089. [Google Scholar]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS Immune Privilege: Hiding in Plain Sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and Vertebral Bone Marrow Are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma. Science 2021, 373, eabf7844. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or More Routes for Leukocyte Migration into the Central Nervous System. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef]
- Albayram, M.S.; Smith, G.; Tufan, F.; Tuna, I.S.; Bostancıklıoğlu, M.; Zile, M.; Albayram, O. Non-Invasive MR Imaging of Human Brain Lymphatic Networks with Connections to Cervical Lymph Nodes. Nat. Commun. 2022, 13, 203. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and Functional Features of Central Nervous System Lymphatic Vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Ahn, J.H.; Cho, H.; Kim, J.-H.; Kim, S.H.; Ham, J.-S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.-H.; Hong, Y.-K.; et al. Meningeal Lymphatic Vessels at the Skull Base Drain Cerebrospinal Fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological Blood–Brain Transport Is Impaired with Age by a Shift in Transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef]
- Török, O.; Schreiner, B.; Schaffenrath, J.; Tsai, H.-C.; Maheshwari, U.; Stifter, S.A.; Welsh, C.; Amorim, A.; Sridhar, S.; Utz, S.G.; et al. Pericytes Regulate Vascular Immune Homeostasis in the CNS. Proc. Natl. Acad. Sci. USA 2021, 118, e2016587118. [Google Scholar] [CrossRef]
- Chen, M.B.; Yang, A.C.; Yousef, H.; Lee, D.; Chen, W.; Schaum, N.; Lehallier, B.; Quake, S.R.; Wyss-Coray, T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020, 30, 4418–4432.e4. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Bieri, G.; Schroer, A.B.; Villeda, S.A. Blood-to-Brain Communication in Aging and Rejuvenation. Nat. Neurosci. 2023, 26, 379–393. [Google Scholar] [CrossRef]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct Vascular Channels Connect Skull Bone Marrow and the Brain Surface Enabling Myeloid Cell Migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef]
- Mazzitelli, J.A.; Smyth, L.C.D.; Cross, K.A.; Dykstra, T.; Sun, J.; Du, S.; Mamuladze, T.; Smirnov, I.; Rustenhoven, J.; Kipnis, J. Cerebrospinal Fluid Regulates Skull Bone Marrow Niches via Direct Access through Dural Channels. Nat. Neurosci. 2022, 25, 555–560. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Drieu, A.; Mamuladze, T.; de Lima, K.A.; Dykstra, T.; Wall, M.; Papadopoulos, Z.; Kanamori, M.; Salvador, A.F.; Baker, W.; et al. Functional Characterization of the Dural Sinuses as a Neuroimmune Interface. Cell 2021, 184, 1000–1016.e27. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional Delivery of CAR T Cells to the Cerebrospinal Fluid for Treatment of Metastatic Medulloblastoma and Ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T Cell Therapy for H3K27M-Mutated Diffuse Midline Gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Morimoto, K.; Nakajima, K. Role of the Immune System in the Development of the Central Nervous System. Front. Neurosci. 2019, 13, 916. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Corriveau, R.A.; Huh, G.S.; Shatz, C.J. Regulation of Class I MHC Gene Expression in the Developing and Mature CNS by Neural Activity. Neuron 1998, 21, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.; Dang, H.; Xu, W.; Gustafson, S.; Jin, Y.; Wickramasinghe, L.; Won, T.; Bobarnac, G.; Middleton, B.; Tian, J.; et al. Major Histocompatibility Complex Class I Molecules Modulate Embryonic Neuritogenesis and Neuronal Polarization. J. Neuroimmunol. 2012, 247, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goddard, C.A.; Butts, D.A.; Shatz, C.J. Regulation of CNS Synapses by Neuronal MHC Class I. Proc. Natl. Acad. Sci. USA 2007, 104, 6828–6833. [Google Scholar] [CrossRef]
- Needleman, L.A.; Liu, X.-B.; El-Sabeawy, F.; Jones, E.G.; McAllister, A.K. MHC Class I Molecules Are Present Both Pre- and Postsynaptically in the Visual Cortex during Postnatal Development and in Adulthood. Proc. Natl. Acad. Sci. USA 2010, 107, 16999–17004. [Google Scholar] [CrossRef]
- Massa, P.T.; Ozato, K.; McFarlin, D.E. Cell Type-Specific Regulation of Major Histocompatibility Complex (MHC) Class I Gene Expression in Astrocytes, Oligodendrocytes, and Neurons. Glia 1993, 8, 201–207. [Google Scholar] [CrossRef]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics During Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef]
