Proteasome Inhibitors against Glioblastoma—Overview of Molecular Mechanisms of Cytotoxicity, Progress in Clinical Trials, and Perspective for Use in Personalized Medicine


Abstract
1. Introduction
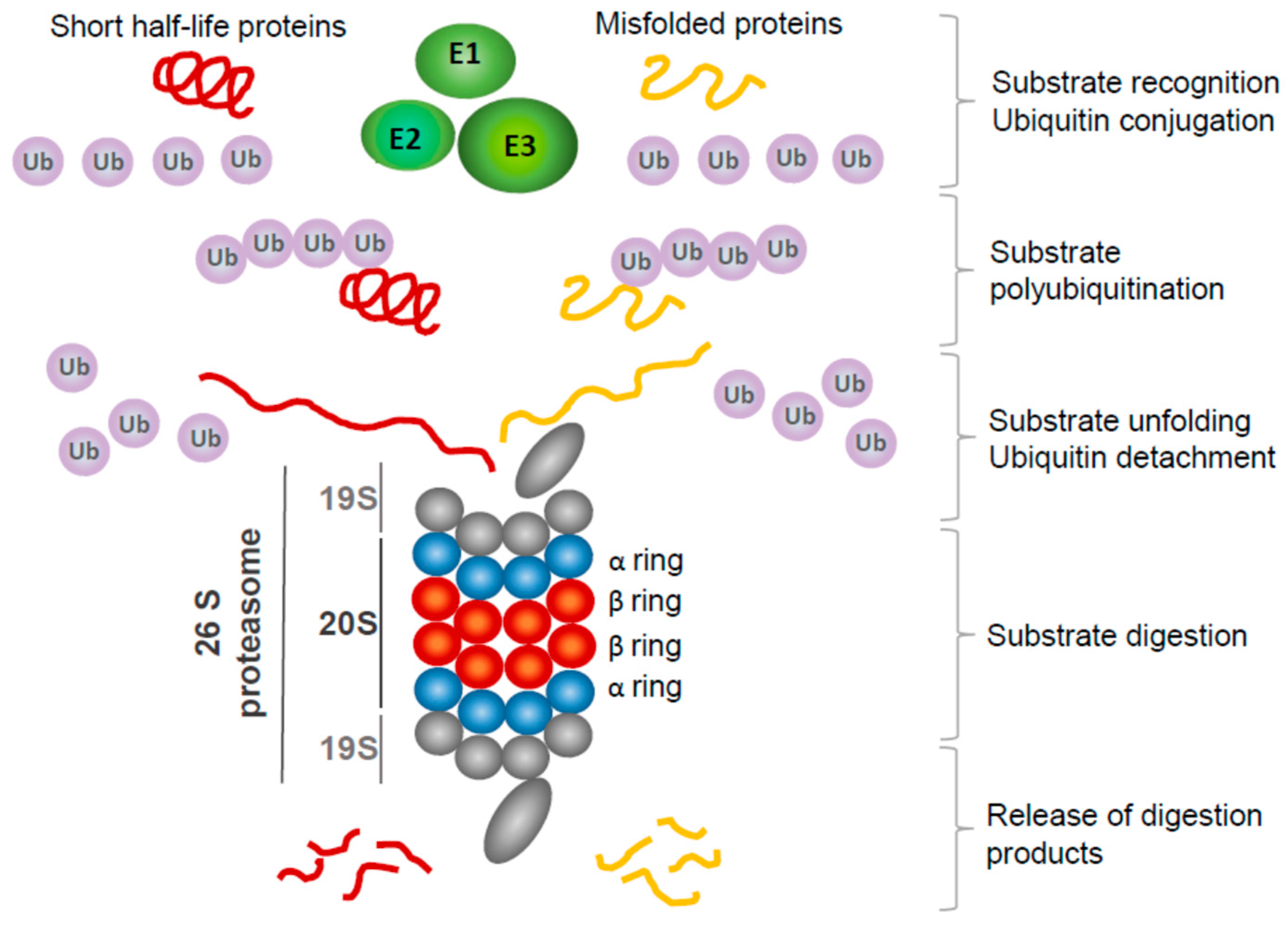
2. Molecular Mechanisms Underlying Proteasome Inhibitor Toxicity in GBM Cells
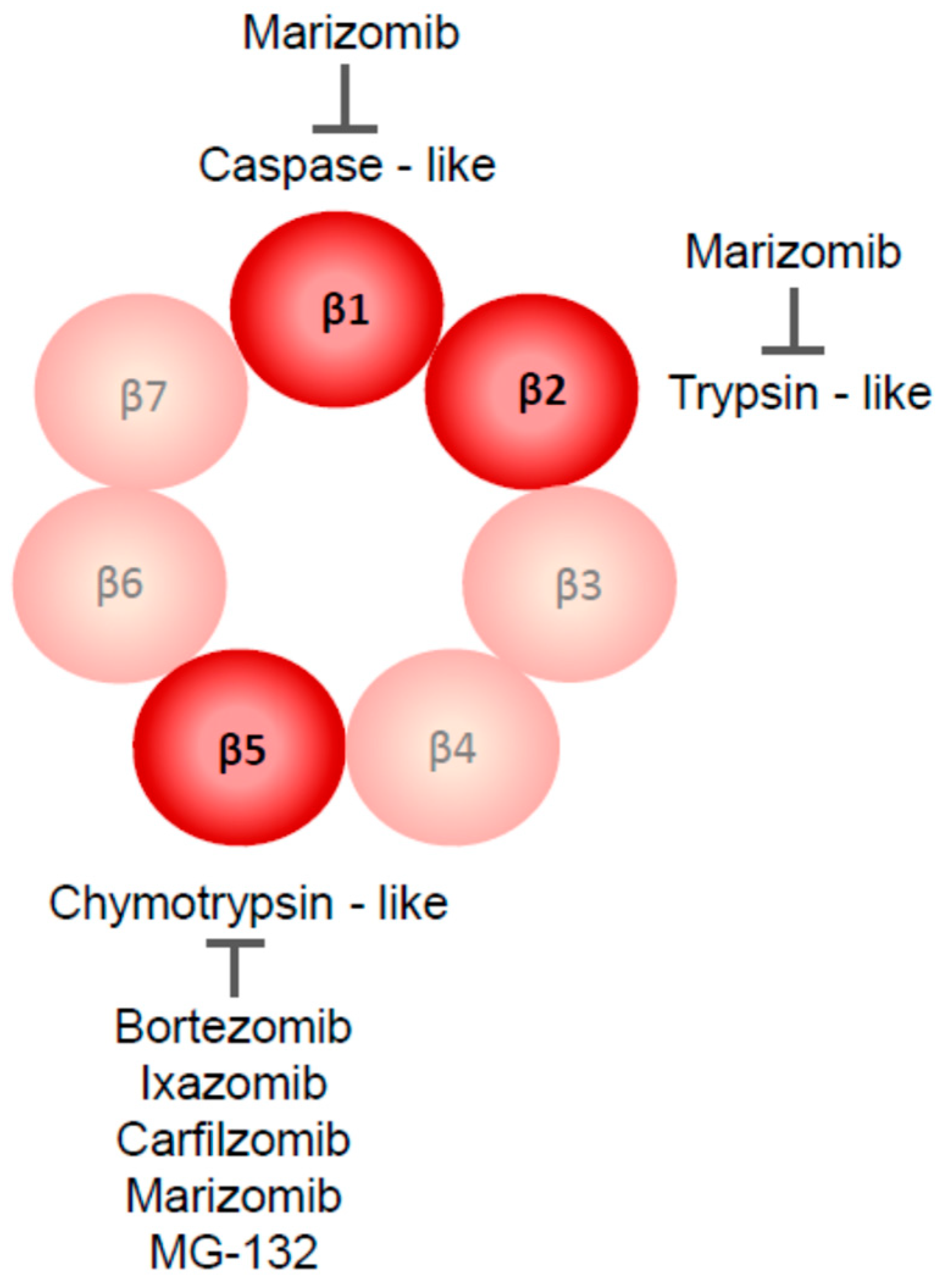
2.1. Proteasome Inhibitors Used in GBM Research
2.2. Proteasome Inhibition Leads to Cell Cycle Arrest in GBM Models
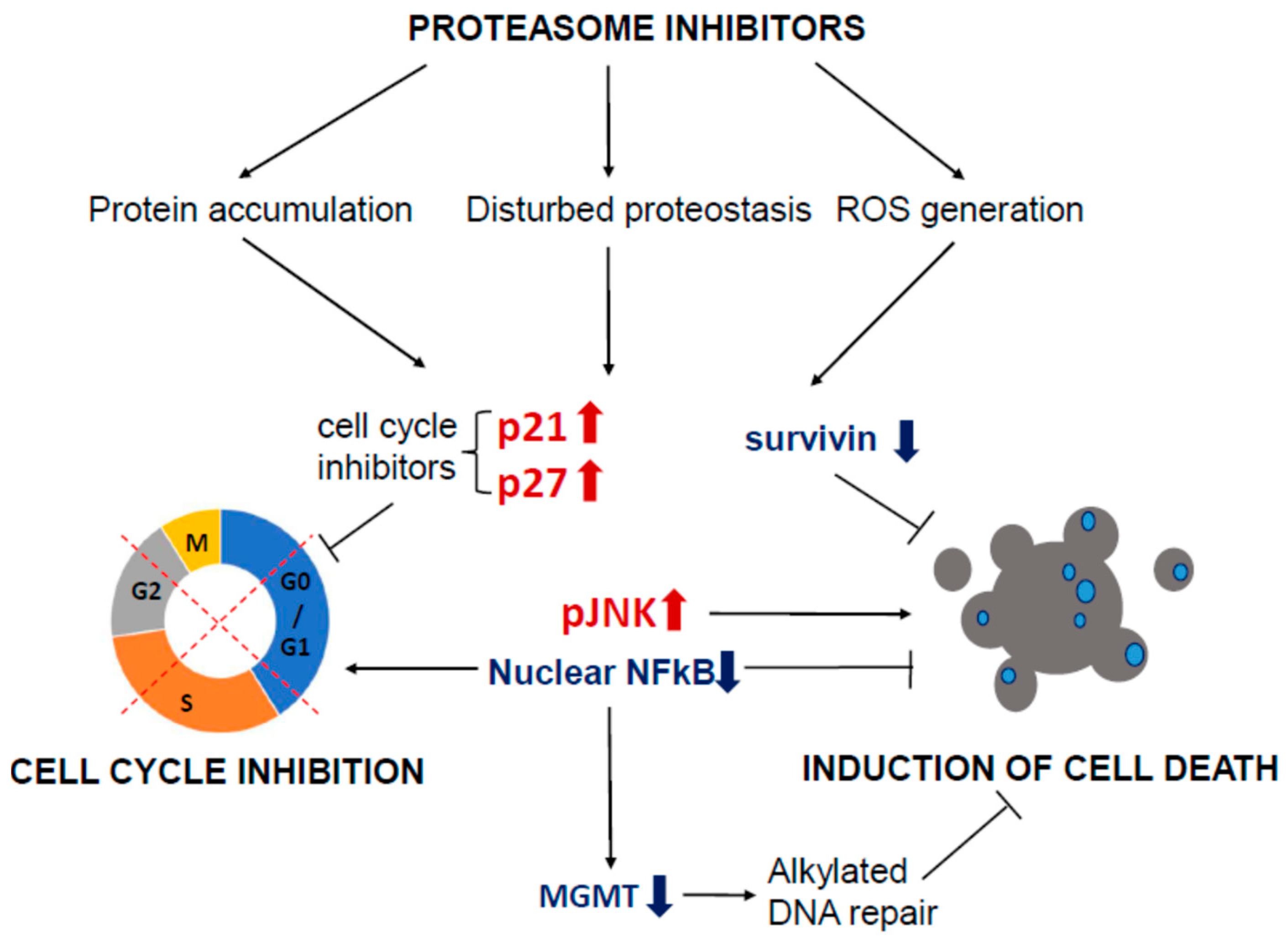
2.3. Mechanisms of Proteasome Inhibitors Triggered GBM Cell Death
2.4. Evaluation of Proteasome Inhibitors’ Potential in Animal GBM Models
2.5. Potentiation of TMZ Toxicity by Proteasome Inhibitors
2.6. Synergistic Action of Proteasome Inhibitors and Other Potential Anti-GBM Drugs
2.7. Potential Mechanisms Impeding Proteasome Efficiency as Anti-GBM Agents
3. Clinical Research on Proteasome Inhibitors as Anti-GBM Agents
3.1. Evaluation of Bortezomib Safety Profile and Therapeutic Efficacy in GBM Patients
3.2. Evaluation of Marizomib Safety Profile and Efficacy in GBM Patients
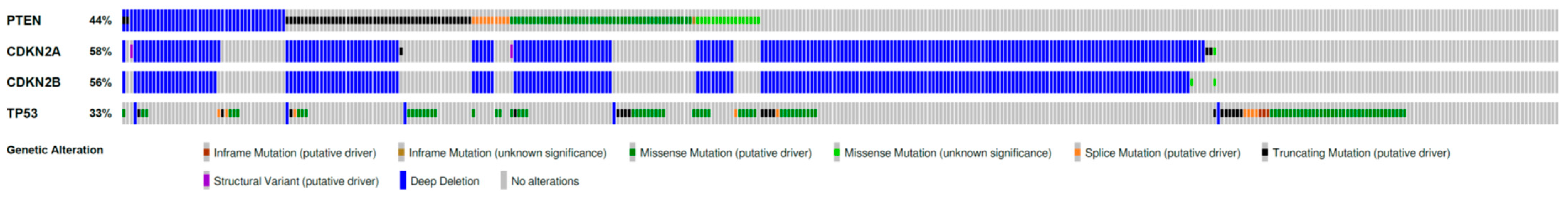
4. Perspectives for Proteasome Inhibitors as Precision Anti-GBM Drugs
5. Concluding Remarks and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Lodi, R.; Bartolini, S.; Brandes, A.A. Glioblastoma: Emerging Treatments and Novel Trial Designs. Cancers 2021, 13, 3750. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Hanna, J.; Guerra-Moreno, A.; Ang, J.; Micoogullari, Y. Protein Degradation and the Pathologic Basis of Disease. Am. J. Pathol. 2019, 189, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–Proteasome System (UPS) as a Target for Anticancer Treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Maksoud, S. The Role of the Ubiquitin Proteasome System in Glioma: Analysis Emphasizing the Main Molecular Players and Therapeutic Strategies Identified in Glioblastoma Multiforme. Mol. Neurobiol. 2021, 58, 3252–3269. [Google Scholar] [CrossRef]
