Tumors of the Digestive System: Comprehensive Review of Ancillary Testing and Biomarkers in the Era of Precision Medicine

 , and
, and
Abstract
1. Introduction
2. Immunotherapeutic Agents Applicable to Digestive System Tumors
2.1. Immune Checkpoint Inhibitors (ICIs)
2.2. Adoptive T-Cell Transfer (ACT)
2.3. Vaccine Based Immunotherapy
2.4. Indolamine 2,3 Dioxygenase Inhibitors (IDOs)
2.5. CCL2–CCR2 Signaling Pathway Inhibitors
3. Ancillary Tests Applicable to Digestive System Tumors
3.1. Immunohistochemistry (IHC)
3.2. Fluorescence In Situ Hybridization (FISH)
3.3. Polymerase Chain Reaction (PCR)
3.4. Next-Generation Sequencing (NGS)
4. Interpretation and Reporting of Ancillary Tests for Relevant Biomarkers of Digestive System Tumors
4.1. HER2
4.2. PD-L1
4.3. Microsatellite Instability (MSI)
4.4. Tumor Mutational Burden (TMB)
5. Immunotherapeutic Biomarkers Per Organ System
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, T.; Cheng, Y.; Bai, Y.; Liang, G. Immune cell infiltration as a biomarker for the diagnosis and prognosis of digestive system cancer. Cancer Sci. 2019, 110, 3639–3649. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.M.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1624. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef]
- Magee, M.S.; Abraham, T.S.; Baybutt, T.R.; Flickinger, J.C.; Ridge, N.A.; Marszalowicz, G.P.; Prajapati, P.; Hersperger, A.R.; Waldman, S.A.; Snook, A.E. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol. Res. 2018, 6, 509–516. [Google Scholar] [CrossRef]
- Siegler, E.L.; Zhu, Y.; Wang, P.; Yang, L. Off-the-Shelf CAR-NK Cells for Cancer Immunotherapy. Cell Stem Cell 2018, 23, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, C.E.; Vogt, A.; Gonzalez-Carmona, M.A.; Schmidt-Wolf, I.G.H. Clinical studies applying cytokine-induced killer cells for the treatment of gastrointestinal tumors. J. Immunol. Res. 2014, 2014, 897214. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Khleif, S.N. Therapeutic vaccines for gastrointestinal cancers. Gastroenterol. Hepatol. 2011, 7, 517–564. [Google Scholar]
- Chudasama, R.; Phung, Q.; Hsu, A.; Almhanna, K. Vaccines in gastrointestinal malignancies: From prevention to treatment. Vaccines 2021, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Pan, L.; Shang, N.; Shangguan, J.; Figini, M.; Xing, W.; Wang, B.; Sun, C.; Yang, J.; Zhang, Y.; Hu, S.; et al. Magnetic resonance imaging monitoring therapeutic response to dendritic cell vaccine in murine orthotopic pancreatic cancer models. Am. J. Cancer Res. 2019, 9, 562–573. [Google Scholar]
- Fujiwara, Y.; Kato, S.; Nesline, M.K.; Conroy, J.M.; DePietro, P.; Pabla, S.; Kurzrock, R. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat. Rev. 2022, 110, 102461. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Zhang, R.; Li, T.; Wang, W.; Gan, W.; Lv, S.; Zeng, Z.; Hou, Y.; Yan, Z.; Yang, M. Indoleamine 2, 3-Dioxygenase 1 and CD8 Expression Profiling Revealed an Immunological Subtype of Colon Cancer with a Poor Prognosis. Front. Oncol. 2020, 10, 594098. [Google Scholar] [CrossRef]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef]
- Dagouassat, M.; Suffee, N.; Hlawaty, H.; Haddad, O.; Charni, F.; Laguillier, C.; Vassy, R.; Martin, L.; Schischmanoff, P.; Gattegno, L.; et al. Monocyte chemoattractant protein-1 (MCP-1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int. J. Cancer 2010, 126, 1095–1108. [Google Scholar] [CrossRef]
- Monti, P.; Leone, B.E.; Marchesi, F.; Balzano, G.; Zerbi, A.; Scaltrini, F.; Pasquali, C.; Calori, G.; Pessi, F.; Sperti, C.; et al. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: Regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003, 63, 7451–7461. [Google Scholar] [PubMed]