- Mosser, C.-A.; Baptista, S.; Arnoux, I.; Audinat, E. Microglia in CNS Development: Shaping the Brain for the Future. Prog. Neurobiol. 2017, 149, 1–20. [Google Scholar] [CrossRef]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Goldman, J.E.; Sekino, Y.; Sato, K. Microglia Enhance Neurogenesis and Oligodendrogenesis in the Early Postnatal Subventricular Zone. J. Neurosci. 2014, 34, 2231–2243. [Google Scholar] [CrossRef]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the Immune System in Humans from Infancy to Old Age. Proc. R. Soc. B Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Havran, W.L.; Allison, J.P. Developmentally Ordered Appearance of Thymocytes Expressing Different T-Cell Antigen Receptors. Nature 1988, 335, 443–445. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Function of Γδ T Cells in Tumor Immunology and Their Application to Cancer Therapy. Exp. Mol. Med. 2021, 53, 318–327. [Google Scholar] [CrossRef]
- Albertsson, A.M.; Zhang, X.; Vontell, R.; Bi, D.; Bronson, R.T.; Supramaniam, V.; Baburamani, A.A.; Hua, S.; Nazmi, A.; Cardell, S.; et al. Γδ T Cells Contribute to Injury in the Developing Brain. Am. J. Pathol. 2018, 188, 757–767. [Google Scholar] [CrossRef]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and Disseminated Tumour Cells—Mechanisms of Immune Surveillance and Escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Vesely, M.D.; Schreiber, R.D. Cancer Immunoediting: Antigens, Mechanisms, and Implications to Cancer Immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New Insights into Cancer Immunoediting and Its Three Component Phases—Elimination, Equilibrium and Escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From Mice to Humans: Developments in Cancer Immunoediting. J. Clin. Investig. 2015, 125, 3338–3346. [Google Scholar] [CrossRef]
- Patel, R.R.; Ramkissoon, S.H.; Ross, J.; Weintraub, L. Tumor Mutational Burden and Driver Mutations: Characterizing the Genomic Landscape of Pediatric Brain Tumors. Pediatr. Blood Cancer 2020, 67, e28338. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The Landscape of Genomic Alterations across Childhood Cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Hinojosa, S.; Grant, M.; Panigrahi, A.; Zhang, H.; Caisova, V.; Bollard, C.M.; Rood, B.R. Proteogenomic Discovery of Neoantigens Facilitates Personalized Multi-Antigen Targeted T Cell Immunotherapy for Brain Tumors. Nat. Commun. 2021, 12, 6689. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.; Nobuta, H.; Li, H.; Kasai, A.; Yong, W.H.; Waschek, J.A. Targeted STAT3 Disruption in Myeloid Cells Alters Immunosuppressor Cell Abundance in a Murine Model of Spontaneous Medulloblastoma. J. Leukocyte Biol. 2014, 95, 357–367. [Google Scholar] [CrossRef]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric Pan-Central Nervous System Tumor Analysis of Immune-Cell Infiltration Identifies Correlates of Antitumor Immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef]
- Terry, R.L.; Meyran, D.; Ziegler, D.S.; Haber, M.; Ekert, P.G.; Trapani, J.A.; Neeson, P.J. Immune Profiling of Pediatric Solid Tumors. J. Clin. Investig. 2020, 130, 3391–3402. [Google Scholar] [CrossRef]
- Li, K.; Duan, W.; Zhao, H.; Wang, L.; Wang, W.; Zhan, Y.; Sun, T.; Zhang, F.; Yu, B.; Bai, Y.; et al. Preoperative Neutrophil to Lymphocyte Ratio and Platelet to Lymphocyte Ratio Are Associated with the Prognosis of Group 3 and Group 4 Medulloblastoma. Sci. Rep. 2019, 9, 13239. [Google Scholar] [CrossRef]
- Arroyo, V.M.; Lupo, P.J.; Scheurer, M.E.; Rednam, S.P.; Murray, J.; Okcu, M.F.; Chintagumpala, M.M.; Brown, A.L. Pilot Study of DNA Methylation-Derived Neutrophil-to-Lymphocyte Ratio and Survival in Pediatric Medulloblastoma. Cancer Epidemiol. 2019, 59, 71–74. [Google Scholar] [CrossRef]
- Patel, S.; Wang, S.; Snuderl, M.; Karajannis, M.A. Pre-Treatment Lymphopenia and Indication of Tumor-Induced Systemic Immunosuppression in Medulloblastoma. J. Neurooncol. 2018, 136, 541–544. [Google Scholar] [CrossRef]