- Thaker, N.G.; Zhang, F.; McDonald, P.R.; Shun, T.Y.; Lewen, M.D.; Pollack, I.F.; Lazo, J.S. Identification of Survival Genes in Human Glioblastoma Cells by Small Interfering RNA Screening. Mol. Pharmacol. 2009, 76, 1246–1255. [Google Scholar] [CrossRef]
- Leonardo-Sousa, C.; Carvalho, A.N.; Guedes, R.A.; Fernandes, P.M.P.; Aniceto, N.; Salvador, J.A.R.; Gama, M.J.; Guedes, R.C. Revisiting Proteasome Inhibitors: Molecular Underpinnings of Their Development, Mechanisms of Resistance and Strategies to Overcome Anti-Cancer Drug Resistance. Molecules 2022, 27, 2201. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Kisselev, A.F. Site-Specific Proteasome Inhibitors. Biomolecules 2021, 12, 54. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Voutsadakis, I.A.; Papandreou, C.N. The Ubiquitin-Proteasome System in Glioma Cell Cycle Control. Cell Div. 2012, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, N.; Mishra, D.P. Therapeutic Targeting of Cancer Cell Cycle Using Proteasome Inhibitors. Cell Div. 2012, 7, 26. [Google Scholar] [CrossRef]
- Yin, D.; Zhou, H.; Kumagai, T.; Liu, G.; Ong, J.M.; Black, K.L.; Koeffler, H.P. Proteasome Inhibitor PS-341 Causes Cell Growth Arrest and Apoptosis in Human Glioblastoma Multiforme (GBM). Oncogene 2005, 24, 344–354. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Braganhol, E.; Battastini, A.M.O.; Moreira, J.C.F. Proteasome Inhibitor MG132 Induces Selective Apoptosis in Glioblastoma Cells through Inhibition of PI3K/Akt and NFkappaB Pathways, Mitochondrial Dysfunction, and Activation of P38-JNK1/2 Signaling. Invest. New Drugs 2012, 30, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-H.; Yang, L.; Chen, J.-X.; Li, Q.-R.; Zhu, L.-R.; Xu, Q.-F.; Huang, G.-H.; Zhang, Z.-X.; Xiang, Y.; Du, L.; et al. Bortezomib Inhibits Growth and Sensitizes Glioma to Temozolomide (TMZ) via down-Regulating the FOXM1–Survivin Axis. Cancer Commun. 2019, 39, 81. [Google Scholar] [CrossRef]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef]
