Management and Prevention of Cellular-Therapy-Related Toxicity: Early and Late Complications


Abstract
1. Introduction
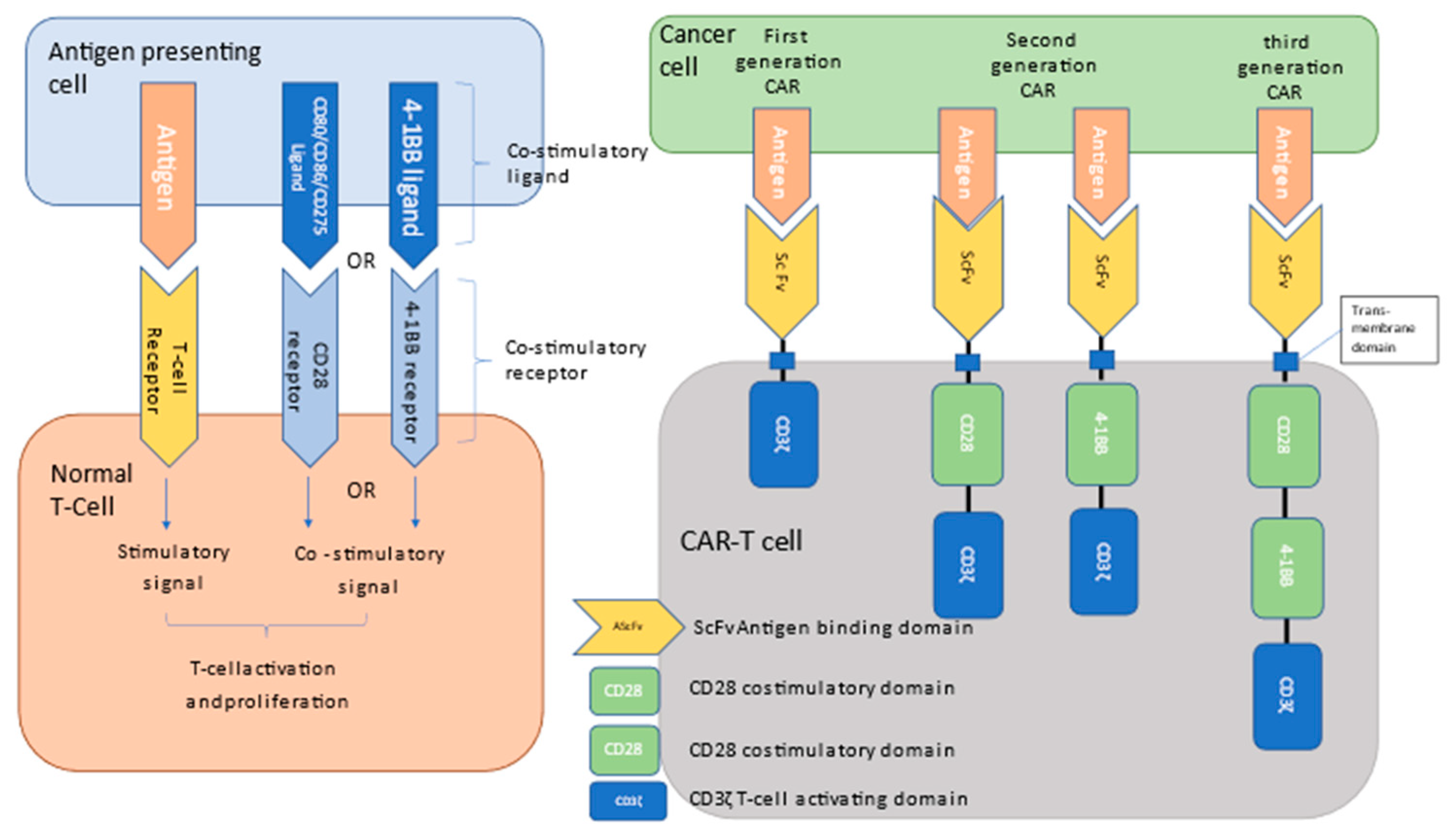
2. Chimeric Antigen Receptor (CAR) Products
3. Chimeric Antigen Receptor-Mediated Toxicities
3.1. Cytokine Release Syndrome (CRS)
3.2. Hemophagocytic Lymphohistiocytosis (HLH)
4. Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
5. Toxicities and Overall Outcomes—Real World Data Explored
6. On Target off Tumor Effects
7. Late Complications
7.1. Hypogammaglobulinemia and Cytopenia
7.2. Late Infections-Prevention and Prophylaxis
7.3. Early and Late Organ Support
8. Resource Utility in CAR-T Therapy Management
9. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, J.; Wickman, E.; De Renzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, H.; Rurik, J.G.; Epstein, J.A. CAR-based therapies: Opportunities for immuno-medicine beyond cancer. Nat. Metab. 2022, 4, 163–169. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Jacobson, C.A.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. 5-Year Follow-Up Supports Curative Potential of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1). Blood 2023, 141, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.D.; Nastoupil, L.J.; Adkins, S.; Lacchetti, C.; Schneider, B.J.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 3978–3992. [Google Scholar] [CrossRef]
- Hoppe, R.T.; Advani, R.H.; Ai, W.Z.; Ambinder, R.F.; Armand, P.; Bello, C.M.; Benitez, C.M.; Chen, W.; Dabaja, B.; Daly, M.E.; et al. NCCN Guidelines® Insights: Hodgkin Lymphoma, Version 2.2022. J. Natl. Compr. Cancer Netw. 2022, 20, 322–334. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242, Erratum in Nat. Rev. Immunol. 2013, 13, 542. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Locke, F.L.; Ma, L.; Asubonteng, J.; Hu, Z.-H.; Siddiqi, T.; Ahmed, S.; Ghobadi, A.; Miklos, D.B.; Lin, Y.; et al. Real-World Evidence of Axicabtagene Ciloleucel for the Treatment of Large B Cell Lymphoma in the United States. Transplant. Cell. Ther. 2022, 28, 581-e1. [Google Scholar] [CrossRef]
- Kwon, M.; Iacoboni, G.; Reguera, J.L.; Corral, L.L.; Morales, R.H.; Ortiz-Maldonado, V.; Guerreiro, M.; Caballero, A.C.; Domínguez, M.L.G.; Pina, J.M.S.; et al. Axicabtagene ciloleucel compared to tisagenlecleucel for the treatment of aggressive B-cell lymphoma. Haematologica 2022, 108, 110–121. [Google Scholar] [CrossRef]
- Bastos-Oreiro, M.; Gutierrez, A.; Reguera, J.L.; Iacoboni, G.; López-Corral, L.; Terol, M.J.; Ortíz-Maldonado, V.; Sanz, J.; Guerra-Dominguez, L.; Bailen, R.; et al. Best Treatment Option for Patients with Refractory Aggressive B-Cell Lymphoma in the CAR-T Cell Era: Real-World Evidence From GELTAMO/GETH Spanish Groups. Front. Immunol. 2022, 13, 855730. [Google Scholar] [CrossRef]
- Riedell, P.A.; Hwang, W.-T.; Nastoupil, L.J.; Pennisi, M.; McGuirk, J.P.; Maziarz, R.T.; Bachanova, V.; Oluwole, O.O.; Brower, J.; Flores, O.A.; et al. Patterns of Use, Outcomes, and Resource Utilization among Recipients of Commercial Axicabtagene Ciloleucel and Tisagenlecleucel for Relapsed/Refractory Aggressive B Cell Lymphomas. Transplant. Cell. Ther. 2022, 28, 669–676. [Google Scholar] [CrossRef]
- Gauthier, J.; Gazeau, N.; Hirayama, A.V.; Hill, J.A.; Wu, V.; Cearley, A.; Perkins, P.; Kirk, A.; Shadman, M.; Chow, V.A.; et al. Impact of CD19 CAR T-cell product type on outcomes in relapsed or refractory aggressive B-NHL. Blood 2022, 139, 3722–3731. [Google Scholar] [CrossRef]
- Kuhnl, A.; Roddie, C.; Kirkwood, A.A.; Tholouli, E.; Menne, T.; Patel, A.; Besley, C.; Chaganti, S.; Sanderson, R.; O’Reilly, M.; et al. A national service for delivering CD19 CAR-T in large B-cell lymphoma—The UK real-world experience. Br. J. Haematol. 2022, 198, 492–502. [Google Scholar] [CrossRef]
- Bachy, E.; Le Gouill, S.; Di Blasi, R.; Sesques, P.; Manson, G.; Cartron, G.; Beauvais, D.; Roulin, L.; Gros, F.X.; Rubio, M.T.; et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat. Med. 2022, 28, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Bethge, W.A.; Martus, P.; Schmitt, M.; Holtick, U.; Subklewe, M.; von Tresckow, B.; Ayuk, F.; Wagner-Drouet, E.M.; Wulf, G.G.; Marks, R.; et al. GLA/DRST real-world outcome analysis of CAR-T cell therapies for large B-cell lymphoma in Germany. Blood 2022, 140, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.A.; Hunter, B.D.; Redd, R.; Rodig, S.J.; Chen, P.-H.; Wright, K.; Lipschitz, M.; Ritz, J.; Kamihara, Y.; Armand, P.; et al. Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J. Clin. Oncol. 