- Grassberger, C.; Shinnick, D.; Yeap, B.Y.; Tracy, M.; Ellsworth, S.G.; Hess, C.B.; Weyman, E.A.; Gallotto, S.L.; Lawell, M.P.; Bajaj, B.; et al. Circulating Lymphocyte Counts Early During Radiation Therapy Are Associated With Recurrence in Pediatric Medulloblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 1044–1052. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα Anti-Phagocytic Axis by a Humanized Anti-CD47 Antibody Is an Efficacious Treatment for Malignant Pediatric Brain Tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef]
- Marques, R.F.; Moreno, D.A.; da Silva, L.; Leal, L.F.; de Paula, F.E.; Santana, I.; Teixeira, G.; Saggioro, F.; Neder, L.; Junior, C.A.; et al. Digital Expression Profile of Immune Checkpoint Genes in Medulloblastomas Identifies CD24 and CD276 as Putative Immunotherapy Targets. Front. Immunol. 2023, 14, 1062856. [Google Scholar] [CrossRef]
- Gate, D.; Danielpour, M.; Rodriguez, J.; Kim, G.-B.; Levy, R.; Bannykh, S.; Breunig, J.J.; Kaech, S.M.; Flavell, R.A.; Town, T. T-Cell TGF-β Signaling Abrogation Restricts Medulloblastoma Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3458–E3466. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the Immune Microenvironment of Diffuse Intrinsic Pontine Glioma: Implications for Development of Immunotherapy. Neuro-Oncology 2019, 21, 83–94. [Google Scholar] [CrossRef]
- Yao, M.; Ventura, P.B.; Jiang, Y.; Rodriguez, F.J.; Wang, L.; Perry, J.S.A.; Yang, Y.; Wahl, K.; Crittenden, R.B.; Bennett, M.L.; et al. Astrocytic Trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 2020, 180, 502–520.e19. [Google Scholar] [CrossRef]
- Fernandez, L.; Portugal, R.; Valentin, J.; Martin, R.; Maxwell, H.; González-Vicent, M.; Díaz, M.Á.; Pérez-Martínez, A. In Vitro Natural Killer Cell Immunotherapy for Medulloblastoma. Front. Oncol. 2013, 3, 94. [Google Scholar] [CrossRef]
- Martin, A.M.; Nirschl, C.J.; Polanczyk, M.J.; Bell, W.R.; Nirschl, T.R.; Harris-Bookman, S.; Phallen, J.; Hicks, J.; Martinez, D.; Ogurtsova, A.; et al. PD-L1 Expression in Medulloblastoma: An Evaluation by Subgroup. Oncotarget 2018, 9, 19177–19191. [Google Scholar] [CrossRef]
- Meister, N.; Shalaby, T.; von Bueren, A.O.; Rivera, P.; Patti, R.; Oehler, C.; Pruschy, M.; Grotzer, M.A. Interferon-γ Mediated up-Regulation of Caspase-8 Sensitises Medulloblastoma Cells to Radio- and Chemotherapy. Eur. J. Cancer 2007, 43, 1833–1841. [Google Scholar] [CrossRef]
- Crotty, E.E.; Smith, S.M.C.; Brasel, K.; Pakiam, F.; Girard, E.J.; Connor, Y.D.; Zindy, F.; Mhyre, A.J.; Roussel, M.F.; Olson, J.M. Medulloblastoma Recurrence and Metastatic Spread Are Independent of Colony-Stimulating Factor 1 Receptor Signaling and Macrophage Survival. J. Neurooncol. 2021, 153, 225–237. [Google Scholar] [CrossRef]
- Dang, M.T.; Gonzalez, M.V.; Gaonkar, K.S.; Rathi, K.S.; Young, P.; Arif, S.; Zhai, L.; Alam, Z.; Devalaraja, S.; To, T.K.J.; et al. Macrophages in SHH Subgroup Medulloblastoma Display Dynamic Heterogeneity That Varies with Treatment Modality. Cell Rep. 2021, 34, 108917. [Google Scholar] [CrossRef]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suvà, M.L.; Vogel, H.; Monje, M. Non-Inflammatory Tumor Microenvironment of Diffuse Intrinsic Pontine Glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef]
- Wu, K.-S.; Jian, T.-Y.; Sung, S.-Y.; Hsieh, C.-L.; Huang, M.-H.; Fang, C.-L.; Wong, T.-T.; Lin, Y.-L. Enrichment of Tumor-Infiltrating B Cells in Group 4 Medulloblastoma in Children. Int. J. Mol. Sci. 2022, 23, 5287. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Poolen, G.C.; van Vliet, L.C.; Schipper, J.G.; Broekhuizen, R.; Monnikhof, M.; Van Hecke, W.; Vermeulen, J.F.; Bovenschen, N. Pediatric Medulloblastoma Express Immune Checkpoint B7-H3. Clin. Transl. Oncol. 2022, 24, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Luther, N.; Ibrahim, G.M.; Hawkins, C.; Vibhakar, R.; Handler, M.H.; Souweidane, M.M. B7-H3, a Potential Therapeutic Target, Is Expressed in Diffuse Intrinsic Pontine Glioma. J. Neurooncol. 2013, 111, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Bandopadhayay, P.; Jenkins, M.R. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol. 2019, 40, 748–761. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The Immune Contexture and Immunoscore in Cancer Prognosis and Therapeutic Efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Nabbi, A.; Beck, P.; Delaidelli, A.; Oldridge, D.A.; Sudhaman, S.; Zhu, K.; Yang, S.Y.C.; Mulder, D.T.; Bruce, J.P.; Paulson, J.N.; et al. Transcriptional Immunogenomic Analysis Reveals Distinct Immunological Clusters in Pediatric Nervous System Tumours. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Vatner, R.E.; Niemierko, A.; Misra, M.; Weyman, E.A.; Goebel, C.P.; Ebb, D.H.; Jones, R.M.; Huang, M.S.; Mahajan, A.; Grosshans, D.R.; et al. Endocrine Deficiency As a Function of Radiation Dose to the Hypothalamus and Pituitary in Pediatric and Young Adult Patients With Brain Tumors. J. Clin. Oncol. 2018, 36, 2854–2862. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Burger, P.C.; Zhou, T.; Holmes, E.J.; Kocak, M.; Onar, A.; Goldwein, J.; Mehta, M.; Packer, R.J.; Tarbell, N.; et al. Outcome of Children With Metastatic Medulloblastoma Treated With Carboplatin During Craniospinal Radiotherapy: A Children’s Oncology Group Phase I/II Study. J. Clin. Oncol. 2012, 30, 2648–2653. [Google Scholar] [CrossRef]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the Age of Molecular Subgroups: A Review: JNSPG 75th Anniversary Invited Review Article. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef]
- Acharya, S.; Guo, Y.; Patni, T.; Li, Y.; Wang, C.; Gargone, M.; Ashford, J.M.; Wilson, L.; Faught, A.; Reddick, W.E.; et al. Association Between Brain Substructure Dose and Cognitive Outcomes in Children with Medulloblastoma Treated on SJMB03: A Step Toward Substructure-Informed Planning. J. Clin. Oncol. 2022, 40, 83–95. [Google Scholar] [CrossRef]
- Oyefiade, A.; Paltin, I.; De Luca, C.R.; Hardy, K.K.; Grosshans, D.R.; Chintagumpala, M.; Mabbott, D.J.; Kahalley, L.S. Cognitive Risk in Survivors of Pediatric Brain Tumors. J. Clin. Oncol. 2021, 39, 1718–1726. [Google Scholar] [CrossRef]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-Specific Immune and Stromal Microenvironment in Medulloblastoma. OncoImmunology 2018, 7, e1462430. [Google Scholar] [CrossRef]
- Vauléon, E.; Tony, A.; Hamlat, A.; Etcheverry, A.; Chiforeanu, D.C.; Menei, P.; Mosser, J.; Quillien, V.; Aubry, M. Immune Genes Are Associated with Human Glioblastoma Pathology and Patient Survival. BMC Med. Genom. 2012, 5, 41. [Google Scholar] [CrossRef]
- Haberthur, K.; Brennan, K.; Hoglund, V.; Balcaitis, S.; Chinn, H.; Davis, A.; Kreuser, S.; Winter, C.; Leary, S.E.S.; Deutsch, G.H.; et al. NKG2D Ligand Expression in Pediatric Brain Tumors. Cancer Biol. Ther. 2016, 17, 1253–1265. [Google Scholar] [CrossRef]
- Hendrikse, L.D.; Haldipur, P.; Saulnier, O.; Millman, J.; Sjoboen, A.H.; Erickson, A.W.; Ong, W.; Gordon, V.; Coudière-Morrison, L.; Mercier, A.L.; et al. Failure of Human Rhombic Lip Differentiation Underlies Medulloblastoma Formation. Nature 2022, 609, 1021–1028. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural Killer Group 2D Receptor and Its Ligands in Cancer Immune Escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Castriconi, R.; Espéli, M.; Anfossi, N.; Juarez, T.; Hue, S.; Conway, H.; Romagné, F.; Dondero, A.; Nanni, M.; et al. Comparative Analysis of Human NK Cell Activation Induced by NKG2D and Natural Cytotoxicity Receptors. Eur. J. Immunol. 2004, 34, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Roe, K. NK-Cell Exhaustion, B-Cell Exhaustion and T-Cell Exhaustion—The Differences and Similarities. Immunology 2022, 166, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef]
- Powell, A.B.; Yadavilli, S.; Saunders, D.; Van Pelt, S.; Chorvinsky, E.; Burga, R.A.; Albihani, S.; Hanley, P.J.; Xu, Z.; Pei, Y.; et al. Medulloblastoma Rendered Susceptible to NK-Cell Attack by TGFβ Neutralization. J. Transl. Med. 2019, 17, 321. [Google Scholar] [CrossRef]
- Nandan, D.; Reiner, N.E. TGF-Beta Attenuates the Class II Transactivator and Reveals an Accessory Pathway of IFN-Gamma Action. J. Immunol. 1997, 158, 1095–1101. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming Growth Factor Β1 Inhibits Expression of NKp30 and NKG2D Receptors: Consequences for the NK-Mediated Killing of Dendritic Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Takami, A.; Yoshioka, K.; Nakata, K.; Sato, T.; Kasahara, Y.; Nakao, S. Human MicroRNA-1245 down-Regulates the NKG2D Receptor in Natural Killer Cells and Impairs NKG2D-Mediated Functions. Haematologica 2012, 97, 1295–1303. [Google Scholar] [CrossRef]