- Xu, W.; Wei, Q.; Han, M.; Zhou, B.; Wang, H.; Zhang, J.; Wang, Q.; Sun, J.; Feng, L.; Wang, S.; et al. CCL2-SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer. Int. J. Biol. Sci. 2018, 14, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Cheng, X.; Xu, Z.; Chen, C.; Shen, X.; Zhao, J.; Lu, A. A positive feedback loop of β-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis. 2019, 10, 643. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.A.; Belt, B.A.; Figueroa, N.M.; Murthy, A.; Patel, A.; Kim, M.; Lord, E.M.; Linehan, D.C.; Gerber, S. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 2016, 7, 86522–86535. [Google Scholar] [CrossRef] [PubMed]
- Duraiyan, J.; Govindarajan, R.; Kaliyappan, K.; Palanisamy, M. Applications of immunohistochemistry. J. Pharm. Bioallied Sci. 2012, 4, S307–S309. [Google Scholar] [CrossRef]
- Rizk, E.M.; Gartrell, R.D.; Barker, L.W.; Esancy, C.L.; Finkel, G.G.; Bordbar, D.D.; Saenger, Y.M. Prognostic and Predictive Immunohistochemistry-Based Biomarkers in Cancer and Immunotherapy. Hematol. Oncol. Clin. N. Am. 2019, 33, 291–299. [Google Scholar] [CrossRef]
- Cui, C.; Shu, W.; Li, P. Fluorescence In Situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016, 4, 89. [Google Scholar] [CrossRef]
- Kwon, S. Single-molecule fluorescence in situ hybridization: Quantitative imaging of single RNA molecules. BMB Rep. 2013, 46, 65–72. [Google Scholar] [CrossRef]
- Matsuda, K. PCR-Based Detection Methods for Single-Nucleotide Polymorphism or Mutation: Real-Time PCR and Its Substantial Contribution Toward Technological Refinement. Adv. Clin. Chem. 2017, 80, 45–72. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; May, T.; Strong, K.; Bilinovich, S.M.; Bupp, C.; Rajasekaran, S.; Worthey, E.A.; Lazar, J. Genome sequencing in the clinic: The past, present, and future of genomic medicine. Physiol. Genom. 2018, 50, 563–579. [Google Scholar] [CrossRef]
- Lincoln, S.E.; Nussbaum, R.L.; Kurian, A.W.; Nielsen, S.M.; Das, K.; Michalski, S.; Yang, S.; Ngo, N.; Blanco, A.; Esplin, E.D. Yield and Utility of Germline Testing Following Tumor Sequencing in Patients With Cancer. JAMA Netw. Open 2020, 3, e2019452. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Arjun Kumar, E.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Bartley, A.N.; Washington, M.K.; Colasacco, C.; Ventura, C.B.; Ismaila, N.; Benson III, A.B.; Carrato, A.; Gulley, M.L.; Jain, D.; Kakar, S.; et al. HER2 Testing and Clinical Decision Making in Gastroesophageal Adenocarcinoma: Guideline From the College of American Pathologists, American Society for Clinical Pathology, and the American Society of Clinical Oncology. J. Clin. Oncol. 2017, 35, 446–464. [Google Scholar] [CrossRef]
- Rong, L.; Wang, B.; Guo, L.; Liu, X.; Wang, B.; Ying, J.; Xue, L.; Lu, N. HER2 expression and relevant clinicopathological features in esophageal squamous cell carcinoma in a Chinese population. Diagn. Pathol. 2020, 15, 27. [Google Scholar] [CrossRef]
- Creemers, A.; Ebbing, E.A.; Hooijer, G.K.J.; Stap, L.; Jibodh-Mulder, R.A.; Gisbertz, S.S.; van Berge Henegouwen, M.I.; van Montfoort, M.L.; Hulshof, M.C.C.M.; Krishnadath, K.K.; et al. The dynamics of HER2 status in esophageal adenocarcinoma. Oncotarget 2018, 9, 26787–26799. [Google Scholar] [CrossRef]
- Patruni, S.; Fayyaz, F.; Bien, J.; Phillip, T.; King, D.A. Immunotherapy in the Management of Esophagogastric Cancer: A Practical Review. JCO Oncol. Pract 2022. [Google Scholar] [CrossRef]