2020, 38, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Iacoboni, G.; Villacampa, G.; Martinez-Cibrian, N.; Bailén, R.; Corral, L.L.; Sanchez, J.M.; Guerreiro, M.; Caballero, A.C.; Mussetti, A.; Sancho, J.; et al. Real-world evidence of tisagenlecleucel for the treatment of relapsed or refractory large B-cell lymphoma. Cancer Med. 2021, 10, 3214–3223. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Jain, M.; Feng, L.; Spiegel, J.Y.; Ghobadi, A.; Lin, Y.; Dahiya, S.; Lunning, M.; Lekakis, L.; Reagan, P.; et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results from the US Lymphoma CAR T Consortium. J. Clin. Oncol. 2020, 38, 3119–3128. [Google Scholar] [CrossRef]
- Pasquini, M.C.; Hu, Z.-H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef]
- Iacoboni, G.; Rejeski, K.; Villacampa, G.; van Doesum, J.A.; Chiappella, A.; Bonifazi, F.; Lopez-Corral, L.; van Aalderen, M.; Kwon, M.; Martínez-Cibrian, N.; et al. Real-world evidence of brexucabtagene autoleucel for the treatment of relapsed or refractory mantle cell lymphoma. Blood Adv. 2022, 6, 3606–3610. [Google Scholar] [CrossRef]
- Wang, Y.; Jain, P.; Locke, F.L.; Maurer, M.J.; Frank, M.J.; Munoz, J.L.; Dahiya, S.; Beitinjaneh, A.M.; Jacobs, M.T.; Mcguirk, J.P.; et al. Brexucabtagene Autoleucel for Relapsed or Refractory Mantle Cell Lymphoma in Standard-of-Care Practice: Results from the US Lymphoma CAR T Consortium. J. Clin. Oncol. 2023, 41, 2594–2606. [Google Scholar] [CrossRef]
- Hansen, D.K.; Sidana, S.; Peres, L.C.; Leitzinger, C.C.; Shune, L.; Shrewsbury, A.; Gonzalez, R.; Sborov, D.W.; Wagner, C.; Dima, D.; et al. Idecabtagene Vicleucel for Relapsed/Refractory Multiple Myeloma: Real-World Experience from the Myeloma CAR T Consortium. J. Clin. Oncol. 2023, 41, 2087–2097. [Google Scholar] [CrossRef]
- Pasquini, M.C.; Locke, F.L.; Herrera, A.F.; Siddiqi, T.; Ghobadi, A.; Komanduri, K.V.; Hu, Z.-H.; Dong, H.; Hematti, P.; Nikiforow, S.; et al. Post-Marketing Use Outcomes of an Anti-CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy, Axicabtagene Ciloleucel (Axi-Cel), for the Treatment of Large B Cell Lymphoma (LBCL) in the United States (US). Blood 2019, 134, 764. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2021, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Wat, J.; Barmettler, S. Hypogammaglobulinemia After Chimeric Antigen Receptor (CAR) T-Cell Therapy: Characteristics, Management, and Future Directions. J. Allergy Clin. Immunol. Pract. 2021, 10, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Giavridis, T.; Van Der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Greenbaum, U.; Strati, P.; Saliba, R.M.; Torres, J.; Rondon, G.; Nieto, Y.; Hosing, C.; Srour, S.A.; Westin, J.; Fayad, L.E.; et al. CRP and ferritin in addition to the EASIX score predict CAR-T–related toxicity. Blood Adv. 2021, 5, 2799–2806. [Google Scholar] [CrossRef]
- Yan, Z.; Zhang, H.; Cao, J.; Zhang, C.; Liu, H.; Huang, H.; Cheng, H.; Qiao, J.; Wang, Y.; Wang, Y.; et al. Characteristics and Risk Factors of Cytokine Release Syndrome in Chimeric Antigen Receptor T Cell Treatment. Front. Immunol. 2021, 12, 611366. [Google Scholar] [CrossRef]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef]
- Thompson, J.A.; Schneider, B.J.; Brahmer, J.; Achufusi, A.; Armand, P.; Berkenstock, M.K.; Bhatia, S.; Budde, L.E.; Chokshi, S.; Davies, M.; et al. Management of Immunotherapy-Related Toxicities, Version 1.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 387–405. [Google Scholar] [CrossRef]
- Maus, M.V.; Alexander, S.; Bishop, M.R.; Brudno, J.N.; Callahan, C.; Davila, M.L.; Diamonte, C.; Dietrich, J.; Fitzgerald, J.C.; Frigault, M.J.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J. Immunother. Cancer 2020, 8, e001511. [Google Scholar] [CrossRef]
- Oluwole, O.O.; Bouabdallah, K.; Muñoz, J.; De Guibert, S.; Vose, J.M.; Bartlett, N.L.; Lin, Y.; Deol, A.; McSweeney, P.A.; Goy, A.H.; et al. Prophylactic corticosteroid use in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br. J. Haematol. 2021, 194, 690–700. [Google Scholar] [CrossRef]
- Strati, P.; Ahmed, S.; Kebriaei, P.; Nastoupil, L.J.; Claussen, C.M.; Watson, G.; Horowitz, S.B.; Brown, A.R.T.; Do, B.; Rodriguez, M.A.; et al. Clinical efficacy of anakinra to mitigate CAR T-cell therapy–associated toxicity in large B-cell lymphoma. Blood Adv. 2020, 4, 3123–3127. [Google Scholar] [CrossRef]
- Diorio, C.; Vatsayan, A.; Talleur, A.C.; Annesley, C.; Jaroscak, J.J.; Shalabi, H.; Ombrello, A.K.; Hudspeth, M.; Maude, S.L.; Gardner, R.A.; et al. Anakinra utilization in refractory pediatric CAR T-cell associated toxicities. Blood Adv. 2022, 6, 3398–3403. [Google Scholar] [CrossRef]
