Potential Molecular Targeted Therapy for Unresectable Hepatocellular Carcinoma



Abstract
1. Introduction
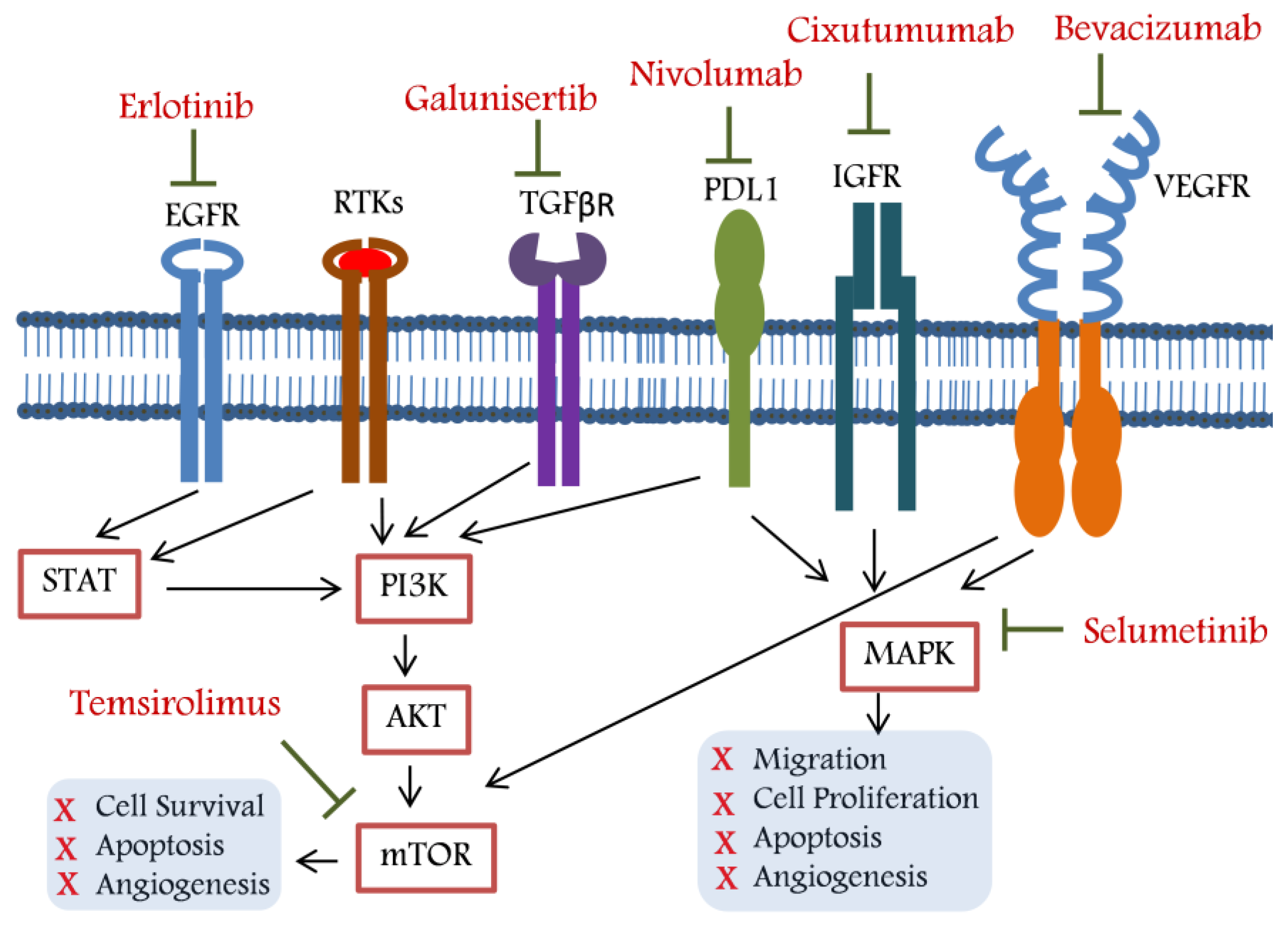
2. Targeted Therapy for Unresectable HCC
2.1. Belinostat: A Histone Deacetylase Inhibitor (HDACi)
2.2. Bevacizumab: A Vascular Endothelial Growth Factor (VEGF) Inhibitor
2.3. Bortezomib: A Proteasome Inhibitor (PI)
2.4. Cixutumumab: An IGF-IR Signalling Inhibitor
2.5. Doxorubicin: A Topoisomerase I and II Inhibitor
2.6. Erlotinib: An Epidermal Growth Factor Receptor (EGFR) Inhibitor
2.7. Galunisertib: A TGF-β Pathway Inhibitor
2.8. Nivolumab: A Programmed Cell Death Protein 1 (PD-1) Inhibitor
2.9. Peretinoin: A Cellular Retinoic Acid-Binding Protein
2.10. Selumetinib: A Mitogen-Activated Protein (MAP) Kinase Inhibitor
2.11. Sofosbuvir: A Nucleotide Analogue
2.12. Temsirolimus: An mTOR Pathway Inhibitor
3. Combination Targeted Therapy—A Way out to HCC Recurrence?
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.M.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: A systematic analysis for the global burden of disease study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Key Statistics About Liver Cancer. Database: American Cancer Society. 2022. Available online: https://www.cancer.org/cancer/liver-cancer/about/what-is-key-statistics.html (accessed on 12 January 2022).