- Manton, C.A.; Johnson, B.; Singh, M.; Bailey, C.P.; Bouchier-Hayes, L.; Chandra, J. Induction of Cell Death by the Novel Proteasome Inhibitor Marizomib in Glioblastoma in Vitro and in Vivo. Sci. Rep. 2016, 6, 18953. [Google Scholar] [CrossRef]
- Mello, S.S.; Attardi, L.D. Deciphering P53 Signaling in Tumor Suppression. Curr. Opin. Cell Biol. 2018, 51, 65–72. [Google Scholar] [CrossRef]
- Manu, K.; Cao, P.; Chai, T.; Casey, P.; Wang, M. P21cip1/Waf1 Coordinates Autophagy, Proliferation and Apoptosis in Response to Metabolic Stress. Cancers 2019, 11, 1112. [Google Scholar] [CrossRef]
- Fan, W.; Hou, Y.; Meng, F.; Wang, X.; Luo, Y.; Ge, P. Proteasome Inhibitor MG-132 Induces C6 Glioma Cell Apoptosis via Oxidative Stress. Acta Pharmacol. Sin. 2011, 32, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Mattes, M.; Lagadec, C.; Donna, L.D.; Phillips, T.M.; Nikolay, P.; McBride, W.H.; Pajonk, F. Differential Effects of the Proteasome Inhibitor NPI-0052 against Glioma Cells. Transl. Oncol. 2010, 3, 50–55. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Li, W.; Wang, C.; Leng, X.; Lian, S.; Feng, J.; Li, J.; Wang, H. Inhibition of Autophagy Enhances Apoptosis Induced by Proteasome Inhibitor Bortezomib in Human Glioblastoma U87 and U251 Cells. Mol. Cell Biochem. 2014, 385, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A. Marizomib Activity as a Single Agent in Malignant Gliomas: Ability to Cross the Blood-Brain Barrier. Neuro Oncol. 2016, 18, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Benitez, J.A.; Finlay, D.; Castanza, A.; Parisian, A.D.; Ma, J.; Longobardi, C.; Campos, A.; Vadla, R.; Izurieta, A.; Scerra, G.; et al. PTEN Deficiency Leads to Proteasome Addiction: A Novel Vulnerability in Glioblastoma. Neuro-Oncol. 