- Raffaghello, L.; Nozza, P.; Morandi, F.; Camoriano, M.; Wang, X.; Garrè, M.L.; Cama, A.; Basso, G.; Ferrone, S.; Gambini, C.; et al. Expression and Functional Analysis of Human Leukocyte Antigen Class I Antigen-Processing Machinery in Medulloblastoma. Cancer Res. 2007, 67, 5471–5478. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Negri, F.; Bellora, F.; Nozza, P.; Carnemolla, B.; Raso, A.; Moretta, L.; Moretta, A.; Bottino, C. Both CD133+ and CD133– Medulloblastoma Cell Lines Express Ligands for Triggering NK Receptors and Are Susceptible to NK-Mediated Cytotoxicity. Eur. J. Immunol. 2007, 37, 3190–3196. [Google Scholar] [CrossRef]
- Kong, Y.; Cao, W.; Xi, X.; Ma, C.; Cui, L.; He, W. The NKG2D Ligand ULBP4 Binds to TCRγ9/Δ2 and Induces Cytotoxicity to Tumor Cells through Both TCRγδ and NKG2D. Blood 2009, 114, 310–317. [Google Scholar] [CrossRef]
- van der Heiden, M.; Björkander, S.; Rahman Qazi, K.; Bittmann, J.; Hell, L.; Jenmalm, M.C.; Marchini, G.; Vermijlen, D.; Abrahamsson, T.; Nilsson, C.; et al. Characterization of the Γδ T-Cell Compartment during Infancy Reveals Clear Differences between the Early Neonatal Period and 2 Years of Age. Immunol. Cell Biol. 2020, 98, 79–87. [Google Scholar] [CrossRef]
- Pistoia, V.; Tumino, N.; Vacca, P.; Veneziani, I.; Moretta, A.; Locatelli, F.; Moretta, L. Human Γδ T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front. Immunol. 2018, 9, 984. [Google Scholar] [CrossRef]
- Khatua, S.; Cooper, L.J.N.; Sandberg, D.I.; Ketonen, L.; Johnson, J.M.; Rytting, M.E.; Liu, D.D.; Meador, H.; Trikha, P.; Nakkula, R.J.; et al. Phase I Study of Intraventricular Infusions of Autologous Ex Vivo Expanded NK Cells in Children with Recurrent Medulloblastoma and Ependymoma. J. Neuro-Oncol. 2020, 22, 1214–1225. [Google Scholar] [CrossRef]
- Gururangan, S.; Reap, E.; Schmittling, R.; Kocak, M.; Reynolds, R.; Grant, G.; Onar-Thomas, A.; Baxter, P.; Pollack, I.F.; Phillips, P.; et al. Regulatory T Cell Subsets in Patients with Medulloblastoma at Diagnosis and during Standard Irradiation and Chemotherapy (PBTC N-11). Cancer Immunol. Immunother. 2017, 66, 1589–1595. [Google Scholar] [CrossRef]
- Vermeulen, J.F.; Van Hecke, W.; Adriaansen, E.J.M.; Jansen, M.K.; Bouma, R.G.; Villacorta Hidalgo, J.; Fisch, P.; Broekhuizen, R.; Spliet, W.G.M.; Kool, M.; et al. Prognostic Relevance of Tumor-Infiltrating Lymphocytes and Immune Checkpoints in Pediatric Medulloblastoma. OncoImmunology 2018, 7, e1398877. [Google Scholar] [CrossRef]
- Murata, D.; Mineharu, Y.; Arakawa, Y.; Liu, B.; Tanji, M.; Yamaguchi, M.; Fujimoto, K.; Fukui, N.; Terada, Y.; Yokogawa, R.; et al. High Programmed Cell Death 1 Ligand–1 Expression: Association with CD8+ T-Cell Infiltration and Poor Prognosis in Human Medulloblastoma. J. Neurosurg. 2017, 128, 710–716. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 Disruption Attenuates Tumor PD-L1 Expression and Promotes Antitumor Immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-γ/PD-L1 Axis between T Cells and Tumor Microenvironment: Hints for Glioma Anti-PD-1/PD-L1 Therapy. J. Neuroinflammation 2018, 15, 290. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC Regulates the Antitumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene —The Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of Oncogenic Pathways on Evasion of Antitumour Immune Responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Smith, C.; Santi, M.; Rajan, B.; Rushing, E.J.; Choi, M.R.; Rood, B.R.; Cornelison, R.; MacDonald, T.J.; Vukmanovic, S. A Novel Role of HLA Class I in the Pathology of Medulloblastoma. J. Transl. Med. 2009, 7, 59. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and Microglia: The Cerberus of Glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef]
- Malawsky, D.S.; Weir, S.J.; Ocasio, J.K.; Babcock, B.; Dismuke, T.; Cleveland, A.H.; Donson, A.M.; Vibhakar, R.; Wilhelmsen, K.; Gershon, T.R. Cryptic Developmental Events Determine Medulloblastoma Radiosensitivity and Cellular Heterogeneity without Altering Transcriptomic Profile. Commun. Biol. 2021, 4, 616. [Google Scholar] [CrossRef]
- Bal, M.M.; Das Radotra, B.; Srinivasan, R.; Sharma, S.C. Expression of C-ErbB-4 in Medulloblastoma and Its Correlation with Prognosis. Histopathology 2006, 49, 92–93. [Google Scholar] [CrossRef]
- Aldaregia, J.; Errarte, P.; Olazagoitia-Garmendia, A.; Gimeno, M.; Uriz, J.J.; Gershon, T.R.; Garcia, I.; Matheu, A. Erbb4 Is Required for Cerebellar Development and Malignant Phenotype of Medulloblastoma. Cancers 2020, 12, 997. [Google Scholar] [CrossRef]