- Tabernero, J.; Bang, Y.J.; van Cutsem, E.; Fuchs, C.S.; Janjigian, Y.Y.; Bhagia, P.; Li, K.; Adelberg, D.; Qin, S.K. KEYNOTE-859: A Phase III study of pembrolizumab plus chemotherapy in gastric/gastroesophageal junction adenocarcinoma. Future Oncol. 2021, 17, 2847–2855. [Google Scholar] [CrossRef] [PubMed]
- Mastracci, L.; Grillo, F.; Parente, P.; Gullo, I.; Campora, M.; Angerilli, V.; Rossi, C.; Sacramento, M.L.; Pennelli, G.; Vanoli, A.; et al. PD-L1 evaluation in the gastrointestinal tract: From biological rationale to its clinical application. Pathologica 2022, 114, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Rashid, S.; Al-Bozom, I.A. PD-L1 immunostaining: What pathologists need to know. Diagn. Pathol. 2021, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.L. Current Microsatellite Instability Testing in Management of Colorectal Cancer. Clin. Color. Cancer 2021, 20, e12–e20. [Google Scholar] [CrossRef]
- Quaas, A.; Rehkaemper, J.; Rueschoff, J.; Pamuk, A.; Zander, T.; Hillmer, A.; Siemanowski, J.; Wittig, J.; Buettner, R.; Plum, P.; et al. Occurrence of High Microsatellite-Instability/Mismatch Repair Deficiency in Nearly 2000 Human Adenocarcinomas of the Gastrointestinal Tract, Pancreas, and Bile Ducts: A Study From a Large German Comprehensive Cancer Center. Front. Oncol. 2021, 11, 569475. [Google Scholar] [CrossRef]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: Part I. The utility of immunohistochemistry. J. Mol. Diagn. 2008, 10, 293–300. [Google Scholar] [CrossRef]
- Benhamida, J.K.; Hechtman, J.F.; Nafa, K.; Villafania, L.; Sadowska, J.; Wang, J.; Wong, D.; Zehir, A.; Zhang, L.; Bale, T.; et al. Reliable Clinical MLH1 Promoter Hypermethylation Assessment Using a High-Throughput Genome-Wide Methylation Array Platform. J. Mol. Diagn. 2020, 22, 368–375. [Google Scholar] [CrossRef]
- Chen, W.; Frankel, W.L. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod. Pathol. 2019, 32, 1–15. [Google Scholar] [CrossRef]
- Engel, K.B.; Moore, H.M. Effects of Preanalytical Variables on the Detection of Proteins by Immunohistochemistry in Formalin-Fixed, Paraffin-Embedded Tissue. Arch. Pathol. Lab. Med. 2011, 135, 537–545. [Google Scholar] [CrossRef]
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Z.D. Phenotypes and Pathology of Drug-Induced Liver Disease. Clin. Liver Dis. 2017, 21, 89–101. [Google Scholar] [CrossRef]
- Salem, M.E.; Bodor, J.N.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Korn, W.M.; Shields, A.F.; Worrilow, W.M.; Kim, E.S.; et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int. J. Cancer 2020, 147, 2948–2956. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Ge, X.; Zhang, Y.; Xue, X. The current management and biomarkers of immunotherapy in advanced gastric cancer. Medicine 2022, 101, E29304. [Google Scholar] [CrossRef]
- Takei, S.; Kawazoe, A.; Shitara, K. The New Era of Immunotherapy in Gastric Cancer. Cancers 2022, 14, 1054. [Google Scholar] [CrossRef]
- Ohmura, H.; Yamaguchi, K.; Hanamura, F.; Ito, M.; Makiyama, A.; Uchino, K.; Shimokawa, H.; Tamura, S.; Esaki, T.; Mitsugi, K.; et al. OX40 and LAG3 are associated with better prognosis in advanced gastric cancer patients treated with anti-programmed death-1 antibody. Br. J. Cancer 2020, 122, 1507–1517. [Google Scholar] [CrossRef]
- Bębnowska, D.; Grywalska, E.; Niedźwiedzka-Rystwe, P.; Sosnowska-Pasiarska, B.; Smok-Kalwat, J.; Pasiarski, M.; Gozdz, S.; Rolinski, J.; Polkowski, W. CAR-T cell therapy—An Overview of Targets in Gastric Cancer. J. Clin. Med. 2020, 9, 1894. [Google Scholar] [CrossRef]