2021, 23, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Alexandru, D.; Keir, S.T.; Bigner, D.; Vredenburgh, J.; Friedman, H.S. Proteasome Inhibition with Bortezomib Induces Cell Death in GBM Stem-like Cells and Temozolomide-Resistant Glioma Cell Lines, but Stimulates GBM Stem-like Cells’ VEGF Production and Angiogenesis: Laboratory Investigation. JNS 2013, 119, 1415–1423. [Google Scholar] [CrossRef]
- Yoo, Y.D.; Lee, D.; Cha-Molstad, H.; Kim, H.; Mun, S.R.; Ji, C.; Park, S.H.; Sung, K.S.; Choi, S.A.; Hwang, J.; et al. Glioma-derived Cancer Stem Cells Are Hypersensitive to Proteasomal Inhibition. EMBO Rep. 2017, 18, 150–168. [Google Scholar] [CrossRef]
- Rahman, M.A.; Gras Navarro, A.; Brekke, J.; Engelsen, A.; Bindesbøll, C.; Sarowar, S.; Bahador, M.; Bifulco, E.; Goplen, D.; Waha, A.; et al. Bortezomib Administered Prior to Temozolomide Depletes MGMT, Chemosensitizes Glioblastoma with Unmethylated MGMT Promoter and Prolongs Animal Survival. Br. J. Cancer 2019, 121, 545–555. [Google Scholar] [CrossRef]
- Gras Navarro, A.; Espedal, H.; Joseph, J.; Trachsel-Moncho, L.; Bahador, M.; Tore Gjertsen, B.; Klæboe Kristoffersen, E.; Simonsen, A.; Miletic, H.; Øyvind Enger, P.; et al. Pretreatment of Glioblastoma with Bortezomib Potentiates Natural Killer Cell Cytotoxicity through TRAIL/DR5 Mediated Apoptosis and Prolongs Animal Survival. Cancers 2019, 11, 996. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The Blood–Brain Barrier and Blood–Tumour Barrier in Brain Tumours and Metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Chandler, J.P.; Ferrarese, R.; Grimm, S.A.; Levy, R.M.; Muro, K.; Rosenow, J.; Helenowski, I.; Rademaker, A.; Paton, M.; et al. A Phase II Trial Evaluating the Effects and Intra-Tumoral Penetration of Bortezomib in Patients with Recurrent Malignant Gliomas. J. Neuro-Oncol. 2016, 129, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cho, H.-Y.; Rosenstein-Sisson, R.; Marín Ramos, N.I.; Price, R.; Hurth, K.