- Sherman, M. Recurrence of hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Toyoda, H.; Kobayashi, M.; Koiwa, Y.; Fujii, H.; Fujita, K.; Maeda, A.; Kaneoka, Y.; Hazama, S.; Nagano, H.; et al. Prediction of early recurrence of hepatocellular carcinoma after resection using digital pathology images assessed by machine learning. Mod. Pathol. 2021, 34, 417–425. [Google Scholar] [CrossRef]
- Campanella, G.; Hanna, M.G.; Geneslaw, L.; Miraflor, A.; Silva, V.W.K.; Busam, K.J.; Brogi, E.; Reuter, V.E.; Klimstra, D.S.; Fuchs, T.J. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 2019, 25, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting histone deacetylases with natural and synthetic agents: An emerging anticancer strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y.; et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; de Bono, J.S. Belinostat: Clinical applications in solid tumors and lymphoma. Expert Opin. Investig. Drugs 2011, 20, 1723–1732. [Google Scholar] [CrossRef]
- Ma, B.B.; Sung, F.; Tao, Q.; Poon, F.F.; Lui, V.W.; Yeo, W.; Chan, S.L.; Chan, A.T. The preclinical activity of the histone deacetylase inhibitor PXD101 (belinostat) in hepatocellular carcinoma cell lines. Investig. New Drugs 2010, 28, 107–114. [Google Scholar] [CrossRef]
- Yeo, W.; Chan, S.L.; Mo, F.K.; Chu, C.M.; Hui, J.W.; Tong, J.H.; Chan, A.W.; Koh, J.; Hui, E.P.; Loong, H.; et al. Phase I/II study of temsirolimus for patients with unresectable Hepatocellular Carcinoma (HCC)- a correlative study to explore potential biokers for response. BMC Cancer 2015, 15, 395. [Google Scholar] [CrossRef]
- Wang, L.-Z.; Ramírez, J.; Yeo, W.; Chan, M.-Y.M.; Thuya, W.-L.; Lau, J.-Y.A.; Wan, S.-C.; Wong, A.L.-A.; Zee, Y.-K.; Lim, R.; et al. Glucuronidation by UGT1A1 is the dominant pathway of the metabolic disposition of belinostat in liver cancer patients. PLoS ONE 2013, 8, e54522. [Google Scholar] [CrossRef]
- Liu, X.; Lu, Y.; Qin, S. Atezolizumab and bevacizumab for hepatocellular carcinoma: Mechanism, pharmacokinetics and future treatment strategies. Futur. Oncol. 2021, 17, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Holen, K.D.; Northfelt, D.W.; Pitot, H.C.; Picus, J.; Flynn, P.J.; Erlichman, C. Phase 2 study of bevacizumab plus erlotinib in patients with advanced hepatocellular cancer. Cancer 2012, 118, 2424–2430. [Google Scholar] [CrossRef]
- Niu, Q.; Tang, Z.Y.; Ma, Z.C.; Qin, L.X.; Zhang, L.H. Serum vascular endothelial growth factor is a potential bioker of metastatic recurrence after curative resection of hepatocellular carcinoma. World J. Gastroenterol. 2000, 6, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.H.; Zhu, X.; Li, X.T.; Yang, R.J. Impact of serum vascular endothelial growth factor on prognosis in patients with unresectable hepatocellular carcinoma after transarterial chemoembolization. Chin. J. Cancer. Res. 2012, 24, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.B.; Han, H.J.; Kim, W.B.; Song, T.J.; Choi, S.Y. VEGF Overexpression Predicts Poor Survival in Hepatocellular Carcinoma. Open Med. (Wars) 2017, 12, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Hu, J.-H.; Cheng, Z.-G.; Liu, Z.-F.; Wang, J.-L.; Jiao, S.-C. Efficacy and safety of bevacizumab for the treatment of advanced hepatocellular carcinoma: A systematic review of phase II trials. PLoS ONE 2012, 7, e49717. [Google Scholar] [CrossRef]
- Atezolizumab Plus Bevacizumab Approved to Treat Liver Cancer. In: NCI Staff. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2020/fda-atezolizumab-bevacizumab-liver-cancer (accessed on 4 June 2020).