- Grom, A.A.; Horne, A.; De Benedetti, F. Macrophage activation syndrome in the era of biologic therapy. Nat. Rev. Rheumatol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Gadoury-Levesque, V.; Dong, L.; Su, R.; Chen, J.; Zhang, K.; Risma, K.A.; Marsh, R.A.; Sun, M. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. 2020, 4, 2578–2594. [Google Scholar] [CrossRef]
- Machaczka, M.; Vaktnäs, J.; Klimkowska, M.; Hägglund, H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: A retrospective population-based analysis from a single center. Leuk. Lymphoma 2011, 52, 613–619. [Google Scholar] [CrossRef]
- Daver, N.; McClain, K.; Allen, C.E.; Parikh, S.A.; Otrock, Z.; Rojas-Hernandez, C.; Blechacz, B.; Wang, S.; Minkov, M.; Jordan, M.B.; et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer 2017, 123, 3229–3240. [Google Scholar] [CrossRef]
- Tong, H.; Ren, Y.; Liu, H.; Xiao, F.; Mai, W.; Meng, H.; Qian, W.; Huang, J.; Mao, L.; Tong, Y.; et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: Comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk. Lymphoma 2008, 49, 81–87. [Google Scholar] [CrossRef]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2006, 48, 124–131. [Google Scholar] [CrossRef]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef]
- Kim, D.W.; Bukhari, A.; Lutfi, F.; Zafforoni, F.; Merechi, F.; Ali, M.K.M.; Gottlieb, D.; Lee, S.T.; Kocoglu, M.H.; Hardy, N.M.; et al. Low utility of the H-Score and HLH-2004 criteria to identify patients with secondary hemophagocytic lymphohistiocytosis after CAR-T cell therapy for relapsed/refractory diffuse large B-Cell lymphoma. Leuk. Lymphoma 2022, 63, 1339–1347. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2017, 15, 47–62. [Google Scholar] [CrossRef]
- Hines, M.R.; Keenan, C.; Alfaro, G.M.; Cheng, C.; Zhou, Y.; Sharma, A.; Hurley, C.; Nichols, K.E.; Gottschalk, S.; Triplett, B.M.; et al. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br. J. Haematol. 2021, 194, 701–707. [Google Scholar] [CrossRef]
- Sandler, R.D.; Tattersall, R.S.; Schoemans, H.; Greco, R.; Badoglio, M.; Labopin, M.; Alexander, T.; Kirgizov, K.; Rovira, M.; Saif, M.; et al. Diagnosis and Management of Secondary HLH/MAS Following HSCT and CAR-T Cell Therapy in Adults; A Review of the Literature and a Survey of Practice Within EBMT Centres on Behalf of the Autoimmune Diseases Working Party (ADWP) and Transplant Complications Working Party (TCWP). Front. Immunol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Priyadarshini, S.; Harris, A.; Treisman, D.; Cupac, J.N.; Li, N.; Yan, D.; Munker, R. Hemophagocytic lymphohistiocytosis secondary to CAR-T cells: Update from the FDA and Vizient databases. Am. J. Hematol. 2022, 97, E374–E376. [Google Scholar] [CrossRef]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results from a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Lichtenstein, D.A.; Schischlik, F.; Shao, L.; Steinberg, S.M.; Yates, B.; Wang, H.-W.; Wang, Y.; Inglefield, J.; Dulau-Florea, A.; Ceppi, F.; et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood 2021, 138, 2469–2484. [Google Scholar] [CrossRef]
- Vallurupalli, M.; Berliner, N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood 2019, 134, 1783–1786. [Google Scholar] [CrossRef]
- Ishii, K.; Pouzolles, M.; Chien, C.D.; Erwin-Cohen, R.A.; Kohler, M.E.; Qin, H.; Lei, H.; Kuhn, S.; Ombrello, A.K.; Dulau-Florea, A.; et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J. Clin. Investig. 2020, 130, 5425–5443. [Google Scholar] [CrossRef]
- Holtzman, N.G.; Xie, H.; Bentzen, S.; Kesari, V.; Bukhari, A.; El Chaer, F.; Lutfi, F.; Siglin, J.; Hutnick, E.; Gahres, N.; et al. Immune effector cell–associated neurotoxicity syndrome after chimeric antigen receptor T-cell therapy for lymphoma: Predictive biomarkers and clinical outcomes. Neuro-Oncology 2020, 23, 112–121. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Butt, O.H.; Zhou, A.Y.; Ances, B.M.; DiPersio, J.F.; Ghobadi, A. A systematic framework for predictive biomarkers in immune effector cell-associated neurotoxicity syndrome. Front. Neurol. 2023, 14, 1110647. [Google Scholar] [CrossRef]
- Möhn, N.; Bonda, V.; Grote-Levi, L.; Panagiota, V.; Fröhlich, T.; Schultze-Florey, C.; Wattjes, M.P.; Beutel, G.; Eder, M.; David, S.; et al. Neurological management and work-up of neurotoxicity associated with CAR T cell therapy. Neurol. Res. Pract. 2022, 4, 1. [Google Scholar] [CrossRef]