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. Intratumoral Delivery of Bortezomib: Impact on Survival in an Intracranial Glioma Tumor Model. J. Neurosurg. 2018, 128, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Nam, J.Y.; de Groot, J.F. Treatment of Glioblastoma. JOP 2017, 13, 629–638. [Google Scholar] [CrossRef]
- Cabrini, G.; Fabbri, E.; Nigro, C.L.; Dechecchi, M.C.; Gambari, R. Regulation of Expression of O6-Methylguanine-DNA Methyltransferase and the Treatment of Glioblastoma (Review). Int. J. Oncol. 2015, 47, 417–428. [Google Scholar] [CrossRef]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT Promoter Methylation Status Testing to Guide Therapy for Glioblastoma: Refining the Approach Based on Emerging Evidence and Current Challenges. Neuro-Oncol. 2019, 21, 167–178. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Hatzidaki, E.; Befani, C.D.; Liakos, P.; Papandreou, C.N. Bortezomib Overcomes MGMT-Related Resistance of Glioblastoma Cell Lines to Temozolomide in a Schedule-Dependent Manner. Investig. New Drugs 2013, 31, 1169–1181. [Google Scholar] [CrossRef]
- Rahman, M.A.; Brekke, J.; Arnesen, V.; Hannisdal, M.H.; Navarro, A.G.; Waha, A.; Herfindal, L.; Rygh, C.B.; Bratland, E.; Brandal, P.; et al. Sequential Bortezomib and Temozolomide Treatment Promotes Immunological Responses in Glioblastoma Patients with Positive Clinical Outcomes: A Phase 1B Study. Immun. Inflamm. Dis. 2020, 8, 342–359. [Google Scholar] [CrossRef]
- Jane, E.P.; Premkumar, D.R.; Pollack, I.F. Bortezomib Sensitizes Malignant Human Glioma Cells to TRAIL, Mediated by Inhibition of the NF-κB Signaling Pathway. Mol. Cancer Ther. 2011, 10, 198–208. [Google Scholar] [CrossRef]