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Ganten, T.M.; Koschny, R.; Haas, T.L.; Sykora, J.; Li-Weber, M.; Herzer, K.; Walczak, H. Proteasome inhibition sensitizes hepatocellular carcinoma cells, but not human hepatocytes, to TRAIL. Hepatology 2005, 42, 588–597. [Google Scholar] [CrossRef]
- Dawson, S.P. Hepatocellular carcinoma and the ubiquitin-proteasome system. Biochim. Biophys. Acta 2008, 1782, 775–784. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer. Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Huang, I.-T.; Dhungel, B.; Shrestha, R.; Bridle, K.R.; Crawford, D.H.G.; Jayachandran, A.; Steel, J.C. Spotlight on Bortezomib: Potential in the treatment of hepatocellular carcinoma. Expert Opin. Investig. Drugs 2019, 28, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Feng, Y.; Benson, A.B.; Su, Y.; Horton, L.; Short, S.P.; Kauh, J.S.; Staley, C.; Mulcahy, M.; Powell, M.; et al. Phase II trial of bortezomib plus doxorubicin in hepatocellular carcinoma (E6202): A trial of the Eastern Cooperative Oncology Group. Investig. New Drugs 2014, 32, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef]
- Sedlaczek, N.; Hasilik, A.; Neuhaus, P.; Schuppan, D.; Herbst, H. Focal overexpression of insulin-like growth factor 2 by hepatocytes and cholangiocytes in viral liver cirrhosis. Br. J. Cancer 2003, 88, 733–739. [Google Scholar] [CrossRef]
- Granata, R.; Trovato, L.; Lupia, E.; Sala, G.; Settanni, F.; Camussi, G.; Ghidoni, R.; Ghigo, E. Insulin-like growth factor binding protein-3 induces angiogenesis through IGF-I- and SphK1-dependent mechanisms. J. Thromb. Haemost. 2007, 5, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Burtrum, D.; Zhu, Z.; Lu, D.; Anderson, D.M.; Prewett, M.; Pereira, D.S.; Bassi, R.; Abdullah, R.; Hooper, A.T.; Koo, H.; et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003, 63, 8912–8921. [Google Scholar]
- Tovar, V.; Alsinet, C.; Villanueva, A.; Hoshida, Y.; Chiang, D.Y.; Solé, M.; Thung, S.; Moyano, S.; Toffanin, S.; Mínguez, B.; et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J. Hepatol. 2010, 52, 550–559. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; O’Donnell, R.; Semrad, T.J.; Mack, P.; Blanchard, S.; Bahary, N.; Jiang, Y.; Yen, Y.; Wright, J.; Chen, H.; et al. A phase I trial of escalating doses of cixutumumab (IMC-A12) and sorafenib in the treatment of advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2018, 81, 957–963. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Gansukh, B.; Shia, J.; Kalin, M.; Katz, S.; Abad, L.; et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J. Hepatol. 2014, 60, 319–324. [Google Scholar] [CrossRef]
- Liu, L.X.; Zhu, A.L.; Jiang, H.C.; Wang, X.Q.; Zhou, J.; Wu, M. Expression of DNA-dependent protein kinase and DNA topoisomerase I in human hepatocellular carcinoma and adjacent normal liver tissues. Chin. J. Dig. Dis. 2002, 3, 138–144. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, X.; Zhang, K.; He, X.; Zhang, Q.; Song, H.; Xu, M.; Lu, H.; Ren, R. Type IIA topoisomerase (TOP2A) triggers epithelial-mesenchymal transition and facilitates HCC progression by regulating Snail expression. Bioengineered 2021, 12, 12967–12979. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943, Erratum J. Hepatol. 2012, 56, 1430. [Google Scholar] [CrossRef]
- Lewis, A.L.; Holden, R.R. DC Bead embolic drug-eluting bead: Clinical application in the locoregional treatment of tumours. Expert Opin. Drug Deliv. 2011, 8, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and el drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Dubbelboer, I.R.; Lilienberg, E.; Ahnfelt, E.; Sjögren, E.; Axén, N.; Lennernäs, H. Treatment of intermediate stage hepatocellular carcinoma: A review of intrahepatic doxorubicin drug-delivery systems. Ther. Deliv. 2014, 5, 447–466. [Google Scholar] [CrossRef] [PubMed]
- Gish, R.G.; Porta, C.; Lazar, L.; Ruff, P.; Feld, R.; Croitoru, A.; Feun, L.; Jeziorski, K.; Leighton, J.; Knox, J.; et al. Phase III randomized controlled trial comparing the survival of patients with unresectable hepatocellular carcinoma treated with nolatrexed or doxorubicin. J. Clin. Oncol. 2007, 25, 3069–3075. [Google Scholar] [CrossRef]