- Gofshteyn, J.S.; Shaw, P.A.; Teachey, D.; Grupp, S.A.; Maude, S.; Banwell, B.; Chen, F.; Lacey, S.F.; Melenhorst, J.J.; Edmonson, M.J.; et al. Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann. Neurol. 2018, 84, 537–546. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Gust, J.; Perna, F. How I treat unique and difficult to manage cases of CAR T-cell therapy associated neurotoxicity. Blood 2023. [Google Scholar] [CrossRef]
- Gust, J.; Ishak, G. Chimeric Antigen Receptor T-Cell Neurotoxicity Neuroimaging: More Than Meets the Eye. Am. J. Neuroradiol. 2019, 40, E50–E51. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Finney, O.C.; Li, D.; Brakke, H.M.; Hicks, R.M.; Futrell, R.B.; Gamble, D.N.; Bs, S.D.R.; Khalatbari, H.; Ishak, G.E.; et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann. Neurol. 2019, 86, 42–54. [Google Scholar] [CrossRef]
- Strati, P.; Nastoupil, L.J.; Westin, J.; Fayad, L.E.; Ahmed, S.; Fowler, N.H.; Hagemeister, F.B.; Lee, H.J.; Iyer, S.P.; Nair, R.; et al. Clinical and radiologic correlates of neurotoxicity after axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020, 4, 3943–3951. [Google Scholar] [CrossRef]
- Gutierrez, C.; Brown, A.R.T.P.; May, H.P.; Beitinjaneh, A.; Stephens, R.S.; Rajendram, P.; Nates, J.L.; Pastores, S.M.; Dharshan, A.; de Moraes, A.G.; et al. Critically Ill Patients Treated for Chimeric Antigen Receptor-Related Toxicity: A Multicenter Study*. Crit. Care Med. 2021, 50, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Saw, J.-L.; Sidiqi, M.H.; Ruff, M.; Hocker, S.; Alkhateeb, H.; Ansell, S.M.; Bennani, N.N.; Dingli, D.; Hayman, S.R.; Johnston, P.B.; et al. Acute seizures and status epilepticus in immune effector cell associated neurotoxicity syndrome (ICANS). Blood Cancer J. 2022, 12, 62. [Google Scholar] [CrossRef]
- Wehrli, M.; Gallagher, K.; Chen, Y.-B.; Leick, M.B.; McAfee, S.L.; El-Jawahri, A.R.; DeFilipp, Z.; Horick, N.; O’Donnell, P.; Spitzer, T.; et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J. Immunother. Cancer 2022, 10, e003847. [Google Scholar] [CrossRef]
- Shah, N.N.; Johnson, B.D.; Fenske, T.S.; Raj, R.V.; Hari, P. Intrathecal chemotherapy for management of steroid-refractory CAR T-cell–associated neurotoxicity syndrome. Blood Adv. 2020, 4, 2119–2122. [Google Scholar] [CrossRef]
- Asawa, P.; Vusqa, U.; Khan, C.; Samhouri, Y.; Fazal, S. Intrathecal Chemotherapy as a Potential Treatment for Steroid-refractory Immune Effector Cell-associated Neurotoxicity Syndrome. Anticancer. Res. 2022, 42, 3853–3856. [Google Scholar] [CrossRef]
- Amini, L.; Silbert, S.K.; Maude, S.L.; Nastoupil, L.J.; Ramos, C.A.; Brentjens, R.J.; Sauter, C.S.; Shah, N.N.; Abou-El-Enein, M. Preparing for CAR T cell therapy: Patient selection, bridging therapies and lymphodepletion. Nat. Rev. Clin. Oncol. 2022, 19, 342–355. [Google Scholar] [CrossRef]
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA -1, and TRANSCEND trials. Am. J. Hematol. 2021, 96, 1295–1312. [Google Scholar] [CrossRef]
- Schuster, S.J.; Zhang, J.; Yang, H.; Agarwal, A.; Tang, W.; Martinez-Prieto, M.; Bollu, V.; Kuzan, D.; Maziarz, R.T.; Kersten, M.J. Comparative efficacy of tisagenlecleucel and lisocabtagene maraleucel among adults with relapsed/refractory large B-cell lymphomas: An indirect treatment comparison. Leuk. Lymphoma 2022, 63, 845–854. [Google Scholar] [CrossRef]
- Cartron, G.; Fox, C.P.; Liu, F.F.; Kostic, A.; Hasskarl, J.; Li, D.; Bonner, A.; Zhang, Y.; Maloney, D.G.; Kuruvilla, J. Matching-adjusted indirect treatment comparison of chimeric antigen receptor T-cell therapies for third-line or later treatment of relapsed or refractory large B-cell lymphoma: Lisocabtagene maraleucel versus tisagenlecleucel. Exp. Hematol. Oncol. 2022, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; Kuruvilla, J.; Liu, F.F.; Kostic, A.; Kim, Y.; Bonner, A.; Zhang, Y.; Fox, C.P.; Cartron, G. Matching-adjusted indirect treatment comparison of liso-cel versus axi-cel in relapsed or refractory large B cell lymphoma. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Oluwole, O.O.; Chen, J.M.; Chan, K.; Patel, A.R.; Jansen, J.P.; Keeping, S.; Zheng, Y.; Snider, J.T.; Locke, F.L. Matching-adjusted indirect comparison of axi-cel and liso-cel in relapsed or refractory large B-cell lymphoma. Leuk. Lymphoma 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maziarz, R.T.; Gauthier, J. Clash of the titans: Axi-cel versus tisa-cel for advanced-stage DLBCL. Nat. Rev. Clin. Oncol. 2022, 20, 5–6. [Google Scholar] [CrossRef]