- Boccellato, C.; Kolbe, E.; Peters, N.; Juric, V.; Fullstone, G.; Verreault, M.; Idbaih, A.; Lamfers, M.L.M.; Murphy, B.M.; Rehm, M. Marizomib Sensitizes Primary Glioma Cells to Apoptosis Induced by a Latest-Generation TRAIL Receptor Agonist. Cell Death Dis. 2021, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II Trial of Vorinostat in Combination with Bortezomib in Recurrent Glioblastoma: A North Central Cancer Treatment Group Study. Neuro-Oncol. 2012, 14, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib Alone or in Combination with Bevacizumab in Patients with Recurrent Glioblastoma: Phase I/II Clinical Trial Data. Neuro-Oncol. Adv. 2021, 3, vdab142. [Google Scholar] [CrossRef]
- Lin, L.; Gaut, D.; Hu, K.; Yan, H.; Yin, D.; Koeffler, H.P. Dual Targeting of Glioblastoma Multiforme with a Proteasome Inhibitor (Velcade) and a Phosphatidylinositol 3-Kinase Inhibitor (ZSTK474). Int. J. Oncol. 2014, 44, 557–562. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, C.; Chen, S.; Yue, P.; Deng, X.; Lonial, S.; Khuri, F.R.; Sun, S.-Y. Proteasome Inhibitor PS-341 (Bortezomib) Induces Calpain-Dependent IκBα Degradation. J. Biol. Chem. 2010, 285, 16096–16104. [Google Scholar] [CrossRef]
- Leone, A.; Colamaria, A.; Fochi, N.P.; Sacco, M.; Landriscina, M.; Parbonetti, G.; de Notaris, M.; Coppola, G.; De Santis, E.; Giordano, G.; et al. Recurrent Glioblastoma Treatment: State of the Art and Future Perspectives in the Precision Medicine Era. Biomedicines 2022, 10, 1927. [Google Scholar] [CrossRef]
- Kubicek, G.J.; Werner-Wasik, M.; Machtay, M.; Mallon, G.; Myers, T.; Ramirez, M.; Andrews, D.; Curran, W.J.; Dicker, A.P. Phase I Trial Using Proteasome Inhibitor Bortezomib and Concurrent Temozolomide and Radiotherapy for Central Nervous System Malignancies. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 433–439. [Google Scholar] [CrossRef]
- Phuphanich, S.; Supko, J.G.; Carson, K.A.; Grossman, S.A.; Burt Nabors, L.; Mikkelsen, T.; Lesser, G.; Rosenfeld, S.; Desideri, S.; Olson, J.J. Phase 1 Clinical Trial of Bortezomib in Adults with Recurrent Malignant Glioma. J. Neuro-Oncol. 2010, 100, 95–103. [Google Scholar] [CrossRef]