- Brabender, J.; Danenberg, K.D.; Metzger, R.; Schneider, P.M.; Park, J.; Salonga, D.; Hölscher, A.H.; Danenberg, P.V. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer Is correlated with survival. Clin. Cancer Res. 2001, 7, 1850–1855. [Google Scholar]
- Buckley, A.F.; Burgart, L.J.; Sahai, V.; Kakar, S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am. J. Clin. Pathol. 2008, 129, 245–251. [Google Scholar] [CrossRef]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Martens, U.M. Small Molecules in Oncology; Springer Berlin Heidelberg Pub: Berlin/Heidelberg, Germany, 2010; Volume 1, p. 238. [Google Scholar]
- Van den Eynde, M.; Baurain, J.F.; Mazzeo, F.; Machiels, J.P. Epidermal growth factor receptor targeted therapies for solid tumours. Acta Clin. Belg. 2011, 66, 10–17. [Google Scholar] [CrossRef]
- Hirte, H.W. Profile of erlotinib and its potential in the treatment of advanced ovarian carcinoma. Onco Targets Ther. 2013, 6, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J. Clin. Oncol. 2005, 23, 6657–6663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zong, Y.; Xu, G.Z.; Xing, K. Erlotinib for advanced hepatocellular carcinoma. A systematic review of phase II/III clinical trials. Saudi. Med. J. 2016, 37, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Cantelli, G.; Crosas-Molist, E.; Georgouli, M.; Sanz-Moreno, V. TGFΒ-induced transcription in cancer. Semin. Cancer Biol. 2017, 42, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; Dijke, P.T.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The rationale for targeting TGF-β in chronic liver diseases. Eur. J. Clin. Investig. 2016, 46, 349–361. [Google Scholar] [CrossRef]
- Rani, B.; Malfettone, A.; Dituri, F.; Soukupova, J.; Lupo, L.; Mancarella, S.; Fabregat, I.; Giannelli, G. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. 2018, 9, 373. [Google Scholar] [CrossRef]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Harding, J.J.; Do, R.K.; Yaqubie, A.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; Lahn, M.; Benhadji, K.A.; Kelley, R.K.; Abou-Alfa, G.K. Phase 1b study of galunisertib and ramucirumab in patients with advanced hepatocellular carcinoma. Cancer Med. 2021, 10, 3059–3067. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.-Z.; Liu, Z.-W.; Zhang, J.-Y.; Yang, Y.-P.; Tien, P.; Wang, F.-S. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, B.; Kirstein, M.M.; Hucke, F.; Finkelmeier, F.; Schulze, K.; Von Felden, J.; Koch, S.; Schwabl, P.; Hinrichs, J.B.; Waneck, F.; et al. Programmed cell death protein-1 (PD-1)-targeted immunotherapy in advanced hepatocellular carcinoma: Efficacy and safety data from an international multicentre real-world cohort. Aliment. Pharmacol. Ther. 2019, 49, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-S.; Li, J.-W.; Li, H.; Jiang, T. Prognostic value of programmed cell death ligand 1 (PD-L1) for hepatocellular carcinoma: A meta-analysis. Biosci. Rep. 2020, 40, BSR20200459. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. REACH Trial Investigators. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Bruix, J.; Tak, W.-Y.; Gasbarrini, A.; Santoro, A.; Colombo, M.; Lim, H.-Y.; Mazzaferro, V.; Wiest, R.; Reig, M.; Wagner, A.; et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur. J. Cancer 2013, 49, 3412–3419. [Google Scholar] [CrossRef]
- Onuma, A.E.; Zhang, H.; Huang, H.; Williams, T.M.; Noonan, A.; Tsung, A. Immune Checkpoint Inhibitors in Hepatocellular Cancer: Current Understanding on Mechanisms of Resistance and Biokers of Response to Treatment. Gene. Expr. 2020, 20, 53–65. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Kang, Y.-K.; Hou, M.-M.; Numata, K.; Yeo, W.; Chopra, A.; Ikeda, M.; et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenibexperienced Asian cohort analysis. J. Hepatol. 2019, 71, 543–552. [Google Scholar] [CrossRef]