- Ghashghaei, H.T.; Weber, J.; Pevny, L.; Schmid, R.; Schwab, M.H.; Lloyd, K.C.K.; Eisenstat, D.D.; Lai, C.; Anton, E.S. The Role of Neuregulin–ErbB4 Interactions on the Proliferation and Organization of Cells in the Subventricular Zone. Proc. Natl. Acad. Sci. USA 2006, 103, 1930–1935. [Google Scholar] [CrossRef]
- Margol, A.S.; Robison, N.J.; Gnanachandran, J.; Hung, L.T.; Kennedy, R.J.; Vali, M.; Dhall, G.; Finlay, J.L.; Erdreich-Epstein, A.; Krieger, M.D.; et al. Tumor-Associated Macrophages in SHH Subgroup of Medulloblastomas. Clin. Cancer Res. 2015, 21, 1457–1465. [Google Scholar] [CrossRef]
- Hume, D.A.; MacDonald, K.P.A. Therapeutic Applications of Macrophage Colony-Stimulating Factor-1 (CSF-1) and Antagonists of CSF-1 Receptor (CSF-1R) Signaling. Blood 2012, 119, 1810–1820. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Tan, I.-L.; Arifa, R.D.N.; Rallapalli, H.; Kana, V.; Lao, Z.; Sanghrajka, R.M.; Sumru Bayin, N.; Tanne, A.; Wojcinski, A.; Korshunov, A.; et al. CSF1R Inhibition Depletes Tumor-Associated Macrophages and Attenuates Tumor Progression in a Mouse Sonic Hedgehog-Medulloblastoma Model. Oncogene 2021, 40, 396–407. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-Associated Macrophages Exhibit Anti-Tumoural Properties in Sonic Hedgehog Medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef]
- Diao, S.; Gu, C.; Zhang, H.; Yu, C. Immune Cell Infiltration and Cytokine Secretion Analysis Reveal a Non-Inflammatory Microenvironment of Medulloblastoma. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.; Choi, S.A.; Kim, S.-K.; Wang, K.-C.; Park, S.-H.; Kim, S.H.; Lee, J.Y.; Phi, J.H. M1 Macrophage Recruitment Correlates with Worse Outcome in SHH Medulloblastomas. BMC Cancer 2018, 18, 535. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-Associated Microglia/Macrophages Predict Poor Prognosis in High-Grade Gliomas and Correlate with an Aggressive Tumour Subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yang, K.; Wang, Z.; Zhao, M.; Deng, Y.; Ji, W.; Zou, Y.; Qian, C.; Liu, Y.; Xiao, H.; et al. CD44-Mediated Poor Prognosis in Glioma Is Associated With M2-Polarization of Tumor-Associated Macrophages and Immunosuppression. Front. Surg. 2022, 8, 785. [Google Scholar] [CrossRef] [PubMed]
- Vidyarthi, A.; Agnihotri, T.; Khan, N.; Singh, S.; Tewari, M.K.; Radotra, B.D.; Chatterjee, D.; Agrewala, J.N. Predominance of M2 Macrophages in Gliomas Leads to the Suppression of Local and Systemic Immunity. Cancer Immunol. Immunother. 2019, 68, 1995–2004. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Beatty, G.L.; Paterson, Y. IFN-γ Can Promote Tumor Evasion of the Immune System In Vivo by Down-Regulating Cellular Levels of an Endogenous Tumor Antigen1. J. Immunol. 2000, 165, 5502–5508. [Google Scholar] [CrossRef]
- Beatty, G.L.; Paterson, Y. Regulation of Tumor Growth by IFN-γ in Cancer Immunotherapy. Immunol. Res. 2001, 24, 201–210. [Google Scholar] [CrossRef]
- Wang, J.; Lin, W.; Popko, B.; Campbell, I.L. Inducible Production of Interferon-γ in the Developing Brain Causes Cerebellar Dysplasia with Activation of the Sonic Hedgehog Pathway. Mol. Cell. Neurosci. 2004, 27, 489–496. [Google Scholar] [CrossRef]
- Sun, L.; Tian, Z.; Wang, J. A Direct Cross-Talk between Interferon-γ and Sonic Hedgehog Signaling That Leads to the Proliferation of Neuronal Precursor Cells. Brain Behav. Immun. 2010, 24, 220. [Google Scholar] [CrossRef]
- Wang, J.; Pham-Mitchell, N.; Schindler, C.; Campbell, I.L. Dysregulated Sonic Hedgehog Signaling and Medulloblastoma Consequent to IFN-α–Stimulated STAT2-Independent Production of IFN-γ in the Brain. J. Clin. Investig. 2003, 112, 535–543. [Google Scholar] [CrossRef]
- Lin, W.; Kemper, A.; McCarthy, K.D.; Pytel, P.; Wang, J.-P.; Campbell, I.L.; Utset, M.F.; Popko, B. Interferon-γ Induced Medulloblastoma in the Developing Cerebellum. J. Neurosci. 2004, 24, 10074–10083. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C.; et al. Pediatric High-Grade Glioma: Biologically and Clinically in Need of New Thinking. Neuro-Oncology 2017, 19, 153–161. [Google Scholar] [CrossRef]
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse Brainstem Glioma in Children: Critical Review of Clinical Trials. Lancet Oncol. 2006, 7, 241–248. [Google Scholar] [CrossRef]