- Hwang, I.; Kang, Y.N.; Kim, J.Y.; Do, Y.R.; Song, H.S.; Park, K.U. Prognostic significance of membrane-associated mucins 1 and 4 in gastric adenocarcinoma. Exp. Ther. Med. 2012, 4, 311–316. [Google Scholar] [CrossRef]
- Knödler, M.; Körfer, J.; Kunzmann, V.; Trojan, J.; Daum, S.; Schenk, M.; Kullmann, F.; Schroll, S.; Behringer, D.; Stahl, M.; et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br. J. Cancer 2018, 119, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Faghfuri, E.; Shadbad, M.A.; Faghfouri, A.H.; Soozangar, N. Cellular immunotherapy in gastric cancer: Adoptive cell therapy and dendritic cell-based vaccination. Immunotherapy 2022, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, O.; Manikhas, G.; Lordick, F.; Rusyn, A.; Vynnychenko, I.; Dudov, A.; Bazin, I.; Bondarenko, I.; Melichar, B.; et al. FAST: A randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann. Oncol. 2021, 32, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Weng, J.; Li, S.; Zhu, Z.; Liu, Q.; Zhang, R.; Yang, Y.; Li, X. Exploring immunotherapy in colorectal cancer. J. Hematol. Oncol. 2022, 15, 95. [Google Scholar] [CrossRef]
- Wang, C.; Yan, J.; Yin, P.; Gui, L.; Ji, L.; Ma, B.; Gao, W. β-Catenin inhibition shapes tumor immunity and synergizes with immunotherapy in colorectal cancer. Oncoimmunology 2020, 9, 1809947. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Strickler, J.H.; Bekaii-Saab, T.S.; Yaeger, R. BRAF-Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022, 40, 2706–2715. [Google Scholar] [CrossRef]
- Lal, N.; White, B.S.; Goussous, G.; Pickles, O.; Mason, M.J.; Beggs, A.D.; Taniere, P.; Wilcox, B.E.; Guinney, J.; Middleton, G.W. KRAS mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin. Cancer Res. 2018, 24, 224–233. [Google Scholar] [CrossRef]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gurusamy, D.; et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, Q.; Wang, Y.N.; Jin, Y.; He, M.; Liu, Z.; Xu, R. Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncol. 2019, 5, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; Hazama, S.; Maeda, Y.; Matsui, H.; Iida, M.; Suzuki, N.; Yoshimura, Y.; Ueno, T.; Yoshino, S.; Sakai, K.; et al. Adoptive immunotherapy with MUC1-mRNA transfected dendritic cells and cytotoxic lymphocytes plus gemcitabine for unresectable pancreatic cancer. J. Transl. Med. 2014, 12, 175. [Google Scholar] [CrossRef]
- Le, K.; Wang, J.; Zhang, T.; Guo, Y.; Chang, H.; Wang, S.; Zhu, B. Overexpression of mesothelin in pancreatic ductal adenocarcinoma (PDAC). Int. J. Med. Sci. 2020, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- DeSelm, C.J.; Tano, Z.E.; Varghese, A.M.; Adusumilli, P.S. CAR T-cell therapy for pancreatic cancer. J. Surg. Oncol. 2017, 116, 63–74. [Google Scholar] [CrossRef]
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. BRCA mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943–1958. [Google Scholar] [CrossRef]
- Wu, W.; Liu, Y.; Jin, Y.; Liu, L.; Guo, Y.; Xu, M.; Hao, Q.; Li, D.; Fang, W.; Zhang, A.; et al. Case Report: Effectiveness of Targeted Treatment in a Patient With Pancreatic Cancer Harboring PALB2 Germline Mutation and KRAS Somatic Mutation. Front. Med. 2022, 8, 746637. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Hechtman, J.F. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 2019, 30, 36–40. [Google Scholar] [CrossRef]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef]