- Strati, P. CAR T-cell therapy: Which product for which patient? Blood 2022, 139, 3673–3674. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Van Oekelen, O.; Aleman, A.; Upadhyaya, B.; Schnakenberg, S.; Madduri, D.; Gavane, S.; Teruya-Feldstein, J.; Crary, J.F.; Fowkes, M.E.; Stacy, C.B.; et al. Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat. Med. 2021, 27, 2099–2103. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Cordeiro, A.; Bezerra, E.D.; Hirayama, A.V.; Hill, J.A.; Wu, Q.V.; Voutsinas, J.; Sorror, M.L.; Turtle, C.J.; Maloney, D.G.; Bar, M. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol. Blood Marrow Transplant. 2019, 26, 26–33. [Google Scholar] [CrossRef]
- Cappell, K.M.; Sherry, R.M.; Yang, J.C.; Goff, S.L.; Vanasse, D.A.; McIntyre, L.; Rosenberg, S.A.; Kochenderfer, J.N. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2020, 38, 3805–3815. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Hill, B.T.; Majeed, A.; Majhail, N.S. Late Effects after Chimeric Antigen Receptor T Cell Therapy for Lymphoid Malignancies. Transplant. Cell. Ther. 2020, 27, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Wudhikarn, K.; Perales, M.-A. Infectious complications, immune reconstitution, and infection prophylaxis after CD19 chimeric antigen receptor T-cell therapy. Bone Marrow Transplant. 2022, 57, 1477–1488. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Hill, J.; Li, D.; Hay, K.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef]
- Otani, I.M.; Lehman, H.K.; Jongco, A.M.; Tsao, L.R.; Azar, A.E.; Tarrant, T.K.; Engel, E.; Walter, J.E.; Truong, T.Q.; Khan, D.A.; et al. Practical guidance for the diagnosis and management of secondary hypogammaglobulinemia: A Work Group Report of the AAAAI Primary Immunodeficiency and Altered Immune Response Committees. J. Allergy Clin. Immunol. 2022, 149, 1525–1560. [Google Scholar] [CrossRef]
- Los-Arcos, I.; Iacoboni, G.; Aguilar-Guisado, M.; Alsina-Manrique, L.; de Heredia, C.D.; Fortuny-Guasch, C.; García-Cadenas, I.; García-Vidal, C.; González-Vicent, M.; Hernani, R.; et al. Recommendations for screening, monitoring, prevention, and prophylaxis of infections in adult and pediatric patients receiving CAR T-cell therapy: A position paper. Infection 2020, 49, 215–231. [Google Scholar] [CrossRef]
- Jain, T.; Knezevic, A.; Pennisi, M.; Chen, Y.; Ruiz, J.D.; Purdon, T.J.; Devlin, S.M.; Smith, M.; Shah, G.L.; Halton, E.; et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020, 4, 3776–3787. [Google Scholar] [CrossRef]
- Fried, S.; Avigdor, A.; Bielorai, B.; Meir, A.; Besser, M.J.; Schachter, J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019, 54, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.M.; Zucchetti, E.; Bachmeier, C.A.; Krivenko, G.S.; Larson, V.; Ninh, D.; Grillo, G.; Cao, B.; Kim, J.; Chavez, J.C.; et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica 2020, 106, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Juluri, K.R.; Wu, Q.V.; Voutsinas, J.M.; Hou, J.; Hirayama, A.V.; Mullane, E.; Miles, N.; Maloney, D.G.; Turtle, C.J.; Bar, M.; et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv. 2022, 6, 2055–2068. [Google Scholar] [CrossRef]
- Frigault, M.J.; Nikiforow, S.; Mansour, M.K.; Hu, Z.-H.; Horowitz, M.M.; Riches, M.L.; Hematti, P.; Turtle, C.J.; Zhang, M.-J.; Perales, M.-A.; et al. Tocilizumab not associated with increased infection risk after CAR T-cell therapy: Implications for COVID-19? Blood 2020, 136, 137–139. [Google Scholar] [CrossRef]
- Baird, J.H.; Epstein, D.J.; Tamaresis, J.S.; Ehlinger, Z.; Spiegel, J.Y.; Craig, J.; Claire, G.K.; Frank, M.J.; Muffly, L.; Shiraz, P.; et al. Immune reconstitution and infectious complications following axicabtagene ciloleucel therapy for large B-cell lymphoma. Blood Adv. 2021, 5, 143–155. [Google Scholar] [CrossRef]
- Hingorani, S. Renal Complications of Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2016, 374, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Parikh, C.R.; Inrig, J.K.; Kambay, M.; Patel, U.D. Chronic Kidney Disease After Hematopoietic Cell Transplantation: A Systematic Review. Am. J. Transplant. 2008, 8, 2378–2390. [Google Scholar] [CrossRef]
- Gupta, S.; Seethapathy, H.; Strohbehn, I.A.; Frigault, M.J.; O’donnell, E.K.; Jacobson, C.A.; Motwani, S.S.; Parikh, S.M.; Curhan, G.C.; Reynolds, K.L.; et al. Acute Kidney Injury and Electrolyte Abnormalities After Chimeric Antigen Receptor T-Cell (CAR-T) Therapy for Diffuse Large B-Cell Lymphoma. Am. J. Kidney Dis. 2020, 76, 63–71. [Google Scholar] [CrossRef]