- Griesinger, A.M.; Birks, D.K.; Donson, A.M.; Amani, V.; Hoffman, L.M.; Waziri, A.; Wang, M.; Handler, M.H.; Foreman, N.K. Characterization of Distinct Immunophenotypes across Pediatric Brain Tumor Types. J. Immunol. 2013, 191, 4880–4888. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-Brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842.e5. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, X.; Gao, L.; Wang, Y.; Guo, Y.; Xing, B.; Ma, W. Classification of Pediatric Gliomas Based on Immunological Profiling: Implications for Immunotherapy Strategies. Mol. Ther. Oncolytics 2021, 20, 34–47. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The Role of Neoantigens in Response to Immune Checkpoint Blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef]
- Plant, A.S.; Koyama, S.; Sinai, C.; Solomon, I.H.; Griffin, G.K.; Ligon, K.L.; Bandopadhayay, P.; Betensky, R.; Emerson, R.; Dranoff, G.; et al. Immunophenotyping of Pediatric Brain Tumors: Correlating Immune Infiltrate with Histology, Mutational Load, and Survival and Assessing Clonal T Cell Response. J. Neurooncol. 2018, 137, 269–278. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Valvi, S.; Gottardo, N.G.; Valvi, S.; Gottardo, N.G. Diffuse Intrinsic Pontine Glioma; IntechOpen: London, UK, 2018; ISBN 978-1-78923-557-9. [Google Scholar]
- Vitanza, N.A.; Monje, M. Diffuse Intrinsic Pontine Glioma: From Diagnosis to Next-Generation Clinical Trials. Curr. Treat. Options Neurol. 2019, 21, 37. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M Mutation in Histone H3.3 Defines Clinically and Biologically Distinct Subgroups of Pediatric Diffuse Intrinsic Pontine Gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Anastas, J.N.; Zee, B.M.; Kalin, J.H.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-Programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, 528–544.e10. [Google Scholar] [CrossRef]
- Bailey, C.P.; Figueroa, M.; Gangadharan, A.; Yang, Y.; Romero, M.M.; Kennis, B.A.; Yadavilli, S.; Henry, V.; Collier, T.; Monje, M.; et al. Pharmacologic Inhibition of Lysine-Specific Demethylase 1 as a Therapeutic and Immune-Sensitization Strategy in Pediatric High-Grade Glioma. Neuro-Oncology 2020, 22, 1302–1314. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.-O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent Somatic Mutations in ACVR1 in Pediatric Midline High-Grade Astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.S.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent Activating ACVR1 Mutations in Diffuse Intrinsic Pontine Glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic Analysis of Diffuse Intrinsic Pontine Gliomas Identifies Three Molecular Subgroups and Recurrent Activating ACVR1 Mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-β Downregulates the Activating Receptor NKG2D on NK Cells and CD8+ T Cells in Glioma Patients. Neuro-Oncology 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA Interference Targeting Transforming Growth Factor-β Enhances NKG2D-Mediated Antiglioma Immune Response, Inhibits Glioma Cell Migration and Invasiveness, and Abrogates Tumorigenicity In Vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef]
- Hwang, E.I.; Sayour, E.J.; Flores, C.T.; Grant, G.; Wechsler-Reya, R.; Hoang-Minh, L.B.; Kieran, M.W.; Salcido, J.; Prins, R.M.; Figg, J.W.; et al. The Current Landscape of Immunotherapy for Pediatric Brain Tumors. Nat. Cancer 2022, 3, 11–24. [Google Scholar] [CrossRef]
- Foster, J.B.; Madsen, P.J.; Hegde, M.; Ahmed, N.; Cole, K.A.; Maris, J.M.; Resnick, A.C.; Storm, P.B.; Waanders, A.J. Immunotherapy for Pediatric Brain Tumors: Past and Present. Neuro-Oncology 2019, 21, 1226–1238. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune Checkpoint Inhibitors: Recent Progress and Potential Biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current Approaches to Increase CAR T Cell Potency in Solid Tumors: Targeting the Tumor Microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-Cell Exhaustion in the Tumor Microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- McBride, J.A.; Striker, R. Imbalance in the Game of T Cells: What Can the CD4/CD8 T-Cell Ratio Tell Us about HIV and Health? PLoS Pathog. 2017, 13, e1006624. [Google Scholar] [CrossRef]
- Chow, A.; Schad, S.; Green, M.D.; Hellmann, M.D.; Allaj, V.; Ceglia, N.; Zago, G.; Shah, N.S.; Sharma, S.K.; Mattar, M.; et al. Tim-4+ Cavity-Resident Macrophages Impair Anti-Tumor CD8+ T Cell Immunity. Cancer Cell 2021, 39, 973–988.e9. [Google Scholar] [CrossRef]