- Schram, A.M.; Odintsov, I.; Espinosa-Cotton, M.; Khodos, I.; Sisso, W.J.; Mattar, M.S.; Lui, A.J.W.; Vojnic, M.; Shameem, S.H.; Chauhan, T.; et al. Zenocutuzumab, a HER2xHER3 Bispecific Antibody, Is Effective Therapy for Tumors Driven by NRG1 Gene Rearrangements. Cancer Discov. 2022, 12, 1233–1247. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, X.; Liang, J.; Liu, Y.; Hou, X.; Zhang, M.; Li, Y.; Jiang, X. Immunotherapy for Hepatocellular Carcinoma: Current Status and Future Prospects. Front. Immunol. 2021, 12, 765101. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, M.; Kishida, N.; Itano, O.; Shigenori, E.; Ueno, A.; Kitago, M.; Abe, Y.; Hibi, T.; Yagi, H.; Masugi, Y.; et al. Long-term complete response of advanced hepatocellular carcinoma treated with multidisciplinary therapy including reduced dose of sorafenib: Case report and review of the literature. World J. Surg. Oncol. 2015, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yuan, F.; Qi, F.; Sun, J.; Rao, Q.; Zhao, Z.; Huang, P.; Fang, T.; Yang, B.; Xia, J. Expression and clinical significance of LAG-3, FGL1, PD-L1 and CD8+T cells in hepatocellular carcinoma using multiplex quantitative analysis. J. Transl. Med. 2020, 18, 306. [Google Scholar] [CrossRef]
- He, Y.; Lu, M.; Che, J.; Chu, Q.; Zhang, P.; Chen, Y. Biomarkers and Future Perspectives for Hepatocellular Carcinoma Immunotherapy. Front. Oncol. 2021, 11, 716844. [Google Scholar] [CrossRef]
- Sun, B.; Yang, D.; Dai, H.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of hepatocellular carcinoma by NKG2D-based CAR-T cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef]
- Nakagawa, H.; Mizukoshi, E.; Kobayashi, E.; Tamai, T.; Hamana, H.; Ozawa, T.; Kishi, H.; Kitahara, M.; Yamashita, T.; Arai, K.; et al. Association Between High-Avidity T-Cell Receptors, Induced by α-Fetoprotein−Derived Peptides, and Anti-Tumor Effects in Patients With Hepatocellular Carcinoma. Gastroenterology 2017, 152, 1395–1406. [Google Scholar] [CrossRef]
- Spear, T.T.; Callender, G.G.; Roszkowski, J.J.; Moxley, K.M.; Simms, P.E.; Foley, K.C.; Murray, D.C.; Scurti, G.M.; Li, M.; Thomas, J.T.; et al. TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer Immunol. Immunother. 2016, 65, 293–304. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef]
- Li, Y.; Song, Y.; Liu, S. The New Insight of Treatment in Cholangiocarcinoma. J. Cancer 2022, 13, 450–464. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sakabe, T.; Abe, H.; Tanii, M.; Takahashi, H.; Chiba, A.; Yanagida, E.; Shibamoto, Y.; Ogasawara, M.; Tsujitani, S.; et al. Dendritic Cell-Based Immunotherapy Targeting Synthesized Peptides for Advanced Biliary Tract Cancer. J. Gastrointest. Surg. 2013, 17, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Li, X.; Cao, D. Case Report: Trastuzumab Treatment in Adenosquamous Carcinoma of the Extrahepatic Biliary Tract With Her-2 Amplification. Front. Oncol. 2021, 11, 538328. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Lee, K.; Kim, J.W.; Sun, K.J.; Nam, A.; Bang, J.; Bang, Y.; Oh, D. Enhanced Antitumor Effect of Binimetinib in Combination With Capecitabine for Biliary Tract Cancer Patients With Mutations in the RAS/RAF/MEK/ERK Pathway: Phase Ib Study. Br. J. Cancer 2019, 121, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and Trastuzumab for HER2-Positive, Metastatic Biliary Tract Cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef]
- Song, X.; Hu, Y.; Li, Y.; Shao, R.; Liu, F.; Liu, Y. Overview of current targeted therapy in gallbladder cancer. Signal Transduct. Target. Ther. 2020, 5, 230. [Google Scholar] [CrossRef] [PubMed]
- Makower, D.; Rozenblit, A.; Kaufman, H.; Edelman, M.; Lane, M.E.; Zwiebel, J.; Haynes, H.; Wadler, S. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. 2003, 9, 693–702. [Google Scholar]