- Gutgarts, V.; Jain, T.; Zheng, J.; Maloy, M.A.; Ruiz, J.D.; Pennisi, M.; Jaimes, E.A.; Perales, M.-A.; Sathick, J. Acute Kidney Injury after CAR-T Cell Therapy: Low Incidence and Rapid Recovery. Biol. Blood Marrow Transplant. 2020, 26, 1071–1076. [Google Scholar] [CrossRef]
- Ansari, R.; Caimi, P.; Lee, H.J.; Chen, Z.; Rashidi, A. Renal outcomes after chimeric antigen receptor T-cell therapy: A single-center perspective. Nephrol. Dial. Transplant. 2022. [Google Scholar] [CrossRef]
- Hanna, P.; Strohbehn, I.; Moreno, D.; Harden, D.; Seethapathy, H.; Gupta, S.; Lee, M.; Ouyang, T.; Frigault, M.; Sise, M.E. Comparison of short- and long-term adverse kidney outcomes after chimeric antigen receptor T cell therapy and autologous hematopoietic stem cell transplant for diffuse large B cell lymphoma. Bone Marrow Transplant. 2022, 57, 1623–1625. [Google Scholar] [CrossRef]
- Alvi, R.M.; Frigault, M.J.; Fradley, M.G.; Jain, M.; Mahmood, S.S.; Awadalla, M.; Lee, D.H.; Zlotoff, D.A.; Zhang, L.; Drobni, Z.D.; et al. Cardiovascular Events Among Adults Treated With Chimeric Antigen Receptor T-Cells (CAR-T). J. Am. Coll. Cardiol. 2019, 74, 3099–3108. [Google Scholar] [CrossRef]
- Lancellotti, P.; Moonen, M.; Galderisi, M. Chimeric Antigen Receptor T-Cells and Cardiovascular Toxicity. J. Am. Coll. Cardiol. 2019, 74, 3109–3111. [Google Scholar] [CrossRef]
- Ruark, J.; Mullane, E.; Cleary, N.; Cordeiro, A.; Bezerra, E.D.; Wu, V.; Voutsinas, J.; Shaw, B.E.; Flynn, K.E.; Lee, S.J.; et al. Patient-Reported Neuropsychiatric Outcomes of Long-Term Survivors after Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. 2019, 26, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, É.; Castro, P.; Maamar, A.; Metaxa, V.; de Moraes, A.G.; Voigt, L.; Wallet, F.; Klouche, K.; Picard, M.; Moreau, A.-S.; et al. Outcomes in patients treated with chimeric antigen receptor T-cell therapy who were admitted to intensive care (CARTTAS): An international, multicentre, observational cohort study. Lancet Haematol. 2021, 8, e355–e364. [Google Scholar] [CrossRef]
- Yang, H.; Hao, Y.; Chai, X.; Qi, C.Z.; Wu, E.Q. Estimation of total costs in patients with relapsed or refractory diffuse large B-cell lymphoma receiving tisagenlecleucel from a US hospital’s perspective. J. Med. Econ. 2020, 23, 1016–1024. [Google Scholar] [CrossRef]
- Ring, A.; Grob, B.; Aerts, E.; Ritter, K.; Volbracht, J.; Schär, B.; Greiling, M.; Müller, A.M.S. Resource utilization for chimeric antigen receptor T cell therapy versus autologous hematopoietic cell transplantation in patients with B cell lymphoma. Ann. Hematol. 2022, 101, 1755–1767. [Google Scholar] [CrossRef]
- Brown, A.R.T.; Jindani, I.; Melancon, J.; Erfe, R.; Westin, J.; Feng, L.; Gutierrez, C. ICU Resource Use in Critically III Patients following Chimeric Antigen Receptor T-Cell Therapy. Am. J. Respir. Crit. Care Med. 2020, 202, 1184–1187. [Google Scholar] [CrossRef]
- Palomba, M.L.; Jun, M.P.; Lymp, J.; Nguyen, A.; McGarvey, N.; Gitlin, M.; Pelletier, C.; Keating, S.J.; Godwin, J. Postinfusion monitoring costs by site of care for patients with relapsed/refractory large B-cell lymphoma receiving third- or later-line treatment with lisocabtagene maraleucel in the TRANSCEND NHL 001 and OUTREACH trials. Leuk. Lymphoma 2021, 62, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.J.; Gu, T.; Jun, M.P.; McBride, A. Health Care Resource Utilization and Total Costs of Care Among Patients with Diffuse Large B Cell Lymphoma Treated with Chimeric Antigen Receptor T Cell Therapy in the United States. Transplant. Cell. Ther. 2022, 28, 404-e1–404-e6. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Siddiqi, T.; Garcia, J.; Dehner, C.; Kim, Y.; Nguyen, A.; Snyder, S.; McGarvey, N.; Gitlin, M.; Pelletier, C.; et al. Cytokine release syndrome and neurological event costs in lisocabtagene maraleucel–treated patients in the TRANSCEND NHL 001 trial. Blood Adv. 2021, 5, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Maziarz, R.T.; Yang, H.; Liu, Q.; Wang, T.; Zhao, J.; Lim, S.; Lee, S.; Dalal, A.; Bollu, V. Real-world healthcare resource utilization and costs associated with tisagenlecleucel and axicabtagene ciloleucel among patients with diffuse large B-cell lymphoma: An analysis of hospital data in the United States. Leuk. Lymphoma 2022, 63, 2052–2062. [Google Scholar] [CrossRef]