- Roderburg, C.; Berres, M.-L.; Wree, A.; Loosen, S.H.; Luedde, T.; Trautwein, C. Excellent response to anti-PD-1 therapy in a patient with hepatocellular carcinoma intolerant to sorafenib. Visc. Med. 2019, 35, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, doubleblind, phase iii trial. J. Clin. Oncol. 2020, 3, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, C.-W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.-C.; Hsu, J.-M.; Dong, Q.; et al. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology 2019, 156, 1849–1861.e13. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Zhang, N.; Ge, Y.; Jia, W. Lenvatinib combined nivolumab injection followed by extended right hepatectomy is a feasible treatment for patients with massive hepatocellular carcinoma: A case report. Onco Targets Ther. 2019, 12, 7355–7359. [Google Scholar] [CrossRef]
- Liu, Z.; Li, X.; He, X.; Xu, Y.; Wang, X. Complete response to the combination of Lenvatinib and Pembrolizumab in an advanced hepatocellular carcinoma patient: A case report. BMC Cancer 2019, 19, 1062. [Google Scholar] [CrossRef]
- Joerger, M.; Guller, U.; Bastian, S.; Driessen, C.; von Moos, R. Prolonged tumor response associated with sequential immune checkpoint inhibitor combination treatment and regorafenib in a patient with advanced pretreated hepatocellular carcinoma. J. Gastrointest. Oncol. 2019, 10, 373–378. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Waidmann, O.; Tro, J. Nivolumab for the treatment of hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2018, 18, 1169–1175. [Google Scholar] [CrossRef]
- Akateh, C.; Black, S.M.; Conteh, L.; Miller, E.D.; Noonan, A.; Elliott, E.; Pawlik, T.M.; Tsung, A.; Cloyd, J.M. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3704–3721. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Nagaoki, Y.; Hyogo, H.; Aikata, H.; Tanaka, M.; Naeshiro, N.; Nakahara, T.; Honda, Y.; Miyaki, D.; Kawaoka, T.; Takaki, S.; et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol. Res. 2012, 42, 368–375. [Google Scholar] [CrossRef] [PubMed]
- TTateishi, R.; Okanoue, T.; Fujiwara, N.; Okita, K.; Kiyosawa, K.; Omata, M.; Kumada, H.; Hayashi, N.; Koike, K. Clinical characteristics, treatment, and prognosis of non-B, non-C hepatocellular carcinoma: A large retrospective multicenter cohort study. J. Gastroenterol. 2015, 50, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Kanematsu, T.; Kawano, T.; Takenaka, K.; Matsumata, T.; Sugimichi, K.; Kuwano, M. Levels of vitamin A and cellular retinol binding protein in human hepatocellular carcinoma and adjacent normal tissue. Nutr. Cancer 1989, 12, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Gräff, A.; Ertelt, V.; Allgaier, H.P.; Koelble, K.; Olschewski, M.; Nitschke, R.; Bochaton-Piallat, M.L.; Gabbiani, G.; Blum, H.E. Cellular retinol-binding protein-1 in hepatocellular carcinoma correlates with beta-catenin, Ki-67 index, and patient survival. Hepatology 2003, 38, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yoon, J.-H.; Yu, S.J.; Chung, G.E.; Jung, E.U.; Kim, H.Y.; Kim, B.H.; Choi, D.H.; Myung, S.J.; Kim, Y.J.; et al. Retinoic acid and its binding protein modulate apoptotic signals in hypoxic hepatocellular carcinoma cells. Cancer Lett. 2010, 295, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shan, W.; Li, T.; Gao, X.; Kong, F.; You, H.; Kong, D.; Qiao, S.; Tang, R. Cellular retinol binding protein-1 inhibits cancer stemness via upregulating WIF1 to suppress Wnt/β-catenin pathway in hepatocellular carcinoma. BMC Cancer 2021, 21, 1224. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, R.; Pandey, A.K. Hepatocellular carcinoma: Causes, mechanism of progression and biomarkers. Curr. Chem. Genom. Transl. Med. 2018, 12, 9–26. [Google Scholar] [CrossRef]
- Miyake, T.; Abe, M.; Furukawa, S.; Tokumoto, Y.; Toshimitsu, K.; Ueda, T.; Yamamoto, S.; Hirooka, M.; Kumagi, T.; Hiasa, Y.; et al. Long-term branched-chain amino acid supplementation improves glucose tolerance in patients with nonalcoholic steatohepatitis-related cirrhosis. Intern. Med. 2012, 51, 2151–2155. [Google Scholar] [CrossRef]
- Ohno, T.; Tanaka, Y.; Sugauchi, F.; Orito, E.; Hasegawa, I.; Nukaya, H.; Kato, A.; Matunaga, S.; Endo, M.; Tanaka, Y.; et al. Suppressive effect of oral administration of branched-chain amino acid granules on oxidative stress and inflammation in HCV-positive patients with liver cirrhosis. Hepatol. Res. 2008, 38, 683–688. [Google Scholar] [CrossRef]