- Saúde-Conde, R.; Parisi, A.; Giunta, E.F.; Meyers, M.; Sclafani, F. The emerging role of immunotherapy in the treatment of anal cancer. Curr. Opin. Pharmacol. 2022, 67, 102309. [Google Scholar] [CrossRef]
- Indio, V.; Astolfi, A.; Urbini, M.; Nannini, M.; Pantaleo, M.A. Genetics and treatment of gastrointestinal stromal tumors with immune checkpoint inhibitors: What do we know? Pharmacogenomics 2020, 21, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Arshad, J.; Costa, P.A.; Barreto-Coelho, P.; Valdes, B.N.; Trent, J.C. Immunotherapy Strategies for Gastrointestinal Stromal Tumor. Cancers 2021, 13, 3525. [Google Scholar] [CrossRef]
- Fanciulli, G.; Modica, R.; La Salvia, A.; Campolo, F.; Florio, T.; Mikovic, N.; Plebani, A.; Di Vito, V.; Colao, A.; Faggiano, A.; et al. Immunotherapy of Neuroendocrine Neoplasms: Any Role for the Chimeric Antigen Receptor T Cells? Cancers 2022, 14, 3991. [Google Scholar] [CrossRef]


{kind=link}
| SCORE | STAINING PATTERN (S) | INTERPRETATION |
|---|---|---|
| 0 | No reactivity OR Membranous staining in <10% of tumor cells | Negative—no ISH required |
| 1+ | Faint staining in >10% of tumor cells Reactivity limited to only part of membrane | Negative—no ISH required |
| 2+ | Weak to moderate complete, basolateral or lateral membranous staining in ≥10% of tumor cells | Equivocal—order ISH testing |
| 3+ | Strong complete, basolateral or lateral membranous staining in ≥10% of tumor cells | Positive—no ISH required |
| SCORE | STAINING PATTERN (S) | INTERPRETATION |
|---|---|---|
| 0 | No reactivity OR Weak/partial staining in <10% of tumor cells | Negative—no ISH required |
| 1+ | Weak membranous staining in >10% of tumor cells | Negative—no ISH required |
| 2+ | Weak to moderate complete membranous staining in >10% of tumor cells | Equivocal—order ISH testing |
| 3+ | Strong complete membranous staining in >10% of tumor cells | Positive—no ISH required |
| HER2/CEP17 RATIO | HER2 SIGNALS/CELL | INTERPRETATION |
|---|---|---|
| <2.0 | <4.0 | Negative |
| <2.0 | ≥4.0 to <6.0 | Equivocal |
| <2.0 | ≥6.0 | Positive |
| ≥2.0 | ≥4.0 to <6.0 | |
| ≥2.0 | <4.0 |
| SUBTYPE | CPS/TPS FOR PEMBROLIZUMAB | CPS/TPS FOR NIVOLUMAB |
|---|---|---|
| Esophageal/GEJ adenocarcinoma | CPS ≥ 10 | CPS ≥ 5 |
| Esophageal SCC | CPS ≥ 10 | TPS ≥ 1% |
| Gastric adenocarcinoma | No recommendations | CPS ≥ 5 |
| Preanalytical factors (e.g., type of fixative used and amount of time in fixative) |
| Diffuse, focal staining |
| Lack of internal positive control |
| Cytoplasm-restricted staining |
| Missense mutation with retained protein antigenicity (nuclear staining) |
| MLH1 (obligatory partner) binds another secondary protein such as MSH3 |
| MSH6 staining lost following neoadjuvant chemotherapy |
| BIOMARKER | ANCILLARY TEST (S) | APPLICATION (S) |
|---|---|---|
| HER2 | IHC, FISH | HER2+ esophageal cancers may be managed with HER2 inhibitors such as trastuzumab or pembrolizumab [40] |
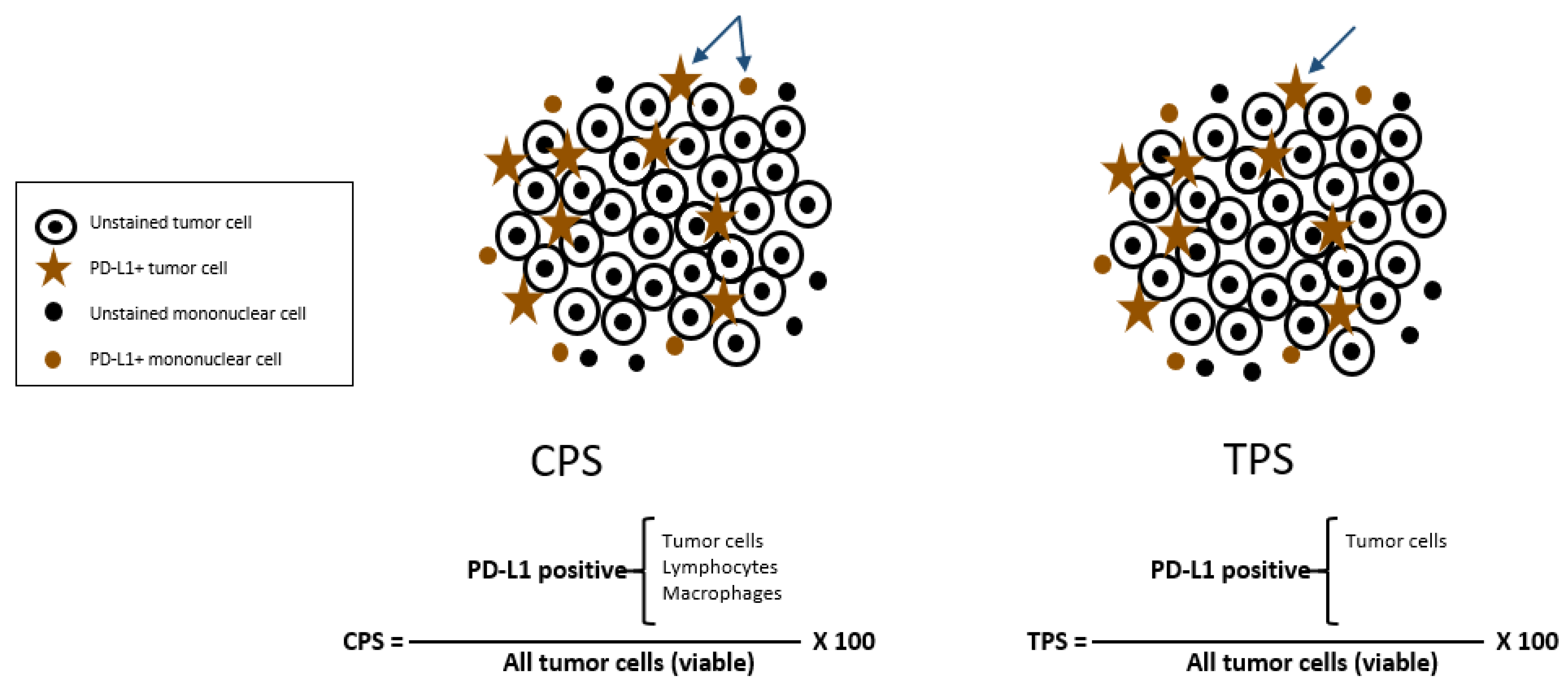
| PD-L1 | IHC | Adenocarcinomas with CPS ≥5 and ≥10 approved for treatment with nivolumab and pembrolizumab, respectively [40] ESCC with TPS ≥ 1% and CPS ≥ 10 approved for treatment with nivolumab and pembrolizumab, respectively [40] |
| MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] |
| BIOMARKER | ANCILLARY TEST (S) | APPLICATION (S) |
|---|---|---|
| HER2 | IHC, FISH | HER2+ gastric cancer may be treated with HER2 inhibitors such as trastuzumab |
| PD-L1 | IHC | CPS ≥ 5: Approved for treatment with HER2 inhibitor trastuzumab [44,56,57] |
| MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] |