- Myers, G.D.; Verneris, M.R.; Goy, A.; Maziarz, R.T. Perspectives on outpatient administration of CAR-T cell therapy in aggressive B-cell lymphoma and acute lymphoblastic leukemia. J. Immunother. Cancer 2021, 9, e002056. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Rossi, J.M.; Chou, J.; Wang, L.; Poddar, S.; Han, G.; Wang, Z.; Kuang, S.-Q.; Chu, F.; Davis, R.E.; et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood 2021, 138, 1081–1085. [Google Scholar] [CrossRef]
- Zurko, J.C.; Fenske, T.S.; Johnson, B.D.; Bucklan, D.; Szabo, A.; Xu, H.; Chaney, K.; Hamadani, M.; Hari, P.; Shah, N.N. Long-term outcomes and predictors of early response, late relapse, and survival for patients treated with bispecific LV20.19 CAR T-cells. Am. J. Hematol. 2022, 97, 1580–1588. [Google Scholar] [CrossRef]
- Watanabe, N.; Mo, F.; McKenna, M.K. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front. Immunol. 2022, 13, 876339. [Google Scholar] [CrossRef]
- Finney, O.C.; Brakke, H.M.; Rawlings-Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Futrell, R.B.; Orentas, R.J.; Li, D.; Gardner, R.A.; et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Investig. 2019, 129, 2123–2132. [Google Scholar] [CrossRef]
- Lee, I.D.W.; Stetler-Stevenson, M.; Yuan, C.M.; Shah, N.N.; Delbrook, R.C.; Yates, R.B.; Zhang, H.; Zhang, L.; Kochenderfer, J.N.; Rosenberg, S.A.; et al. Long-Term Outcomes Following CD19 CAR T Cell Therapy for B-ALL Are Superior in Patients Receiving a Fludarabine/Cyclophosphamide Preparative Regimen and Post-CAR Hematopoietic Stem Cell Transplantation. Blood 2016, 128, 218. [Google Scholar] [CrossRef]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor–positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- A Morgan, R.; Yang, J.C.; Kitano, M.; E Dudley, M.; Laurencot, C.M.; A Rosenberg, S. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.-X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 1–21. [Google Scholar] [CrossRef]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef]


{kind=link}
| CAR-T Product | Antigen Target | Co-Stimulatory Domain | Indication | CR | CRS Any Grade | CRS Grade ≥ 3 | ICANS AnyGrade | ICANS Grade ≥ 3 |
|---|---|---|---|---|---|---|---|---|
| axicabtagene ciloleucel (YESCARTA®) [5] | CD19 | CD28 | DLBCL Mediastinal B-cell lymphomaFollicular lymphoma | 54% | 85% | 13% | 64% | 28% |
| tisagenlecleucel (KYMRIAH®) [13] | CD19 | 4-1BB | DLBCL ALL | 40% | 58% | 22% | 21% | 12% |
| lisocabtagene maraleucel (BREYANZI®) [12] | CD19 | 4-1BB | DLBCL High-grade B-cell lymphoma Mediastinal B-cell lymphomaFollicular lymphoma | 53% | 42% | 2% | 30% | 10% |
| brexucabtagene autoleucel (TECARTUS®) [11] | CD19 | CD28 | Mantle cell lymphoma ALL | 67% | 91% | 15% | 63% | 31% |
| idecabtagene vicleucel (ABECMA®) [14] | BCMA | 4-1BB | Multiple myeloma | 33% | 85% | 5% | 18% | 3% |
| ciltacabtagene autoleucel (CARVYKTI®) [15] | BCMA | 4-1BB | Multiple myeloma | 67% | 51% | 4% | 21% | 9% |
| Study (First Author, Year) | Product | Number of Patients Infused | ORR/CR (%) | CRS (%) Any/Gr 3+ | ICANS (%) Any/Gr 3+ |
|---|---|---|---|---|---|
| Jacobson, 2022 [16] | Axi-cel | 1297 | 73/56 | 83/8 | 55/24 |
| Kwon, 2023 [17] | Axi-cel | 134 | 60/42 | 88/8 | 42/18 |
| Tisa-cel | 127 | 54/34 | 73/6 | 16/5 | |
| Kuhnl, 2022 [21] | Axi-cel | 224 | 77/52 | 93/8 | 37/16 |
| Tisa-cel | 76 | 57/44 | 56/8 | 15/4 | |
| Bachy, 2022 [22] | Axi-cel | 213 | 80/60 | 86/5 | 48/14 |
| Tisa-cel | 419 | 66/42 | 75/9 | 22/3 | |
| Riedell, 2022 [19] | Axi-cel | 156 | 52/44 | 85/9 | 56/39 |
| Tisa-cel | 84 | 41/35 | 39/1 | 11/1 | |
| Bethge, 2022 [23] | Axi-cel | 173 | 74/42 | 81/10 | 44/16 |
| Tisa-cel | 183 | 53/32 | 65/13 | 22/7 | |
| Gauthier, 2022 [20] | Axi-cel | 68 | 75/43 | 87/7 | 62/29 |
| Tisa-cel | 31 | 58/32 | 70/0 | 23/13 | |
| Jacobson, 2020 [24] | Axi-cel | 122 | 70/50 | 93/16 | 70/35 |
| Iacoboni, 2021 [25] | Tisa-cel | 75 | 60/32 | 71/5 | 15/1 |
| Nastoupil, 2020 [26] | Axi-cel | 275 | 82/64 | 91/7 | 69/31 |
| Pasquini, 2019 [31] | Axi-cel | 533 | 74/54 | 83/9 | 53/17 |
| Iacoboni, 2022 [28] | Brexu-cel | 33 | 91/79 | 91/3 | 64/36 |
| Wang, 2023 [29] | Brexu-cel | 168 | 90/82 | 90/8 | 61/32 |
| Hansen, 2023 [30] | Ide-cel | 108 | 64/34 | 82/4 | 15/5 |
| Management | |
| G1:Fever: ≥38 °C without hypotension or hypoxia | Supportive care Empiric antibiotics |
| G2:Fever: ≥38 °C AND Hypotension: not requiring vasopressors And/or Hypoxia: Oxygen ≤6 L/min | Supplemental oxygen IV fluid bolus Tocilizumab 8 mg/kg IV, repeat q8h if no improvement, max 3 doses Dexamethasone 10 mg IV (or equivalent) every 12 h if hypotension persists after 2 fluid boluses and 1–2 doses of tocilizumab Manage per G3 if there is no improvement after 24 h |