- Portnow, J.; Frankel, P.; Koehler, S.; Twardowski, P.; Shibata, S.; Martel, C.; Morgan, R.; Cristea, M.; Chow, W.; Lim, D.; et al. A Phase I Study of Bortezomib and Temozolomide in Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2012, 69, 505–514. [Google Scholar] [CrossRef][Green Version]
- Kong, X.-T.; Nguyen, N.T.; Choi, Y.J.; Zhang, G.; Nguyen, H.N.; Filka, E.; Green, S.; Yong, W.H.; Liau, L.M.; Green, R.M.; et al. Phase 2 Study of Bortezomib Combined with Temozolomide and Regional Radiation Therapy for Upfront Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Safety and Efficacy Assessment. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1195–1203. [Google Scholar] [CrossRef]
- Roth, P.; Gorlia, T.; Reijneveld, J.C.; De Vos, F.Y.F.L.; Idbaih, A.; Frenel, J.-S.; Le Rhun, E.; Sepulveda Sánchez, J.M.; Perry, J.R.; Masucci, L.; et al. EORTC 1709/CCTG CE.8: A Phase III Trial of Marizomib in Combination with Temozolomide-Based Radiochemotherapy versus Temozolomide-Based Radiochemotherapy Alone in Patients with Newly Diagnosed Glioblastoma. JCO 2021, 39, 2004-2004. [Google Scholar] [CrossRef]
- Utecht, K.N.; Kolesar, J. Bortezomib: A Novel Chemotherapeutic Agent for Hematologic Malignancies. Am. J. Health-Syst. Pharm. 2008, 65, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG Island Methylator Phenotype (G-CIMP): Biological and Clinical Implications. Neuro-Oncol. 2018, 20, 608–620. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef]
- Maksoud, S. The DNA Double-Strand Break Repair in Glioma: Molecular Players and Therapeutic Strategies. Mol. Neurobiol. 2022, 59, 5326–5365. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Trial Phase | Number of Patients | Tumor Type/Status | Treatment Investigated | Year of the Publication | Trial #No. d, Publication |
|---|---|---|---|---|---|---|
| 1. | I | 19 a | Newly diagnosed and recurrent GBM b | Bortezomib (dose escalation study), RT, TMZ | 2009 | [47] |