- ASPEN Board of Directors and the Clinical Guidelines Task Force. Guidelines for the use of parenteral and enteral nutrition in adult and pediatric patients. JPEN J. Parenter Enter. Nutr. 2002, 26 (Suppl. 1), 1SA–138SA. [Google Scholar] [CrossRef]
- Plauth, M.; Merli, M.; Kondrup, J.; Weimann, A.; Ferenci, P.; Müller, M. ESPEN guidelines for nutrition in liver disease and transplantation. Clin. Nutr. 1997, 16, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.Y.; Yoo, S.Y.; Heo, J. Peretinoin, an Acyclic Retinoid, for the Secondary Prevention of Hepatocellular Carcinoma. Molecules 2021, 26, 295. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Cox, A.D.; Kloog, Y. Inhibitors of chronically active ras: Potential for treatment of human malignancies. Recent Pat. Anticancer. Drug Discov. 2008, 3, 31–47. [Google Scholar] [CrossRef]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell Biochem. 1998, 182, 31–48. [Google Scholar] [CrossRef]
- Schmidt, C.M.; McKillop, I.H.; Cahill, P.A.; Sitzmann, J.V. Increased MAPK expression and activity in priy human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 1997, 236, 54–58. [Google Scholar] [CrossRef]
- Huynh, H.; Soo, K.C.; Chow, P.K.; Tran, E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol. Cancer Ther. 2007, 6, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.J.; Schmidt, C.M.; Wiesenauer, C.A.; Choi, J.N.; Gage, E.A.; Yip-Schneider, M.T.; Wiebke, E.A.; Wang, Y.; Omer, C.; Sebolt-Leopold, J.S. The effects of a el MEK inhibitor PD184161 on MEK-ERK signaling and growth in human liver cancer. Neoplasia 2006, 8, 1–8. [Google Scholar] [CrossRef]
- Adjei, A.A.; Cohen, R.B.; Franklin, W.; Morris, C.; Wilson, D.; Molina, J.R.; Hanson, L.J.; Gore, L.; Chow, L.; Leong, S.; et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J. Clin. Oncol. 2008, 26, 2139–2146. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Goff, L.W.; Kauh, J.S.; Strosberg, J.R.; Bekaii-Saab, T.S.; Lee, R.M.; Kazi, A.; Moore, D.T.; Learoyd, M.; Lush, R.M.; et al. Phase II study of the mitogen-activated protein kinase 1/2 inhibitor selumetinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2011, 29, 2350–2356. [Google Scholar] [CrossRef]
- Facciorusso, A.; Licinio, R.; Carr, B.I.; Di Leo, A.; Barone, M. MEK 1/2 inhibitors in the treatment of hepatocellular carcinoma. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 993–1003. [Google Scholar] [CrossRef]
- Henry, B. Drug pricing & challenges to hepatitis c treatment access. J. Health Biomed. Law 2018, 14, 265–283. [Google Scholar] [PubMed]
- Irekeola, A.A.; Ear, E.N.S.; Amin, N.A.Z.M.; Mustaffa, N.; Shueb, R.H. Antivirals against HCV infection: The story thus far. J. Infect. Dev. Ctries. 2022, 16, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, W.; Ma, B.; Li, M.; Peppelenbosch, M.P.; Pan, Q. Sofosbuvir directly promotes the clonogenic capability of human hepatocellular carcinoma cells. Clin. Res. Hepatol. Gastroenterol. 2019, 43, e79–e81. [Google Scholar] [CrossRef] [PubMed]
- Atif, M.; Mustaan, M.A.; Falak, S.; Ghaffar, A.; Munir, B. Targeting the effect of sofosbuvir on selective oncogenes expression level of hepatocellular carcinoma Ras/Raf/MEK/ERK pathway in Huh7 cell line. Saudi J. Biol. Sci. 2022, 29, 103332. [Google Scholar] [CrossRef]
- Karam, P.; Powdrill, M.H.; Liu, H.-W.; Vasquez, C.; Mah, W.; Bernatchez, J.; Götte, M.; Cosa, G. Dynamics of hepatitis C virus (HCV) RNA-dependent RNA polymerase NS5B in complex with RNA. J. Biol. Chem. 2014, 289, 14399–14411. [Google Scholar] [CrossRef] [PubMed]
- Muzica, C.M.; Stanciu, C.; Huiban, L.; Singeap, A.M.; Sfarti, C.; Zeia, S.; Cojocariu, C.; Trifan, A. Hepatocellular carcinoma after direct-acting antiviral hepatitis C virus therapy: A debate near the end. World J. Gastroenterol. 2020, 26, 6770–6781. [Google Scholar] [CrossRef]
- Grandhe, S.; Frenette, C.T. Occurrence and Recurrence of Hepatocellular Carcinoma After Successful Direct-Acting Antiviral Therapy for Patients with Chronic Hepatitis C Virus Infection. Gastroenterol. Hepatol. 2017, 13, 421–425. [Google Scholar]
- Li, S.; Liang, Y.; Wu, M.; Wang, X.; Fu, H.; Chen, Y.; Wang, Z. The el mTOR inhibitor CCI-779 (temsirolimus) induces antiproliferative effects through inhibition of mTOR in Bel-7402 liver cancer cells. Cancer Cell Int. 2013, 13, 30. [Google Scholar] [CrossRef]