| G3:Fever: ≥38 °C AND Hypotension: requiring vasopressors And/or Hypoxia: requiring high-flow oxygen, face mask, nonrebreather mask | Admit patient to ICU Tocilizumab as per G2 if maximum dose is not reached within 24 h period dexamethasone 10 mg IV every 6 h (or equivalent); taper once improved to G1 |
| G4:Fever: ≥38 °C AND Hypotension: requiring multiple vasopressors And/or Hypoxia: requiring positive pressure ventilation (CPAP, BiPAP, mechanical ventilation) | Tocilizumab as per G2 if maximum is not reached within 24 h Consider anakinra, siltuximab, ruxolitinib High-dose methylprednisolone If not improving, consider methylprednisolone 1000 mg IV 2 times a day or alternate/rescue therapy |
| Criteria | HLH 2004 Criteria | H-Score | CAR-T HLH/MAS |
| Fever | Fever > 38.4 | Fever > 38.4 | N/A |
| Organ assessment | Splenomegaly | Organomegaly
| ≥2 organ systems with grade ≥ 3 organ failure * after CRS
|
| Cytopenias (≥2 of 3 lines) | Hemoglobin < 90 g/L Platelets < 100 × 109/L Neutrophils < 1.0 × 109/L | Hemoglobin < 9.2 g/dL Platelets < 110,000/mm3 Leukocytes < 50,000/mm3 | N/A |
| Triglycerides | ≥3.0 mmol/L | >1.5 mmol/L | N/A |
| Fibrinogen | ≤1.5 g/L | ≤2.5 g/L | |
| Ferritin | Ferritin ≥ 500 ng/mL | Ferritin > 2000 ng/mL | Ferritin > 10,000 ng/mL (during CRS) |
| Additional laboratory considerations | Low or absent NK-cell activity (according to local laboratory reference) | Serum GOT ≥ 30 IU/L | |
| Soluble CD25 (i.e., soluble IL-2 receptor) ≥ 2400 U/mL | |||
| Histopathology | Hemophagocytosis | Hemophagocytosis | Hemophagocytosis on histopathology or CD68 immunohistochemistry |
| Grading | No Concurrent CRS | Additional Therapy if Concurrent CRS |
| G1: ICE score: 7–9 Normal consciousness | Supportive care a | Tocilizumab 8 mg/kg IV Dexamethasone 10 mg IV if more than 1 dose of tocilizumab is required for ongoing CRS |
| G2: ICE score: 3–6 And/or Mild somnolence awaking to voice | Dexamethasone 10 mg IV and reassess Repeat every 6–12 h if no improvement. Taper once symptoms improve to G1 | Consider ICU if CRS and ICANS ≥ 2 Tocilizumab per grade 1 |
| G3: ICE score: 0–2 And/or Awakening only to tactile stimulus And/or Any clinical seizure or non-convulsive seizures on EEG that resolve with intervention And/or Focal edema on neuroimaging | Consider transfer to ICU Dexamethasone 10 mg IV every 6–12 h or methylprednisolone equivalent (1 mg/kg IV every 12 h). Consider repeat imaging if persistent ≥ G3 | Tocilizumab as per grade 1 |
| G4: ICE score: 0 And/or Stupor or coma And/or Prolonged seizure (>5 min) or repetitive clinical or electrical seizures without return to baseline And/or Diffuse cerebral edema on neuroimaging, clinical signs of elevated intracranial pressure (decelerate posturing, papilledema, Cushing’s triad) | Admit patient to ICU Consider mechanical ventilation for airway protection High-dose methylprednisolone IV 1000 mg once a day, up to 3 times per day if refractory Consider anakinra and/or additional rescue therapy for persistent or worsening symptoms b Repeat CNS imaging Treat status epilepticus per institutional guidelines | Tocilizumab as per grade 1 |
| ICE score: Orientation: orientation to year, month, city, hospital: 4 points Naming: ability to name 3 objects (e.g., point to clock, pen, button): 3 points Following commands: ability to follow simple commands (e.g., “Show me 2 fingers” or “Close your eyes and stick out your tongue”): 1 point Writing: ability to write a standard sentence (e.g., “Our national bird is the bald eagle”): 1 point Attention: ability to count backward from 100 by 10: 1 point 0 points if patient is unresponsive or unable to perform | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucha, S.R.; Rajendram, P. Management and Prevention of Cellular-Therapy-Related Toxicity: Early and Late Complications. Curr. Oncol. 2023, 30, 5003-5023. https://doi.org/10.3390/curroncol30050378
Mucha SR, Rajendram P. Management and Prevention of Cellular-Therapy-Related Toxicity: Early and Late Complications. Current Oncology. 2023; 30(5):5003-5023. https://doi.org/10.3390/curroncol30050378
Chicago/Turabian StyleMucha, Simon R., and Prabalini Rajendram. 2023. "Management and Prevention of Cellular-Therapy-Related Toxicity: Early and Late Complications" Current Oncology 30, no. 5: 5003-5023. https://doi.org/10.3390/curroncol30050378
APA StyleMucha, S. R., & Rajendram, P. (2023). Management and Prevention of Cellular-Therapy-Related Toxicity: Early and Late Complications. Current Oncology, 30(5), 5003-5023. https://doi.org/10.3390/curroncol30050378