| EBV | IHC, PCR | EBV expression associated with increased tumor-infiltrating lymphocytes (TILs) and increased response to ICIs [56,57] |
| LAG3 | IHC | LAG3 expression may indicate treatment with novel LAG3 inhibitor relatlimab [58] |
| MUC1 | IHC | Positive (cytoplasmic) staining may indicate CAR-T therapy [59,60] |
| CEA | IHC | Possible CAR-T target [59] |
| EPCAM | IHC | EpCAM inhibitor catumaxomab may be used to treat peritoneal carcinomatosis of EpCAm+ gastric cancer [61,62] |
| MESOTHELIN | IHC | Positive staining for mesothelin may indicate CAR-T therapy [59] |
| CLDN 18.2 | IHC | Positive staining for CLDN 18.2 may indicate treatment with CL18.2 inhibitor zolbetuximab or CAR-T therapy [59,63] |
| BIOMARKER | ANCILLARY TEST (S) | APPLICATION (S) |
|---|---|---|
| MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H CRC [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] |
| CEA | IHC | CEA-T cell specific antibody (CEA-TCB) [64] |
| B2M | PCR | MSI-H patients with mutations of the gene encoding this protein may be resistant to management by ICIs [65] |
| B-CATENIN | IHC | Nuclear β-catenin inversely proportional to TIL density → poor response to ICIs → may be overcome by ICRT14 β-catenin inhibitor [66] |
| PTEN | IHC, PCR, NGS | Loss of PTEN expression associated with reduced CD8+ TILs → poor response to ICIs [67] |
| JAK1/2 | PCR, NGS | JAK1/2 mutations associated with ICI resistance in TMB-high CRC [68] |
| BRAF | PCR, NGS | BRAF V600E mutations associated with poor response to chemotherapy and increased response to ICIs [69] BRAF K601E mutations associated with better response to chemotherapy [69] |
| KRAS | PCR, NGS | KRAS mutations indicative of primary resistance to immunotherapy [70] |
| CD8+ TILS | IHC | High CD8+ TIL count associated with good response to ICIs [65] |
| CD39+ TILS | IHC | High CD39+ TIL count associated with good response to ICIs [71] |
| POLE/POLD1 | PCR, NGS | POLE and POLD1 mutations associated with MSI, high TMB, high PD-L1 expression, and increased TILs → Good response to ICIs [72] |
| BIOMARKER | ANCILLARY TEST (S) | APPLICATION (S) |
|---|---|---|
| MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] |
| MUC1 | IHC | Potential target of adoptive T-cell and dendritic cell therapy [73] |
| MESOTHELIN | IHC | Potential target of adoptive T-cell therapy and peptide vaccines [74] |
| FAP | IHC | Potential target of CAR-T therapy [75] |
| BRCA1/2 | PCR, NGS | BRCA1/2-mutant pancreatic adenocarcinoma may be targeted by poly ADP ribose polymerase (PARP) inhibitor olaparib [76] |
| PALB2 | PCR, NGS | PALB2-mutant pancreatic adenocarcinoma is a plausible target of olaparib [77] |
| NTRK | PCR, NGS | NTRK-mutant pancreatic adenocarcinoma may be targeted by NTRK inhibitors such as larotrectinib [78] |
| KRAS | PCR, NGS | KRAS-mutant pancreatic adenocarcinoma may be associated with poor response to ICIs [79] |
| NRG1 | PCR, NGS | NRG1-mutant pancreatic adenocarcinoma may be targeted by NRG1 inhibitor zenocutuzumab [80] |
| BIOMARKER | ANCILLARY TEST (S) | APPLICATION (S) | |
|---|---|---|---|
| HEPATOCELULAR CARCINOMA (HCC) | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| GLY-3 | IHC | Gly-3 may be targeted by CAR T-cell therapy or peptide vaccine [81,82] | |
| TIM3 | IHC | TIM3 indicates poor response to ICIs [83,84] | |
| LAG3 | IHC | LAG3 expression may serve as a target for LAG3 inhibitors [84] | |
| Beta-Catenin | IHC, PCR, NGS | Beta-catenin+ HCC associated with poor response to immunotherapy [85] | |
| TP53 | IHC, PCR, NGS | Associated with poor response to immunotherapy [85] | |