- Stone, M.J.; Klintmalm, G.B.; Polter, D.; Husberg, B.S.; Mennel, R.G.; Ramsay, M.A.; Flemens, E.R.; Goldstein, R.M. Neoadjuvant chemotherapy and liver transplantation for hepatocellular carcinoma: A pilot study in 20 patients. Gastroenterology 1993, 104, 196–202. [Google Scholar] [CrossRef]
- Treiber, G. mTOR inhibitors for hepatocellular cancer: A forward-moving target. Expert Rev. Anticancer. Ther. 2009, 9, 247–261. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Ojetti, V.; Tortora, A.; Di Maurizio, L.; Purchiaroni, F.; Gasbarrini, A. The metabolic and toxicological considerations for mTOR inhibitors in the treatment of hepatocarcinoma. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1535–1546. [Google Scholar] [CrossRef]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.J.; Qin, R.; Strosberg, J.R.; Tan, B.; Kaubisch, A.; El-Khoueiry, A.B.; Bekaii-Saab, T.S.; Rousey, S.R.; Chen, H.X.; Erlichman, C. A phase II trial of bevacizumab plus temsirolimus in patients with advanced hepatocellular carcinoma. Investig. New Drugs 2015, 33, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Ou, T.M.; Hsu, C.W.; Horng, C.T.; Lee, C.C.; Tsai, Y.Y.; Tsai, C.C.; Liou, Y.S.; Yang, C.C.; Hsueh, C.W.; et al. Current systemic treatment of hepatocellular carcinoma: A review of the literature. World J. Hepatol. 2015, 7, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Hajagos-Tóth, J.; Kormányos, Z.; Falkay, G.; Pál, A.; Gáspár, R. Potentiation of the uterus-relaxing effects of β-adrenergic agonists with nifedipine: Studies on rats and the human myometrium. Acta Obstet. Gynecol. Scand. 2010, 89, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-J.; Baik, I.H.; Ye, S.-K.; Lee, Y.-H. Molecular Targeted Therapy for Hepatocellular Carcinoma: Present Status and Future Directions. Biol. Pharm. Bull. 2015, 38, 986–991. [Google Scholar] [CrossRef]
- Assenat, E.; Pageaux, G.-P.; Thézenas, S.; Peron, J.-M.; Bécouarn, Y.; Seitz, J.-F.; Merle, P.; Blanc, J.-F.; Bouché, O.; Ramdani, M.; et al. Sorafenib alone vs. sorafenib plus GEMOX as 1st-line treatment for advanced HCC: The phase II randomised PRODIGE 10 trial. Br. J. Cancer 2019, 120, 896–902. [Google Scholar] [CrossRef]
- Liu, Y.; Yue, H.; Xu, S.; Wang, F.; Ma, N.; Li, K.; Qiao, L.; Wang, J. First-line gemcitabine and oxaliplatin (GEMOX) plus sorafenib, followed by sorafenib as maintenance therapy, for patients with advanced hepatocellular carcinoma: A preliminary study. Int. J. Clin. Oncol. 2015, 20, 952–959. [Google Scholar] [CrossRef]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.-Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.-H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, E.A.; Margonis, G.A.; Amini, N.; Anastasiou, M.; Angelou, A.; Kim, Y.; Kouraklis, G. Sorafenib as an adjuvant therapy for resectable hepatocellular carcinoma: A single center experience. J. BUON 2016, 21, 1189–1194. [Google Scholar] [PubMed]
- Zhuang, L.; Wen, T.; Xu, M.; Yang, J.; Wang, W.; Wu, H.; Zeng, Y.; Yan, L.; Wei, Y.; Li, B. Sorafenib combined with hepatectomy in patients with intermediate-stage and advanced hepatocellular carcinoma. Arch. Med. Sci. 2017, 13, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, Y.; Cai, X.-B.; Liu, B. Sorafenib after resection improves the outcome of BCLC stage C hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 4034–4040. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Z.; Zhou, Y.; Yang, J.; Hu, K.; Wang, Z. Should we apply sorafenib in hepatocellular carcinoma patients with microvascular invasion after curative hepatectomy? Onco. Targets Ther. 2019, 12, 541–548. [Google Scholar] [CrossRef]
- Zhang, X.P.; Chai, Z.T.; Gao, Y.Z.; Chen, Z.H.; Wang, K.; Shi, J.; Guo, W.X.; Zhou, T.F.; Ding, J.; Cong, W.M.; et al. Postoperative adjuvant sorafenib improves survival outcomes in hepatocellular carcinoma patients with microvascular invasion after R0 liver resection: A propensity score matching analysis. HPB 2019, 21, 1687–1696. [Google Scholar] [CrossRef]
- Kuzuya, T.; Ishigami, M.; Ito, T.; Ishizu, Y.; Honda, T.; Ishikawa, T.; Hirooka, Y.; Fujishiro, M. Clinical characteristics and outcomes of candidates for second-line therapy, including regorafenib and ramucirumab, for advanced hepatocellular carcinoma after sorafenib treatment. Hepatol. Res. 2019, 49, 1054–1065. [Google Scholar] [CrossRef]