- Kang, C.-W.; Dutta, A.; Chang, L.-Y.; Mahalingam, J.; Lin, Y.-C.; Chiang, J.-M.; Hsu, C.-Y.; Huang, C.-T.; Su, W.-T.; Chu, Y.-Y.; et al. Apoptosis of Tumor Infiltrating Effector TIM-3+CD8+ T Cells in Colon Cancer. Sci. Rep. 2015, 5, 15659. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Wilson, A.L.; Huang, W.; Seidel, K.; Brown, C.; Gustafson, J.A.; Yokoyama, J.K.; Johnson, A.J.; Baxter, B.A.; Koning, R.W.; et al. Intraventricular B7-H3 CAR T Cells for Diffuse Intrinsic Pontine Glioma: Preliminary First-in-Human Bioactivity and Safety. Cancer Discov. 2022, 13, 114–131. [Google Scholar] [CrossRef]
- Gnjatic, S.; Bronte, V.; Brunet, L.R.; Butler, M.O.; Disis, M.L.; Galon, J.; Hakansson, L.G.; Hanks, B.A.; Karanikas, V.; Khleif, S.N.; et al. Identifying Baseline Immune-Related Biomarkers to Predict Clinical Outcome of Immunotherapy. J. Immunother. Cancer 2017, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-Macrophage: A New Immunotherapy Candidate against Solid Tumors. Biomed. Pharmacother. 2021, 139, 111605. [Google Scholar] [CrossRef] [PubMed]


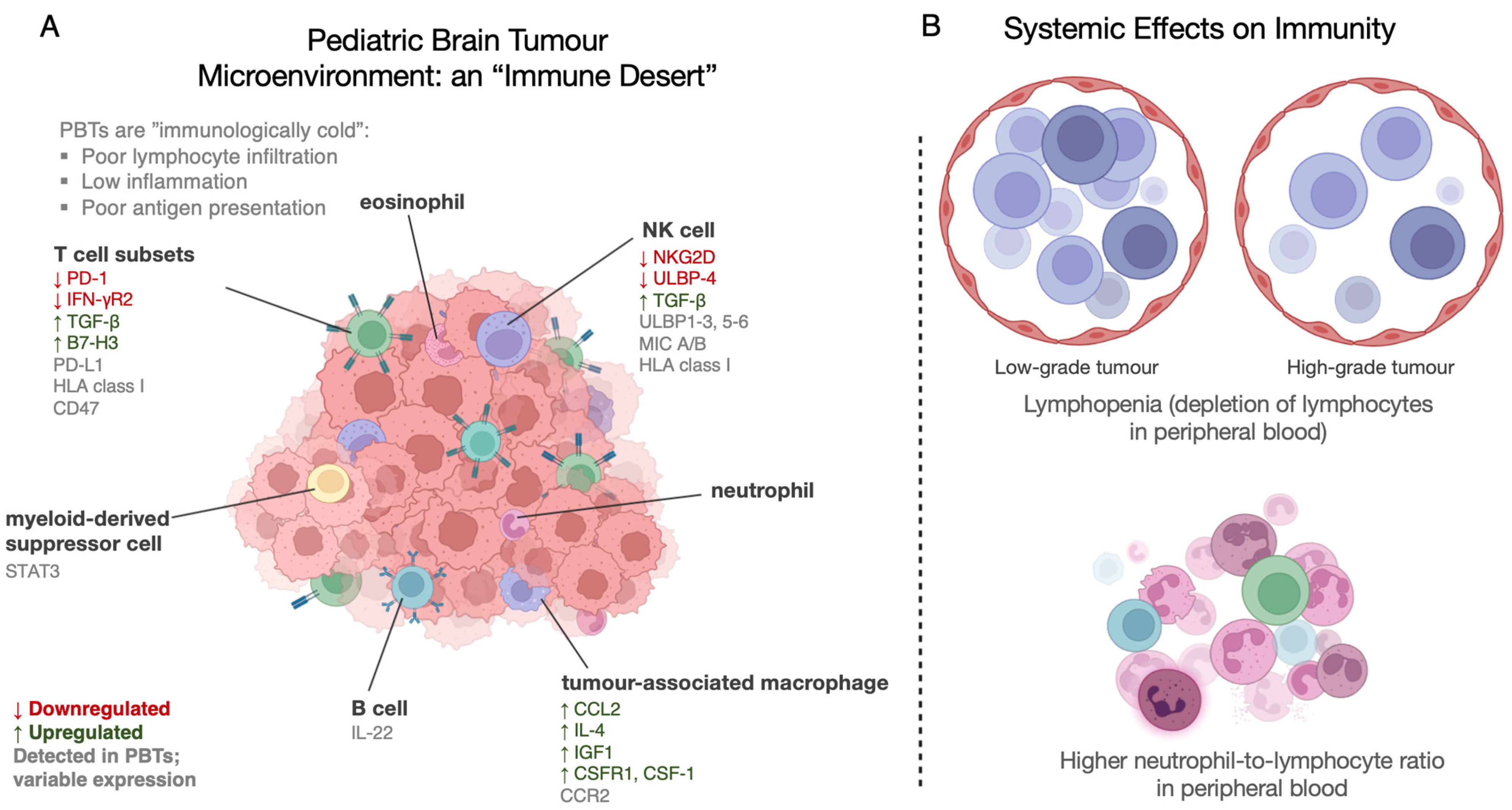{kind=link}
| Gene | Differential Expression | MBL Subtype | Expressed by | Function |
|---|---|---|---|---|
| CD47 | Downregulated | Group 3, Group 4 | CD8+ T cells | Evasion of phagocytosis |
| STAT3 | Upregulated | Not specified | Myeloid-derived suppressor cells | Suppression of pro-inflammatory signalling |
| TGF-β | Upregulated | SHH | Tumour cell | Promotes TReg cell infiltration, abrogation of NKG2D in NK cells |
| ULBP-4 | Downregulated | Not specified | Tumour cell | NKG2D activating ligand |
| IGF1 | Upregulated | SHH | Tumour-associated microglia | Pro-tumourigenic signalling |
| ERBB4 | Upregulated | Group 4 | Tumour-associated microglia | Pro-tumourigenic signalling |
| HLA class I | Upregulated | Not specified | Tumour cell | Antigen recognition by CD8+ T cells, blockade of NKG2DL-mediated NK cell cytotoxicity |
| PD-L1 | Upregulated | SHH | Tumour cell | Immune checkpoint protein; inhibition of T cell activation |
| IFN-γR2 | Downregulated | Not specified | Tumour cell | Interferon signalling; increased apoptosis and HLA class I expression |
| CSFR1 | Upregulated | SHH | Tumour-associated macrophages | Macrophage recruitment and M1 macrophage polarization |
| CCR2 | Upregulated | SHH | Tumour-associated monocytes and macrophages | CCL2-mediated chemotaxis |
| IL-22 | Upregulated | Group 4 | T cells and macrophages | B cell activation |
| B7-H3 | Upregulated | All subgroups | Tumour cell | Immune checkpoint protein; inhibition of T cell activation |
| Gene | Differential Expression | pHGG Type | Expressed by | Function |
|---|---|---|---|---|
| ULBP-2/4/5/6 | Downregulated | Not specified | Tumour cell | NKG2D activating ligand |
| TGF-β | Upregulated | Non-DMG | Tumour cell | Promotes TReg cell infiltration, abrogation of NKG2D in NK cells |
| CCL2 | Downregulated | Non-DMG, DMG | Tumour cell | CCR2-mediated chemotaxis |
| IL-1β | Downregulated | DMG and non-DMG | Tumour-associated macrophages | Pro-inflammatory cytokine signalling |
| PD-L1 | Upregulated | Non-DMG | Tumour cell | Immune checkpoint protein; inhibition of T cell activation |
| HLA class I | Upregulated | DMG | Tumour cell | Antigen recognition by CD8+ T cells, blockade of NKG2DL-mediated NK cell cytotoxicity |
| B7-H3 | Upregulated | DMG and non-DMG | Tumour cell | Immune checkpoint protein; inhibition of T cell activation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, B.; Delaidelli, A.; Vogel, H.; Sorensen, P.H. Pediatric Brain Tumours: Lessons from the Immune Microenvironment. Curr. Oncol. 2023, 30, 5024-5046. https://doi.org/10.3390/curroncol30050379
Yao B, Delaidelli A, Vogel H, Sorensen PH. Pediatric Brain Tumours: Lessons from the Immune Microenvironment. Current Oncology. 2023; 30(5):5024-5046. https://doi.org/10.3390/curroncol30050379
Chicago/Turabian StyleYao, Betty, Alberto Delaidelli, Hannes Vogel, and Poul H. Sorensen. 2023. "Pediatric Brain Tumours: Lessons from the Immune Microenvironment" Current Oncology 30, no. 5: 5024-5046. https://doi.org/10.3390/curroncol30050379
APA StyleYao, B., Delaidelli, A., Vogel, H., & Sorensen, P. H. (2023). Pediatric Brain Tumours: Lessons from the Immune Microenvironment. Current Oncology, 30(5), 5024-5046. https://doi.org/10.3390/curroncol30050379