| NKG2DL | IHC | Potential target of CAR-T therapy [86] | |
| AFP | IHC | Target of novel peptide vaccine [87] | |
| hTERT | IHC | Target of novel peptide vaccine [81] | |
| MRP3 | IHC | Target of novel peptide vaccine [81] | |
| HCV | IHC | Potential target of modified TCR-T cell therapy for a subset of HCCs [88] | |
| INTRAHEPATIC CHOLANGIO-CARCINOMA (ICAC) | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| IDH1/2 | PCR, NGS | IDH inhibitors such as ivosidenib may be used to treat IDH-mutant ICAC [89] | |
| FGFR2 | PCR, NGS | FGFR inhibitor pemigatinib may be used to treat FGFR-mutant ICAC [90] | |
| WT1 | IHC | Target of dendritic cell vaccine [91] | |
| MUC1 | IHC | Target of dendritic cell vaccine [91] | |
| EXTRAHEPATIC CHOLANGIO-CARCINOMA (ECAC) | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| HER2 | IHC, FISH | HER2+ ECAC is a potential target of HER2 inhibitors such as trastuzumab [92] | |
| MEK | PCR, NGS | MEK-mutant ECAC may be managed with MEK1/2 Inhibitor binimetinib [93] | |
| GALLBLADDER CARCINOMA | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| HER2 | IHC, FISH | HER2+ ECAC is a potential target of HER2 inhibitors such as trastuzumab [94] | |
| BRCA1/2 | PCR | Strong correlation with MSI-H status and good response to ICIs [95] | |
| TP53 | IHC, PCR | Confers poor prognosis and may be target of intralesional ONYX-015 oncolytic adenovirus [96] |
| BIOMARKER | ANCILLARY TEST (S) | APPLICATION (S) | |
|---|---|---|---|
| ANAL SQUAMOUS CELL CARCINOMA | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| P16 | IHC | Epitopes of HPV16 E6 and E7 proteins are potential targets of novel vaccines and adoptive T-cell therapy [97] | |
| GASTROINTESTINAL STROMAL TUMOR (GIST) | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| PDGFRA | PCR, NGS | PDGFRA D842V mutation confers increased response to ICIs [98] | |
| cKIT/CD117 | IHC, PCR, NGS | cKIT inhibitor imatinib exhibits immunotherapeutic effects via inhibition of IDO [99] | |
| NEUROENDOCRINE TUMOR (NET) | MSI | IHC, PCR, NGS | Pembrolizumab approved for management of MSI-H solid tumors [55] |
| TMB | NGS | Pembrolizumab approved for all solid tumors with TMB ≥ 10 Mut/Mb [52] | |
| CDH17 | IHC | Possible target of CAR-T therapy [100] | |
| SSTR2/5 | IHC | Possible target of CAR-T Therapy [100] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molnar, A.; Monroe, H.; Basri Aydin, H.; Arslan, M.E.; Lightle, A.; Lee, H.; El Jabbour, T. Tumors of the Digestive System: Comprehensive Review of Ancillary Testing and Biomarkers in the Era of Precision Medicine. Curr. Oncol. 2023, 30, 2388-2404. https://doi.org/10.3390/curroncol30020182
Molnar A, Monroe H, Basri Aydin H, Arslan ME, Lightle A, Lee H, El Jabbour T. Tumors of the Digestive System: Comprehensive Review of Ancillary Testing and Biomarkers in the Era of Precision Medicine. Current Oncology. 2023; 30(2):2388-2404. https://doi.org/10.3390/curroncol30020182
Chicago/Turabian StyleMolnar, Attila, Hunter Monroe, Hasan Basri Aydin, Mustafa Erdem Arslan, Andrea Lightle, Hwajeong Lee, and Tony El Jabbour. 2023. "Tumors of the Digestive System: Comprehensive Review of Ancillary Testing and Biomarkers in the Era of Precision Medicine" Current Oncology 30, no. 2: 2388-2404. https://doi.org/10.3390/curroncol30020182
APA StyleMolnar, A., Monroe, H., Basri Aydin, H., Arslan, M. E., Lightle, A., Lee, H., & El Jabbour, T. (2023). Tumors of the Digestive System: Comprehensive Review of Ancillary Testing and Biomarkers in the Era of Precision Medicine. Current Oncology, 30(2), 2388-2404. https://doi.org/10.3390/curroncol30020182