- Uchikawa, S.; Kawaoka, T.; Aikata, H.; Kodama, K.; Nishida, Y.; Inagaki, Y.; Hatooka, M.; Morio, K.; Nakahara, T.; Murakami, E.; et al. Clinical outcomes of sorafenib treatment failure for advanced hepatocellular carcinoma and candidates for regorafenib treatment in real-world practice. Hepatol. Res. 2018, 48, 814–820. [Google Scholar] [CrossRef]
- Ogasawara, S.; Chiba, T.; Ooka, Y.; Suzuki, E.; Maeda, T.; Yokoyama, M.; Wakamatsu, T.; Inoue, M.; Saito, T.; Kobayashi, K.; et al. Characteristics of patients with sorafenib-treated advanced hepatocellular carcinoma eligible for second-line treatment. Investig. New Drugs 2018, 36, 332–339. [Google Scholar] [CrossRef]


{kind=link}
| Study Design | Clinical Trila.Gov Identifier | Phase/Type | Primary End Point | Secondary End Point | Study Status |
|---|---|---|---|---|---|
| Galunisertib + Nivolumab | NCT02423343 | I/II | MTD | PK/Cmin/PFS/CR/TTR/OS | Completed |
| Huaier Granule | NCT01770431 | IV | IRMAH | PSP | Completed |
| Sofosbuvir + Ribavirin | NCT01559844 | II | pTVR | VF | Completed |
| Peretinoin | NCT01640808 | III | RFS | DFS/TTR | Completed |
| Bevacizumab | NCT00055692 | II | PFS/CR + PR | - | Completed |
| Sorafenib | NCT00692770 | III | RFS | TTR/OS | Completed |
| Doxorubicin + Bortezomib | NCT00083226 | II | RECIST | PFS/OS | Completed |
| Cixutumumab | NCT00639509 | II | PFS/ORR | OS | Completed |
| Selumetinib | NCT00604721 | I | CR + PR | PFS/OS | Completed |
| Belinostat | NCT00321594 | I/II | DLT/MTD | Completed | |
| Bevacizumab + Erlotinib | NCT00365391 | II | ORR | TTP/PFS/OS | Completed |
| Bevacizumab + Temsirolimus | NCT01010126 | II | CR + PR/T | - | Completed |
| Sorafenibtosylate | NCT01502410 | II | CR + PR | T/PK | Completed |
| Biological: -205/NY-ESO-1 Fusion Protein CDX-1401 ± Sirolimus | NCT01522820 | I | - | - | Completed |
| Sustained released 5-FU and cisplatin | NCT00817895 | Interventional | - | - | Completed |
| Statin therapy and the risk of HCC recurrence | NCT03490461 | Observational | - | - | Completed |
| Ginsenoside Rg3 | NCT01717066 | Interventional | - | - | Completed |
| Lenvatinib | NCT04415567 | Observational | - | - | Completed |
| Direct Acting Antivirals against hepatitis C virus infection | NCT03197155 | Observational | - | - | Completed |
| Rapamycin | NCT02724332 | I | - | - | Completed |
| Ispinesib | NCT00095992 | II | - | - | Completed |
| Licartin | NCT00819650 | II | - | - | Completed |
| Epirubicin and lipiodol | NCT00820053 | II | - | - | Completed |
| Ethiodised oil | NCT00870558 | III | - | - | Completed |
| Bortezomib | NCT00077441 | II | - | - | Completed |
| Gefitinib | NCT00071994 | II | - | - | Completed |
| Interferon-alfa-2b and Ribavirin | NCT00375661 | IV | - | - | Completed |
| Cixutumumab and Sorafenib Tosylate | NCT01008566 | I | - | - | Completed |
| Doxorubicin hydrochloride and Nolatrexed dihydrochloride | NCT00012324 | III | - | - | Completed |
| Erlotinib hydrochloride | NCT00047346 | I | - | - | Completed |
| Oxaliplatin | NCT00052364 | II | - | - | Completed |
| Interferon alpha-2b | NCT00273247 | III | - | - | Completed |
| Oxaliplatin | NCT00091182 | II | - | - | Completed |
| Sorafenib tosylate | NCT00844168 | I | - | - | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Pandey, A.K. Potential Molecular Targeted Therapy for Unresectable Hepatocellular Carcinoma. Curr. Oncol. 2023, 30, 1363-1380. https://doi.org/10.3390/curroncol30020105
Kumar S, Pandey AK. Potential Molecular Targeted Therapy for Unresectable Hepatocellular Carcinoma. Current Oncology. 2023; 30(2):1363-1380. https://doi.org/10.3390/curroncol30020105
Chicago/Turabian StyleKumar, Shashank, and Abhay Kumar Pandey. 2023. "Potential Molecular Targeted Therapy for Unresectable Hepatocellular Carcinoma" Current Oncology 30, no. 2: 1363-1380. https://doi.org/10.3390/curroncol30020105
APA StyleKumar, S., & Pandey, A. K. (2023). Potential Molecular Targeted Therapy for Unresectable Hepatocellular Carcinoma. Current Oncology, 30(2), 1363-1380. https://doi.org/10.3390/curroncol30020105