| 2. | I | 51 b | Recurrent GBM | Bortezomib (dose escalation study), EIASDs | 2010 | [48] |
| 3. | II | 37 | Recurrent GBM | Bortezomib, Vorinostat | 2011 | [42] |
| 4. | I | 25 c | Solid tumors | Bortezomib, TMZ, HEIA’s | 2012 | [49] |
| 5. | II | 10 | Recurrent GBM | Bortezomib, TMZ | 2016 | [32] |
| 6. | II | 24 | Newly diagnosed GBM | Bortezomib, RT, TMZ | 2018 | NCT00998010, ref. [50] |
| 7. | I/II | 10 | Recurrent GBM | Bortezomib, TMZ | 2020 | NCT03643549, ref. [39] |
| 8. | I/II | 30 | Recurrent GBM | Marizomib (dose escalation study) | 2021 | NCT02330562, ref. [43] |
| 9. | I/II | 67 | Recurrent GBM | Marizomib, Bevacizumab | 2021 | NCT02330562, ref. [43] |
| 10. | III | 749 | Newly diagnosed GBM | Marizomib, RT, TMZ | 2021 | NCT03345095, ref. [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gozdz, A. Proteasome Inhibitors against Glioblastoma—Overview of Molecular Mechanisms of Cytotoxicity, Progress in Clinical Trials, and Perspective for Use in Personalized Medicine. Curr. Oncol. 2023, 30, 9676-9688. https://doi.org/10.3390/curroncol30110702
Gozdz A. Proteasome Inhibitors against Glioblastoma—Overview of Molecular Mechanisms of Cytotoxicity, Progress in Clinical Trials, and Perspective for Use in Personalized Medicine. Current Oncology. 2023; 30(11):9676-9688. https://doi.org/10.3390/curroncol30110702
Chicago/Turabian StyleGozdz, Agata. 2023. "Proteasome Inhibitors against Glioblastoma—Overview of Molecular Mechanisms of Cytotoxicity, Progress in Clinical Trials, and Perspective for Use in Personalized Medicine" Current Oncology 30, no. 11: 9676-9688. https://doi.org/10.3390/curroncol30110702
APA StyleGozdz, A. (2023). Proteasome Inhibitors against Glioblastoma—Overview of Molecular Mechanisms of Cytotoxicity, Progress in Clinical Trials, and Perspective for Use in Personalized Medicine. Current Oncology, 30(11), 9676-9688. https://doi.org/10.3390/curroncol30110702